
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA macrocycle-assisted nanoparticlization process for bulk Ag2S†‡
Xin
He
a,
Yuechao
Wang
b,
Cai-Yan
Gao
a,
Hong
Jiang
b and
Liang
Zhao
*a
aThe Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: zhaolchem@mail.tsinghua.edu.cn
bBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, Institute of Theoretical and Computational Chemistry, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
First published on 11th September 2014
Abstract
We report herein a new nanoparticlization process for the bulk-to-nano transformation of Ag2S by incorporating both top-down and bottom-up approaches. Bulk Ag2S was dissolved in solution with the assistance of a macrocyclic ligand, hexamethylazacalix[6]pyridine (Py[6]), to produce polynuclear silver sulfide cluster aggregates. All Ag–S cluster aggregates obtained in three crystalline complexes were protected by Py[6] macrocycles. Removing the protective Py[6] macrocycles by protonation led to the generation of unconventional Ag–S nanoparticles with a large energy gap. Theoretical calculations by a hybrid DFT method demonstrated that the silver sulfide clusters with high Ag/S ratio exhibited more localized HOMO–LUMO orbitals, which consequently enlarged their band gap energies. These experimental and theoretical studies broaden our understanding of the fabrication of nanomaterials by virtue of the advantages of both bottom-up and top-down methods and meanwhile provide a viable means of adjusting the band gap of binary nanomaterials independent of their size.
Introduction
Silver sulfide, a well-known type of direct and narrow band gap semiconductor,1 has attracted considerable attention due to its good stability, low toxicity2 and extensive potential applications in photovoltaic cells, photoconductors,3 infrared detectors4 and near-infrared imaging.5 Besides a band gap of 0.9–1.1 eV for bulk α-Ag2S,6 diminishing the size of Ag2S to the nanometer scale provides an efficient way to finely tune the band gap of this material based on the quantum confinement effect, thus resulting in many intriguing size-specific optical and optoelectronic properties.7 In this regard, different synthetic methods6e.g. the microemulsion approach8 and the hot injection method9 have been comprehensively explored during the past two decades with the aim of achieving uniformly sized Ag2S nanocrystals. However, the use of exotic ligand- or surfactant-stabilized silver and sulfide ions or their precursors and the requirement for elevated temperature and high pressure in most cases made these synthetic protocols arduous. Obviously, if bulk Ag2S solid could be directly transformed into its nano-sized prototype, this would be a concise and ideal synthetic strategy. However, to the best of our knowledge, no reported method for the synthesis of silver chalcogenide nanomaterials involves such a direct transformation.Recently, the synthesis of silver chalcogenide nanocluster compounds in crystalline form has been reported in the literature.10,11 During the reaction between various silver thiolates (SR) and silylated sulfide sources in the presence of coordinative phosphane ligands, nanometer-sized Ag/S/SR silver clusters can be generated at room temperature, but amorphous Ag2S is obtained on increasing the reaction temperature. Such biased reaction pathways suggest a possible intermediate role of silver sulfide clusters in the synthesis of silver sulfide nano-objects. Inspired by this understanding, we envision that successful transformation of bulk Ag2S to silver sulfide nanomaterials could be initiated with the synthesis of polynuclear silver sulfide clusters directly from bulk Ag2S. A combination of top-down (from bulk Ag2S to silver sulfide clusters) and bottom-up (from silver sulfide clusters to nanomaterials) approaches may facilitate the bulk-to-nano transformation of Ag2S. However, bulk Ag2S is known for its very poor solubility (Ksp = 8 × 10−51 at 25 °C), which makes it a formidable challenge to dissolve bulk Ag2S solid and complete the bulk-to-nano transformation by means of cluster intermediates. Very recently, we found that a new class of macrocycles, azacalix[n]pyridines (Py[n]), exhibited a positive allosteric effect upon binding with metal ions,12 which significantly enhanced their affinity to a multimetallic aggregate and led to the formation of silver acetylide clusters by using slightly soluble polymeric [RC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CAg]n as starting materials.13 We have thus conceived a synthetic strategy to implement the bulk-to-nano transformation of silver sulfide, as illustrated in Scheme 1. A particular polypyridine macrocyclic ligand Py[n] will be used to initially facilitate the formation of macrocycle-protected silver sulfide clusters based on its positive allosteric effect. Upon the interruption of coordination bonds between polypyridine ligands and silver atoms via protonation, the encapsulated silver sulfide clusters may mutually coalesce to finally produce silver sulfide nanoparticles, which can be stabilized by additional surfactants.
CAg]n as starting materials.13 We have thus conceived a synthetic strategy to implement the bulk-to-nano transformation of silver sulfide, as illustrated in Scheme 1. A particular polypyridine macrocyclic ligand Py[n] will be used to initially facilitate the formation of macrocycle-protected silver sulfide clusters based on its positive allosteric effect. Upon the interruption of coordination bonds between polypyridine ligands and silver atoms via protonation, the encapsulated silver sulfide clusters may mutually coalesce to finally produce silver sulfide nanoparticles, which can be stabilized by additional surfactants.
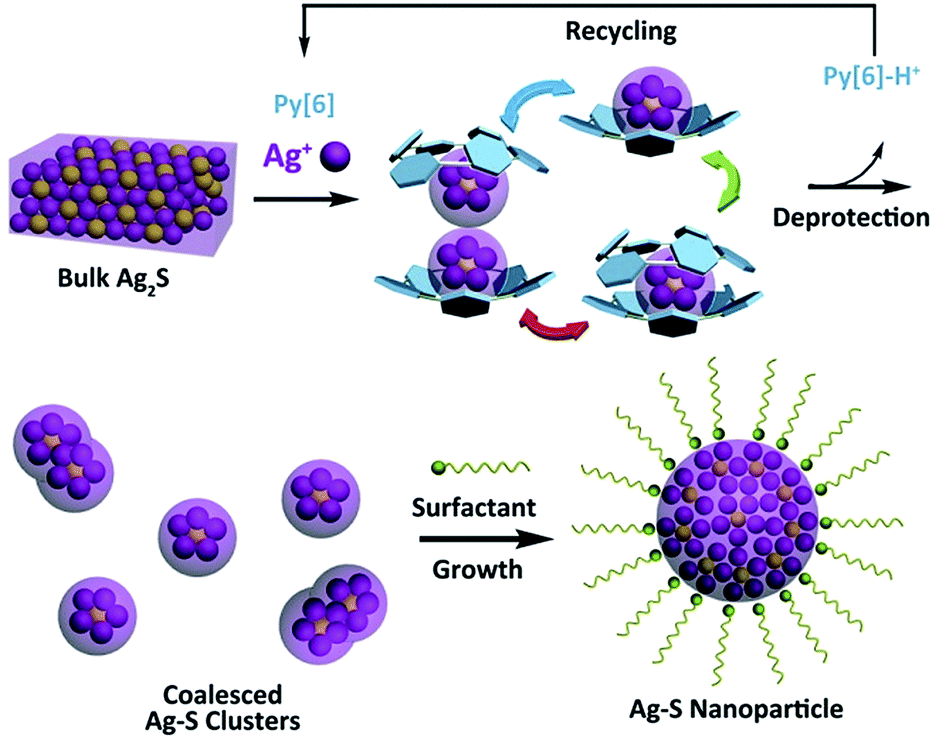 | ||
| Scheme 1 Macrocycle-assisted bulk-to-nano transformation of Ag2S. | ||
Results and discussion
Considering the high efficiency of Py[6] (composed of six 1,3-pyridine rings bridged by six N–CH3 moieties)14 in our previous synthesis of a silver ethynediide cluster-encapsulated supramolecular capsule,13b we firstly attempted to utilize this macrocyclic ligand to dissolve Ag2S solid. When Ag2S solid was added to a methanol solution of AgCF3SO3 (0.2 M), no obvious color change of the solution was observed. However, addition of Py[6] into the Ag2S–AgCF3SO3 mixture led to the appearance of a yellow color very quickly and a gradual decrease in the amount of Ag2S solid, suggesting the dissolution of Ag2S. It should be noted that silver triflate is essential for dissolving Ag2S solid, since mixing only Ag2S with Py[6] does not cause any color change. We next carried out 1H-NMR analysis of the reaction mixture of Ag2S–AgCF3SO3–Py[6]. In the 1H-NMR spectrum, there were three triplet peaks at 8.05 (Ha), 7.87 (Hb) and 7.60 (Hc) ppm corresponding to the pyridyl γ-protons of Py[6] (Fig. 1a). A typical downfield shift of these peaks relative to neat Py[6] (7.46 ppm for pyridyl γ-protons)14 suggested the occurrence of coordination between Py[6] and silver ions. Furthermore, diffusion ordered spectroscopy (DOSY) exhibited two diffusion bands (Fig. 1b), implying the existence of two dominant assembled species in the reaction mixture. As shown in Fig. 1b, the species corresponding to the signal Hb (denoted as A) has a larger diffusion coefficient than the other species (denoted as B) accounting for the signals Ha and Hc. The diameter ratio of A to B in the classical spherical model was deduced as 0.80 based on their respective diffusion coefficients and the Stokes–Einstein equation, which agrees quite well with the ratio (0.78) of the measured distances between the two most separated points of the functional units in the crystal structures of 1 and 2vide infra.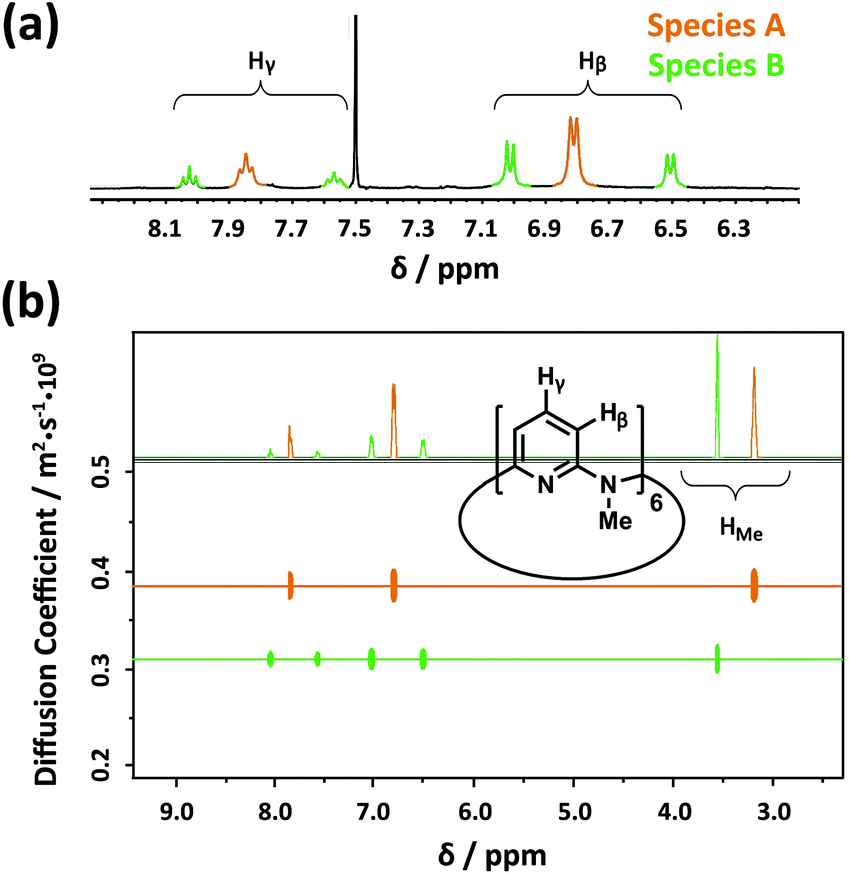 | ||
Fig. 1 (a) Partial 1H NMR spectrum (400 MHz, CDCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol-d4 (v/v) = 1:1, 25.0 °C) and (b) DOSY spectrum of the reaction mixture of AgCF3SO3, Ag2S and Py[6]. :methanol-d4 (v/v) = 1:1, 25.0 °C) and (b) DOSY spectrum of the reaction mixture of AgCF3SO3, Ag2S and Py[6]. | ||
The NMR titration experiment of Py[6] with silver triflate clarified that the species A was actually derived from the assembly of silver triflate and Py[6], due to identical proton NMR spectra (see Fig. S1 in the ESI‡). Single crystals of the species A (denoted as crystalline complex 1) for X-ray crystallographic analysis were deposited from the CH3OH–CH2Cl2 mixed solution of Py[6] and AgCF3SO3. As shown in Fig. 2a, the ratio of silver triflate to Py[6] in complex 1 was determined as 3:1, giving the formula of 1 as {Ag3(Py[6])(CF3SO3)3(H2O)0.5}. The central silver atom Ag1 in 1 adopted a linear coordination geometry to bind with the two opposite pyridines of the Py[6], thus causing the formation of a cage-like structure. This unimolecular folding fashion is similar to our previously reported scenarios for two larger macrocycles Py[8] and Py[9].12
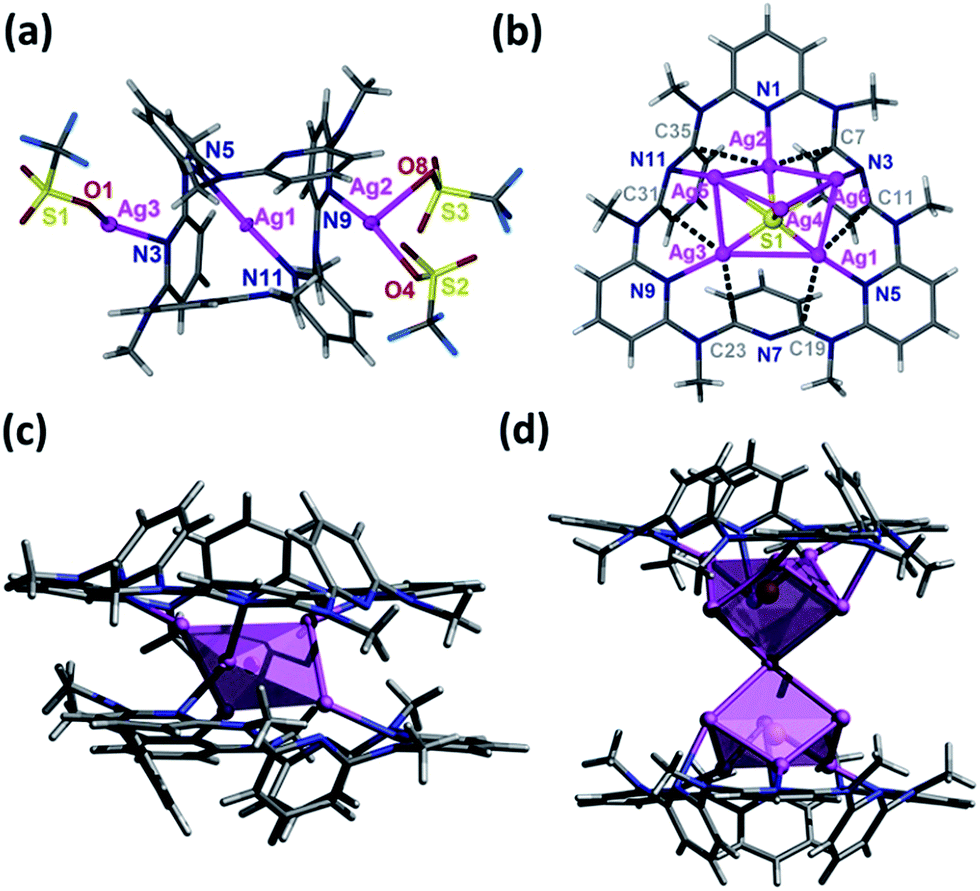 | ||
| Fig. 2 (a) Crystal structure of complex {Ag3(Py[6])(CF3SO3)3(H2O)0.5} (1). Selected bond distances (Å): Ag1–N5 2.154(3); Ag1–N11 2.168(3); Ag2–N9 2.281(4); Ag3–N3 2.284(3). (b) Partial crystal structure of [Ag5S(Py[6])](CF3SO3)3·CH3OH (2). Ag5 and Ag6 each have an occupancy ratio of 0.5. The three triflate groups on the top side of the [Ag5–S] cluster are omitted for clarity. Silver–aromatic π interactions are shown by dashed lines. Ag–C distances (Å): Ag1–C11 2.987; Ag1–C19 2.948; Ag2–C7 2.731; Ag2–C35 2.957; Ag3–C23 2.836; Ag3–C31 3.090. (c) Side view of complex [Ag5S(Py[6])2](CF3SO3)3 (3) with the central silver sulfide cluster represented by a polyhedron. (d) Crystal structure of [Ag12S2(Py[6])2](CF3SO3)8·H2O·CH3OH (4). Two silver atoms each have an occupancy ratio of 0.5. Triflate groups and solvent molecules are omitted for clarity. Color scheme for atoms: Ag, purple; C, gray; H, white; N, blue; S, yellow; F, cyan. | ||
The structural analysis of species B was complicated. High-resolution mass spectroscopy (HR-MS) of the Ag2S–AgCF3SO3–Py[6] yellow reaction mixture revealed two isotopically well-resolved peaks at m/z = 1258.9341 and 554.9862 corresponding to the [(CF3SO3)2Ag3(Py[6])]+ and [(CF3SO3)Ag3(Py[6])]2+ species (Fig. S2‡), confirming the existence of species A. In addition, several peaks corresponding to the species composed of a Py[6] macrocycle and a polynuclear silver sulfide cluster plus some silver triflate groups were found by HR-MS (Fig. S2‡). For example, a strong peak at m/z = 1762.5754 can be ascribed to the species [Ag6S(Py[6])(CF3SO3)3]+ and the peak at m/z = 934.7313 is isotopically in good agreement with the species [Ag7S(Py[6])(CF3SO3)3]2+. We then conducted a crystallization process by adding diethyl ether to the reaction mixture, and a crystalline compound with the formula of [Ag5S(Py[6])](CF3SO3)3·(CH3OH) (2) was finally obtained. The crystal structure of 2 (Fig. 2b) comprises a central sulfur anion that is enclosed by six silver atoms, two of which (Ag5 and Ag6) each have an occupancy ratio of 0.5. This silver sulfide cluster is properly described as a [Ag5–S] aggregate, which is coordinated by a Py[6] macrocycle on one side and is further bonded to three triflate anions on another side. Argentophilic interactions15 and silver–aromatic π interactions both play a significant role in the stabilization of such a [Ag5–S] cluster situated inside a Py[6] macrocycle. Interestingly, a similar crystallization process but with a longer crystallization time than that for complex 2 resulted in a new crystalline complex 3, which has a formula of [Ag5S(Py[6])2](CF3SO3)3 based on its crystal structure analysis. As shown in Fig. 2c, complex 3 also comprises a [Ag5–S] aggregate with C2-axis symmetry, which is protected by two face-to-face Py[6] ligands to give a cluster-embedded supramolecular capsule. To the best of our knowledge, this is the first example of a discrete silver sulfide cluster with an inner penta-coordinated sulfide.16 This [Ag5–S] structural motif is also consistent with the basic structural unit of bulk Ag2S.17 But in contrast, the Ag–S bond lengths in 3 (2.369(3)–2.432(4) Å) are ∼0.2 Å shorter than the values for bulk Ag2S17 and other silver sulfide clusters.11,16,18 Another crystalline complex [Ag12S2(Py[6])2](CF3SO3)8·H2O·CH3OH (4) was serendipitously acquired upon reducing the amount of Py[6] employed in the above synthetic procedure for complex 2. As shown in Fig. 2d, the dumbbell-shaped [Ag12S2] silver sulfide cluster aggregate in 4 can be described as two single sulfide-centered cage-like silver clusters fused by sharing a middle silver atom. In addition, the upper and lower sides of this dumbbell-shaped silver cluster are enclosed by two Py[6] macrocycles similarly to 3. The total seven silver atoms in the asymmetric unit of 4 share a refined site occupancy of six, since silver atom Ag6 is located at a position of special symmetry while Ag7 is disordered, and both have a refined site occupancy ratio of less than one (see ESI for details‡). Despite the structural difference of the silver sulfide clusters between complexes 2–4 both in nuclearity number and cluster configuration, the Py[6] macrocycles in 2–4 all adopt a similar quasi-C3v bowl-shaped conformation. Moreover, we substantiated that most silver sulfide cluster species in the reaction mixture of Ag2S, AgCF3SO3 and Py[6] were stabilized by one or two Py[6] macrocyclic ligands. In addition, based on the HR-MS and 1H-NMR data and the DOSY result that reflected a size ratio (0.80) comparable to the value of 0.78 in the crystal structures of 1 and 2, we hypothesized that complex 2, which is composed of a Py[6] macrocycle and a polynuclear silver sulfide cluster, is probably the second dominant species (species B) in the reaction mixture of Ag2S, AgCF3SO3 and Py[6].
The successful isolation of discrete (in 2 and 3) and joint (in 4) silver sulfide clusters by varying the amount of the protective Py[6] proves the viability of synthesizing Ag–S binary nanoparticles through the coalescence and fusion of silver sulfide clusters. In view of the fact that Py[6] can be easily protonated by a strong acid due to the good Lewis basicity of pyridine, we added CF3COOH into the filtrate of the reaction mixture of Ag2S, AgCF3SO3 and Py[6] to interrupt the coordination interactions between the central silver sulfide cluster and the surrounding Py[6] macrocycles. The dismantlement of Py[6] led to a clear yellow solution. This solution sample retained its yellow color for an hour, but further standing resulted in a black precipitate. The formation and stepwise growth of metal sulfide nanoparticles was affirmed by transmission electron microscopy (TEM) photographs prepared at different intervals (Fig. 3a–b). We subsequently employed oleic amine9 as a surfactant to stabilize the obtained nanoparticles. The resulting yellow solution can retain its solution homogeneity for several days. TEM images of this solution sample (denoted as D-NP) substantiated the formation of metal nanoparticles with an average diameter of 4 ± 0.4 nm (Fig. 3c). Fourier transform IR spectroscopy analysis of a solid sample of D-NP prepared by centrifugation clearly showed the absence of Py[6] and the presence of oleic amine molecules in D-NP (Fig. S3‡). This observation verified our above assumption that Py[6]-protected silver sulfide clusters could indeed undergo a deprotection process to act as nuclei and activated monomers for the fabrication of silver sulfide nanoparticles. Notably, we found that the acidified Py[6] ligands can be recycled after neutralization and extraction and then employed for subsequent bulk-to-nano transformation of silver sulfide.
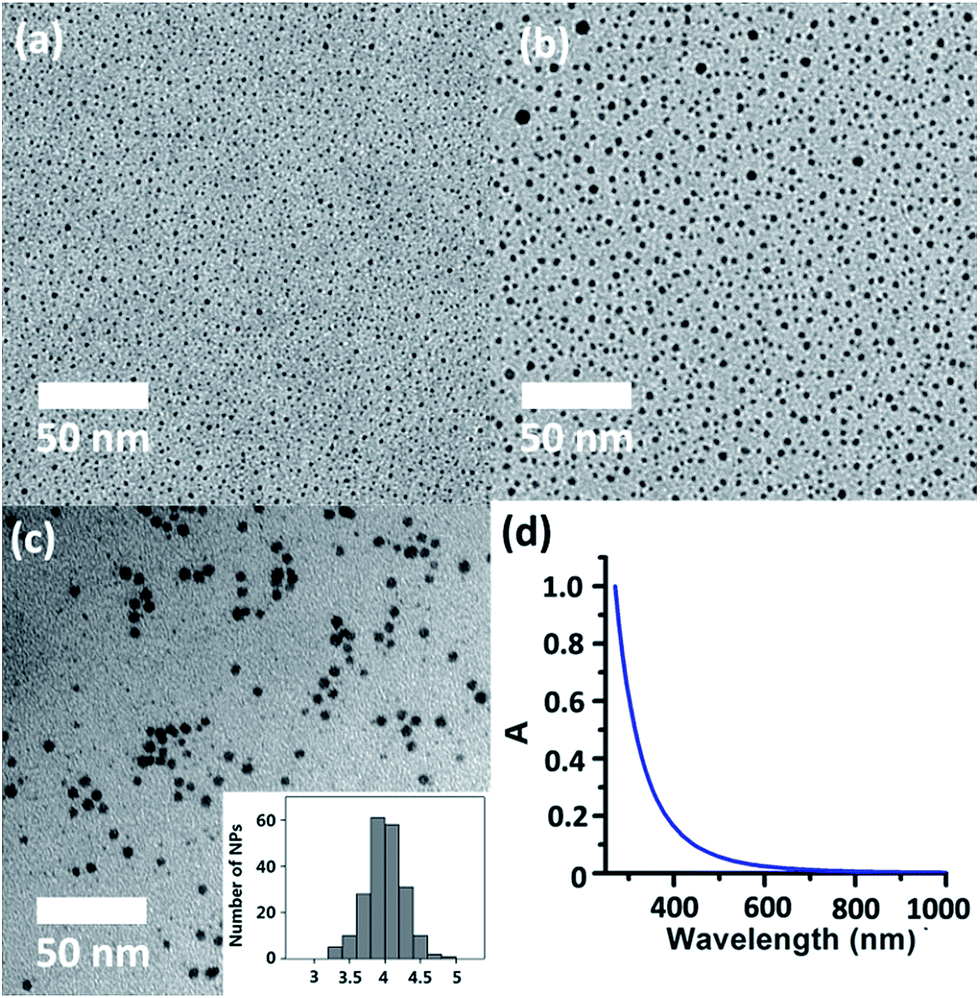 | ||
| Fig. 3 TEM images of deprotected silver sulfide clusters following the addition of CF3COOH (a) after 30 s and (b) after 2 min. (c) TEM images of sample D-NP. (inset) Size-distribution histogram of silver sulfide nanoparticles in D-NP. (d) UV-vis spectrum of D-NP in cyclohexane at 298 K. | ||
On the other hand, high-resolution TEM (HR-TEM) of D-NP showed very ambiguous lattice fringes, in contrast to the clear pattern in previously reported Ag2S nanoclusters,19 suggesting a poorly crystalline form of D-NP. The low crystallinity of D-NP was further substantiated by selected area electron diffraction (SAED) (Fig. S4‡), which exhibited two weak diffraction rings that could be indexed to the (−103) and (232) facets of monoclinic Ag2S (JCPDS card 14-0072). Energy dispersive X-ray spectroscopy (EDX) measurements of D-NP indicated the presence of elements Ag and S and further gave a Ag/S atomic ratio of 3.5 (Fig. S5‡). We also conducted XPS analysis of D-NP. As reflected in the XPS experiment (Fig. S6‡), we confirmed the +1 oxidation state of the silver atoms in D-NP based on their Ag 3d5/2 and Ag 3d3/2 binding energy peaks at 374.5 and 368.5 eV, respectively. In addition, the Ag/S molar ratio was determined to be 3.7 based on the XPS data, which is comparable to the energy dispersive X-ray spectroscopy (EDX) result of 3.5. This Ag/S elemental ratio in D-NP is larger than the values of around 2.0 in bulk Ag2S17 and previously reported silver sulfide nanoclusters.1,3,19 We hypothesized that such a high Ag/S ratio in D-NP may arise from the coalescence and fusion of the silver-rich Ag–S clusters in 2–4 by forming inter-cluster interactions (e.g., argentophilicity) and sharing silver atoms similarly to the above-mentioned scenarios in 4. In addition, the absorption spectrum of the cyclohexane solution of D-NP (Fig. 3d) exhibited a monotonic decrease in the whole recorded range, which is similar to the spectra of a number of reported silver sulfide nanocluster samples.3c,5b The band gap energy could be fitted by using the Bardeen or Tauc equations (Fig. S7‡). The direct transition of D-NP was thus deduced to be 4.0 eV, largely blue-shifted relative to the band gap of bulk α-Ag2S.6 It is noteworthy that modifying the composition ratio of different constituents in nanomaterials in order to perform band gap adjustment has been frequently applied in ternary and quaternary alloyed semiconductor nanomaterials, but rarely in binary systems.20 The method reported herein represents a viable means to tune the band gap of binary nanomaterials independent of their size.
In order to clarify the relationship between the energy gap and the Ag/S ratio in silver sulfide clusters, we carried out a theoretical HOMO–LUMO gap calculation of silver sulfide clusters with different Ag/S ratios via a hybrid density functional theory (DFT) method (see calculation details in the ESI‡). It is highly challenging to construct a structural model and optimize the structure of a 4 nm Ag–S cluster. We thus carried out the calculation on four small Ag–S clusters (Ag8S4, Ag10S5, Ag11S3(OH)5 and Ag12S3(OH)6) with different Ag:S ratios (Fig. 4). The initial structures of the four clusters were built up according to the reported structural motifs of α-Ag2S17 and the crystal structures of silver sulfide clusters.10,11 For the sake of simplifying the structural optimization and energy gap computation, hydroxyl groups were used in substitution of the coordinated peripheral CF3SO3− and CF3CO2− anions in the calculation for silver sulfide clusters with an Ag/S ratio larger than two. As reflected in the calculated results (Table S1‡), the two clusters Ag8S4 and Ag10S5 with an Ag/S ratio of two have HOMO–LUMO energy gaps of 2.45 and 1.86 eV, respectively. In contrast, the other two size-comparable clusters Ag11S3(OH)5 and Ag12S3(OH)6 with an Ag/S ratio larger than 2 have corresponding energy gaps of 3.11 and 3.09 eV. These calculated results agree well with the trend in our above experiment. Compared with the two clusters Ag8S4 and Ag10S5, Ag11S3(OH)5 and Ag12S3(OH)6 with a higher Ag:S elemental ratio exhibited more localized HOMO–LUMO orbitals (Fig. 4 and S8‡). The less dispersed orbitals in these two Ag–S clusters ultimately led to the band gap enlargement.
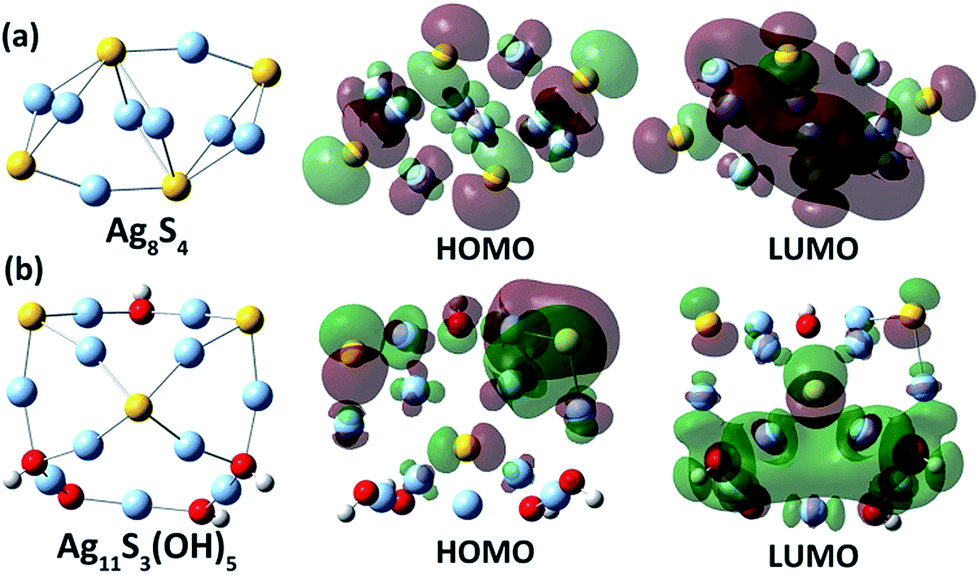 | ||
| Fig. 4 Optimized structures of two Ag–S clusters with different Ag/S elemental ratios, (a) Ag8S4 and (b) Ag11S3(OH)5, and their HOMO and LUMO orbitals. | ||
Conclusions
In summary, we have demonstrated a viable means of synthesizing silver sulfide nanomaterials directly from the bulk form with the assistance of coordinative macrocyclic ligands. This method could be successfully applied in the fabrication of other binary silver nanomaterials such as silver halides, acetylides etc., based on our recent investigation. Considering that the size of macrocyclic ligands can dominate the nuclearity number of the obtained metal cluster aggregates, this approach can also be employed to achieve nanomaterials with different properties dependent on different nucleation centers. Following this study, we foresee the synthesis of numerous new nanomaterials with novel properties based on a synthetic revisiting inspired by the combination of bottom-up and top-down methods reported herein.Acknowledgements
Financial support by the MOST (973 program, 2013CB834501 and 2011CB932501) and NNSFC (91127006, 21132005, 21121004) is gratefully acknowledged. This work is also supported by MOE (NCET-12-0296), Tsinghua University Initiative Scientific Research Program (2011Z02155) and Beijing Higher Education Young Elite Teacher Project (YETP0130). We are grateful to Profs. Mei-Xiang Wang and De-Xian Wang for their helpful discussion.Notes and references
- (a) K. Akamatsu, S. Takei, M. Mizuhata, A. Kajinami, S. Deki, S. Takeoka, M. Fujii, S. Hayashi and K. Yamamoto, Thin Solid Films, 2000, 359, 55 CrossRef CAS; (b) W. P. Lim, Z. Zhang, H. Y. Low and W. S. Chin, Angew. Chem., Int. Ed., 2004, 43, 5685 CrossRef CAS PubMed.
- P. Hirsch, Environ. Toxicol. Chem., 1998, 17, 601 CrossRef.
- (a) K. Terabe, T. Hasegawa, T. Nakayama and M. Aono, Nature, 2005, 433, 47 CrossRef CAS PubMed; (b) F. Gao, Q. Y. Lu and D. Y. Zhao, Nano Lett., 2003, 3, 85 CrossRef CAS; (c) V. M. Huxter, T. Mirkovic, P. S. Nair and G. D. Scholes, Adv. Mater., 2008, 20, 2439 CrossRef CAS; (d) W. J. Lou, X. B. Wang, M. Chen, W. M. Liu and J. C. Hao, Nanotechnology, 2008, 19, 225607 CrossRef PubMed; (e) J. H. Xiang, H. Q. Cao, Q. Z. Wu, S. C. Zhang, X. R. Zhang and A. A. R. Watt, J. Phys. Chem. C, 2008, 112, 3580 CrossRef CAS; (f) Y. Lei, H. Jia, W. He, Y. Zhang, L. Mi, H. Hou, G. Zhu and Z. Zheng, J. Am. Chem. Soc., 2012, 134, 17392 CrossRef CAS PubMed; (g) B. Liu, D. Wang, Y. Zhang, H. Fan, Y. Lin, T. Jiang and T. Xie, Dalton Trans., 2013, 42, 2232 RSC; (h) P.-J. Wu, J.-W. Yu, H.-J. Chao and J.-Y. Chang, Chem. Mater., 2014, 26, 3485 CrossRef CAS.
- G. Hodes, J. Manassen and D. Cahen, Nature, 1976, 261, 403 CrossRef CAS.
- (a) Y. Du, B. Xu, T. Fu, M. Cai, F. Li, Y. Zhang and Q. Wang, J. Am. Chem. Soc., 2010, 132, 1470 CrossRef CAS PubMed; (b) M. Yarema, S. Pichler, M. Sytnyk, R. Seyrkammer, R. T. Lechner, G. Fritz-Popovski, D. Jarzab, K. Szendrei, R. Resel, O. Korovyanko, M. A. Loi, O. Paris, G. Hesser and W. Heiss, ACS Nano, 2011, 5, 3758 CrossRef CAS PubMed; (c) G. Hong, J. T. Robinson, Y. Zhang, S. Diao, A. L. Antaris, Q. Wang and H. Dai, Angew. Chem., Int. Ed., 2012, 51, 9818 CrossRef CAS PubMed.
- S. V. Kershaw, A. S. Susha and A. L. Rogach, Chem. Soc. Rev., 2013, 42, 3033 RSC.
- (a) A. Tubtimtae, K. Wu, H. Tung, M. Lee and G. Wang, Electrochem. Commun., 2010, 12, 1158 CrossRef CAS PubMed; (b) R. Wang, R. Tangirala, S. Raoux, J. Jordan-Sweet and D. Milliron, Adv. Mater., 2012, 24, 99 CrossRef CAS PubMed; (c) E. Kong, Y. Chang, H. Park and H. Jang, Small, 2014, 10, 1300 CrossRef CAS PubMed.
- (a) V. Buschmann, G. Van Tendeloo, P. Monnoyer and J. B. Nagy, Langmuir, 1998, 14, 1528 CrossRef CAS; (b) X. Jiang, Y. Xie, J. Lu, L. Zhu, W. He and Y. Qian, J. Mater. Chem., 2001, 11, 584 RSC.
- D. Wang, T. Xie, Q. Peng and Y. Li, J. Am. Chem. Soc., 2008, 130, 4016 CrossRef CAS PubMed.
- (a) J. F. Corrigan, O. Fuhr and D. Fenske, Adv. Mater., 2009, 21, 1867 CrossRef CAS; (b) O. Fuhr, S. Dehnen and D. Fenske, Chem. Soc. Rev., 2013, 42, 1871 RSC.
- G. Li, Z. Lei and Q.-M. Wang, J. Am. Chem. Soc., 2010, 132, 17678 CrossRef CAS PubMed.
- X. He, X.-B. Xu, X. Wang and L. Zhao, Chem. Commun., 2013, 49, 7153 RSC.
- (a) C.-Y. Gao, L. Zhao and M.-X. Wang, J. Am. Chem. Soc., 2011, 133, 8448 CrossRef CAS PubMed; (b) C.-Y. Gao, L. Zhao and M.-X. Wang, J. Am. Chem. Soc., 2012, 134, 824 CrossRef CAS PubMed; (c) C.-Y. Gao, X. He, L. Zhao and M.-X. Wang, Chem. Commun., 2012, 48, 8368 RSC; (d) X. He, C.-Y. Gao, M.-X. Wang and L. Zhao, Chem. Commun., 2012, 48, 10877 RSC.
- E.-X. Zhang, D.-X. Wang, Q.-Y. Zheng and M.-X. Wang, Org. Lett., 2008, 10, 2565 CrossRef CAS PubMed.
- (a) M. Jansen, Angew. Chem., Int. Ed. Engl., 1987, 26, 1098 CrossRef; (b) P. Pyykkö, Chem. Rev., 1997, 97, 597 CrossRef PubMed.
- Discrete μ4-S silver cluster: V. W.-W. Yam, K. K.-W. Lo, C.-R. Wang and K.-K. Cheung, Inorg. Chem., 1996, 35, 5116 CrossRef CAS.
- D. Santamaría-Pérez, M. Marqués, R. Chuliá-Jordán, J. M. Menendez, O. Gomis, J. Ruiz-Fuertes, J. A. Sans, D. Errandonea and J. M. Recio, Inorg. Chem., 2012, 51, 5289 CrossRef PubMed.
- R. Ahlrichs, A. Eichhöfer, D. Fenske, O. Hampe, M. M. Kappes, P. Nava and J. Olkowska-Oetzel, Angew. Chem., Int. Ed., 2004, 43, 3823 CrossRef CAS PubMed.
- (a) A. Sahu, L. Qi, M. S. Kang, D. Deng and D. J. Norris, J. Am. Chem. Soc., 2011, 133, 6509 CrossRef CAS PubMed; (b) R. Rica and A. H. Velders, J. Am. Chem. Soc., 2011, 133, 2875 CrossRef PubMed; (c) Y. Zhang, Y. Liu, C. Li, X. Chen and Q. Wang, J. Phys. Chem. C, 2014, 118, 4918 CrossRef CAS.
- (a) J. M. Luther and J. M. Pietryga, ACS Nano, 2013, 7, 1845 CrossRef CAS PubMed; (b) S. J. Oh, N. E. Berry, J.-H. Choi, E. A. Gaulding, T. Paik, S.-H. Hong, C. B. Murray and C. R. Kagan, ACS Nano, 2013, 7, 2413 CrossRef CAS PubMed.
Footnotes |
| † This work is dedicated to Professor Thomas C. W. Mak in celebration of his 78th birthday. |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures and crystal structure determination details. Analytical data, spectra, and images. X-ray crystallographic data for 1–4 in CIF format. CCDC 1006732, 1006826, 1006827 and 1006830. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc01884b |
| This journal is © The Royal Society of Chemistry 2015 |