
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Azidophenyl as a click-transformable redox label of DNA suitable for electrochemical detection of DNA–protein interactions†
Jana
Balintová
a,
Jan
Špaček
b,
Radek
Pohl
a,
Marie
Brázdová
b,
Luděk
Havran
bc,
Miroslav
Fojta
*bc and
Michal
Hocek
*ad
aInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Gilead & IOCB Research Center, Flemingovo nam. 2, CZ-16610 Prague 6, Czech Republic. E-mail: hocek@uochb.cas.cz
bInstitute of Biophysics, v.v.i. Academy of Sciences of the Czech Republic, Kralovopolska 135, 61265 Brno, Czech Republic. E-mail: fojta@ibp.cz
cCentral European Institute of Technology, Masaryk University, Kamenice 753/5, CZ-625 00 Brno, Czech Republic
dDepartment of Organic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 8, CZ-12843 Prague 2, Czech Republic
First published on 16th September 2014
Abstract
New redox labelling of DNA by an azido group which can be chemically transformed to nitrophenyltriazole or silenced to phenyltriazole was developed and applied to the electrochemical detection of DNA–protein interactions. 5-(4-Azidophenyl)-2′-deoxycytidine and 7-(4-azidophenyl)-7-deaza-2′-deoxyadenosine nucleosides were prepared by aqueous-phase Suzuki cross-coupling and converted to nucleoside triphosphates (dNTPs) which served as substrates for incorporation into DNA by DNA polymerase. The azidophenyl-modified nucleotides and azidophenyl-modified DNA gave a strong signal in voltammetric studies, at −0.9 V, due to reduction of the azido function. The Cu-catalyzed click reaction of azidophenyl-modified nucleosides or azidophenyl-modified DNA with 4-nitrophenylacetylene gave nitrophenyl-substituted triazoles, exerting a reduction peak at −0.4 V under voltammetry, whereas the click reaction with phenylacetylene gave electrochemically silent phenyltriazoles. The transformation of the azidophenyl label to nitrophenyltriazole was used for electrochemical detection of DNA–protein interactions (p53 protein) since only those azidophenyl groups in the parts of the DNA not shielded by the bound p53 protein were transformed to nitrophenyltriazoles, whereas those covered by the protein were not.
Introduction
Electrochemical detection of redox-labelled DNA1 is an alternative to fluorescence techniques for DNA sequencing and diagnostics. However, despite the extensive research and number of available oxidizable or reducible labels,2 the redox labelling of DNA often suffers from problems with sensitivity, stability and cross-reactivity of the labels. On the other hand, the use of several labels offers access to direct redox coding of DNA.3 To the best of our knowledge, applications of redox labelling and electrochemistry for studying DNA–protein interactions are still relatively scarce, limited to techniques based on changes in DNA-mediated charge transfer upon the protein binding (developed by J. K. Barton’s group4) and our recent studies utilizing immunoprecipitation at magnetic beads.5 Most known methods for detection and footprinting of those interactions6 are based on specific enzymatic or chemical cleavage of DNA.7Copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC or click reaction) is one of the most important bioorthogonal reactions8 and has been widely used for modifications of oligonucleotides (ONs) and DNA.9 Due to better compatibility with phosphoramidite synthesis, triphosphorylation and polymerase incorporations, base-modified nucleotides bearing an acetylene are typically incorporated into ON or DNA and are then clicked with an azido-derivative of the other component.10 Only recently, 5-azidomethyl-dUTP has been synthesized and used for metabolic labelling through polymerase incorporation and click reaction with a fluorescent acetylene.11 We have envisaged the azido group12 as a new redox label suitable for electrochemical detection but also transformable to another redox label through the click reaction.
Results and discussion
Synthesis of modified nucleosides and triphosphates
Our strategy for the synthesis of labelled ONs and DNA relied on polymerase-catalyzed incorporations13 of base-modified nucleotides. The modified dNTPs, required as substrates, are available through triphosphorylation of modified nucleosides. The synthesis of the azidophenyl-modified nucleosides was based on a Suzuki–Miyaura cross-coupling reaction of the unprotected halogenated nucleosides 5-iodocytidine (dCI) and 7-deaza-7-iodoadenosine (dAI) with 4-azidophenyltrifluoroborate (1).14 The reactions were performed in the presence of a PdCl2(dppf) catalyst and Cs2CO3 in MeOH and gave the desired modified nucleosides (dCA and dAA) in good yields of 58 and 63% (Scheme 1, Table 1, entries 1 and 2). A Huisgen–Sharpless CuAAC reaction15 between the azidophenyl-modified nucleosides (dCA and dAA) and an alkyne (phenylacetylene 2 or 1-ethynyl-4-nitrobenzene 3) in the presence of copper(II) sulfate pentahydrate and sodium ascorbate as a reducing agent in tBuOH–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was used for the synthesis of 1,4-disubstituted 1,2,3-triazoles (dNTP and dNTNO2) in good yields of 40–94% (Scheme 1, Table 1, entries 5–8). The phenyltriazole (in dNTP) was chosen as an electrochemically silent group, whereas the nitrophenyltriazole (in dNTNO2) should be reducible at an electrode due to the nitro group.
:1) was used for the synthesis of 1,4-disubstituted 1,2,3-triazoles (dNTP and dNTNO2) in good yields of 40–94% (Scheme 1, Table 1, entries 5–8). The phenyltriazole (in dNTP) was chosen as an electrochemically silent group, whereas the nitrophenyltriazole (in dNTNO2) should be reducible at an electrode due to the nitro group.
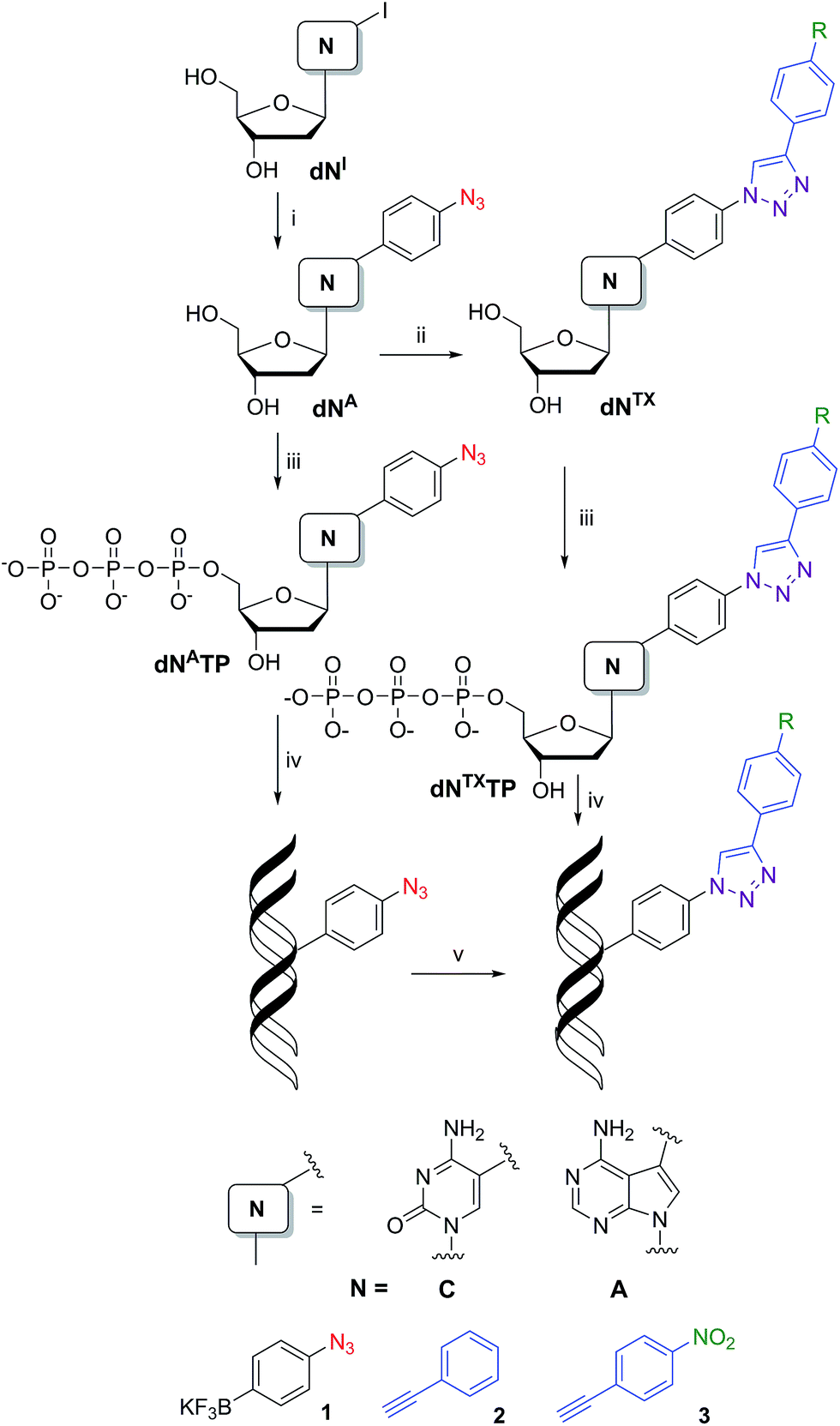 | ||
| Scheme 1 Synthesis of modified nucleosides: (i) Suzuki–Miyaura cross-coupling: 1, PdCl2(dppf), Cs2CO3, MeOH, 2 h, 80 °C; (ii) CuAAC: 2 (or 3), sodium ascorbate, CuSO4·5H2O, tBuOH–H2O (1:1), 12 h, rt; (iii) triphosphorylation of modified nucleosides: (1) PO(OMe)3, POCl3, 0 °C; (2) (NHBu3)2H2P2O7, Bu3N, DMF, 0 °C; (3) TEAB; (iv) PEX experiment; (v) azide–alkyne Huisgen cycloaddition: 2 (or 3), sodium ascorbate, CuBr, TBTA ligand, tBuOH–DMSO (1:3), 2 h, 37 °C. | ||
| Entry | Starting compound | Reagent | Additives | Solvent | Product | Reaction time | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | dAI | 1 | PdCl2(dppf), Cs2CO3 | MeOH | dAA | 2 h, 80 °C | 58 |
| 2 | dCI | 1 | PdCl2(dppf), Cs2CO3 | MeOH | dCA | 2 h, 80 °C | 63 |
| 3 | dAA | 1. PO(OMe)3, POCl3, 0 °C; 2. (NHBu3)2H2P2O7, Bu3N, DMF, 0 °C; 3. TEAB | dAATP | 6 h, 0 °C | 34 | ||
| 4 | dCA | 1. PO(OMe)3, POCl3, 0 °C; 2. (NHBu3)2H2P2O7, Bu3N, DMF, 0 °C; 3. TEAB | dCATP | 6 h, 0 °C | 21 | ||
| 5 | dAA | 2 | Sodium ascorbate, CuSO4·5H2O |
tBuOH–H2O (1:1) |
dATP | 12 h, rt | 72 |
| 6 | dCA | 2 | Sodium ascorbate, CuSO4·5H2O |
tBuOH–H2O (1:1) |
dCTP | 12 h, rt | 40 |
| 7 | dAA | 3 | Sodium ascorbate, CuSO4·5H2O |
tBuOH–H2O (1:1) |
dATNO2 | 12 h, rt | 94 |
| 8 | dCA | 3 | Sodium ascorbate, CuSO4·5H2O |
tBuOH–H2O (1:1) |
dCTNO2 | 12 h, rt | 62 |
| 9 | dATP | 1. PO(OMe)3, POCl3, 0 °C; 2. (NHBu3)2H2P2O7, Bu3N, DMF, 0 °C; 3. TEAB | dATPTP | 18 h, 0 °C | 13 | ||
| 10 | dCTP | 1. PO(OMe)3, POCl3, 0 °C; 2. (NHBu3)2H2P2O7, Bu3N, DMF, 0 °C; 3. TEAB | dCTPTP | 18 h, 0 °C | 52 | ||
| 11 | dATNO2 | 1. PO(OMe)3, POCl3, 0 °C; 2. (NHBu3)2H2P2O7, Bu3N, DMF, 0 °C; 3. TEAB | dATNO2TP | 18 h, 0 °C | 18 | ||
| 12 | dCTNO2 | 1. PO(OMe)3, POCl3, 0 °C; 2. (NHBu3)2H2P2O7, Bu3N, DMF, 0 °C; 3. TEAB | dCTNO2TP | 18 h, 0 °C | 18 | ||
For the preparation of dNATPs, we have applied a triphosphorylation16 of the corresponding nucleosides (dNA). Treatment of dCA or dAA with POCl3 in PO(OMe)3 followed by the addition of (NHBu3)2H2P2O7 and Bu3N, and then treatment with TEAB (Scheme 1) gave the desired dNATPs (Table 1, entries 3 and 4) in 21 and 34% yield after isolation by RP HPLC. Triazole-modified triphosphates dNTPTP and dNTNO2TP were prepared by analogous triphosphorylation of modified nucleosides dNTP and dNTNO2 (Scheme 1, Table 1, entries 9–12) in 13–52% yield.
Electrochemistry of modified dNTPs
All six modified dNTPs dAATP, dCATP, dATPTP, dCTPTP, dATNO2TP and dCTNO2TP were subjected to an electrochemical study using cyclic voltammetry at a hanging mercury drop electrode (HMDE; Fig. 1). The azidophenyl modified nucleotides dAATP and dCATP exerted a strong reduction peak at −0.9 V (peak Nred3), whereas the phenyltriazole derivatives dATPTP and dCTPTP did not produce any redox signals from the label. The nitrophenyltriazole derivatives dATNO2TP and dCTNO2TP gave a strong reduction peak at −0.4 V, due to the reduction of the nitro group (peak NOred2). Since the azidophenyl derivatives are easily transformed to both types of triazole by CuAAC reactions with alkynes, the click reaction with phenylacetylene can be used for silencing of the redox signal of the azido group whereas the click reaction with nitrophenylacetylene can be used for transformation of one redox label (azido) into another (nitro), exerting a different redox potential (vide infra for analytical applications of this finding).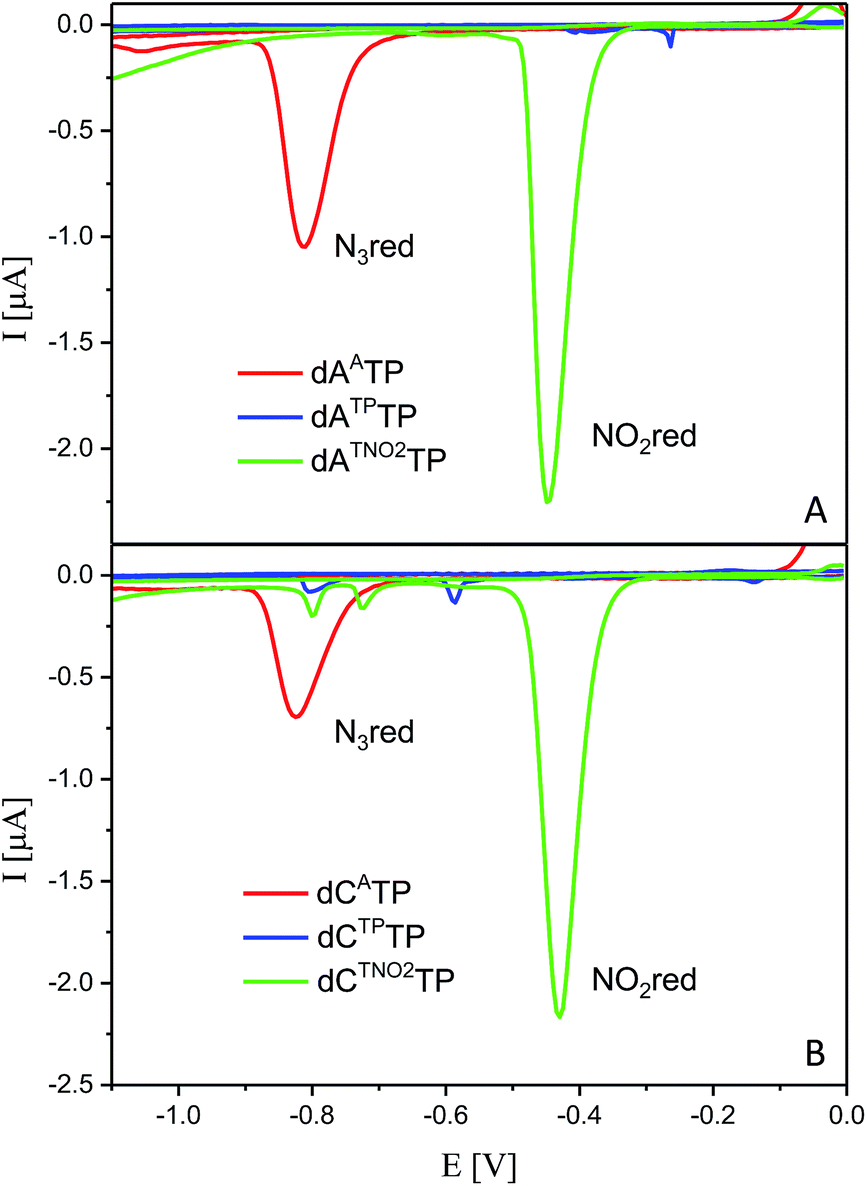 | ||
| Fig. 1 CV responses of dNATPs, dNTPTPs and dNTNOTPs at HMDE. | ||
Enzymatic synthesis of modified DNA
The next goal was the polymerase-catalyzed synthesis of DNA bearing azidophenyl labels and the study of their conversion to (nitro)phenyltriazole groups by CuAAC of the azidophenyl modified DNA with acetylenes 2 or 3. For comparison, the direct incorporation of triazole-modified nucleotides using dNTPTPs and dNTNO2TPs as substrates, leading to the same triazole-modified DNA molecules, was also tried.The enzymatic incorporations of the azidophenyl modified nucleotides were studied using a primer extension (PEX) process, with dNATPs as the substrates together with a 19 nt template, a radiolabeled 15 nt primer and a DNA polymerase, KOD XL (Fig. 2) or Pwo (Fig. S1 in the ESI†), and the products were analyzed by sequencing polyacrylamide gel electrophoresis (PAGE). In all cases fully extended products were obtained.
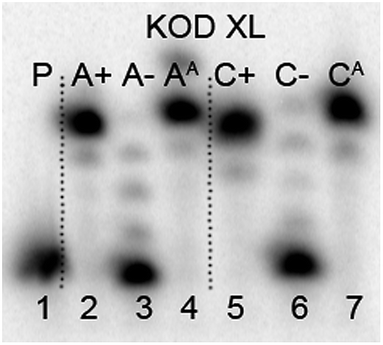 | ||
| Fig. 2 PAGE analysis of PEX single-incorporations into 19 nt DNA using dNATP, tempA and tempC template and KOD XL polymerase. | ||
Then we performed a simple kinetics study to explore the efficiency of the PEX with the modified dNATPs in comparison to natural dNTPs. The rates of the PEX using Pwo DNA polymerase with tempC (for C, without natural dGTP), tempAterm (for A) and primrnd were compared (see Table 2). The reaction mixtures were incubated for the time intervals indicated, and then the reactions were stopped by the addition of PAGE loading buffer and immediate heating. The incorporation of the natural nucleotides was finished in 1–2 minutes whereas the PEX with dNATPs took ca. 1–10 minutes to complete (Fig. S3 and S4 in the ESI†), but the synthesis was always completed within 10 minutes.
| Sequences | |
|---|---|
| a p53 recognition sequences are in italics and nucleotides bearing modification are in bold and underlined. | |
| Primrnd | 5′-CATGGGCGGCATGGG-3′ |
| Temprnd16 | 5′-CTAGCATGAGCTCAGTCCCATGCCGCCCATG-3′ |
| TempA | 5′-CCCTCCCATGCCGCCCATG-3′ |
| TempAterm | 5′-TCCCATGCCGCCCATG-3′ |
| TempC | 5′-CCCGCCCATGCCGCCCATG-3′ |
| Primp53_15 | 5′-GAATTCGATATCAAG-3′ |
| Tempp53_2CON_4 | 5′-TACCTTATCCATAATAGACATGTCTAGACATGTCTCTTGATATCGAATTC-3′ |
| Tempp53_1a2G | 5′-TAGGTTATGGATAATAAACATGTCTAGGCATGTCTCTTGATATCGAATTC-3′ |
| ONp53_2CON_4 | 5′-GAATTCGATATCAAG![[A with combining low line]](https://www.rsc.org/images/entities/b_i_char_0041_0332.gif) GCTGTCTGCTGTCT GCTGTCTGCTGTCT![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) TTTGGTGGT-3′ TTTGGTGGT-3′ |
| ONp53_1a2G | 5′-GAATTCGATATCAAGGCTGCCTGCTGTTTTTTCCTCCT-3′ |
The multiple incorporations of dNA nucleotides into random sequences were tested using a 31 nt template in the presence of KOD XL (Fig. 3), Pwo (Fig. S5 in the ESI†) or Vent (exo-) (Fig. S6 in the ESI†). PEX reactions with both modified dNATPs in the presence of any of these polymerases were successful, giving full-length products in PAGE analyses (Fig. 3, lane 5 and 8).
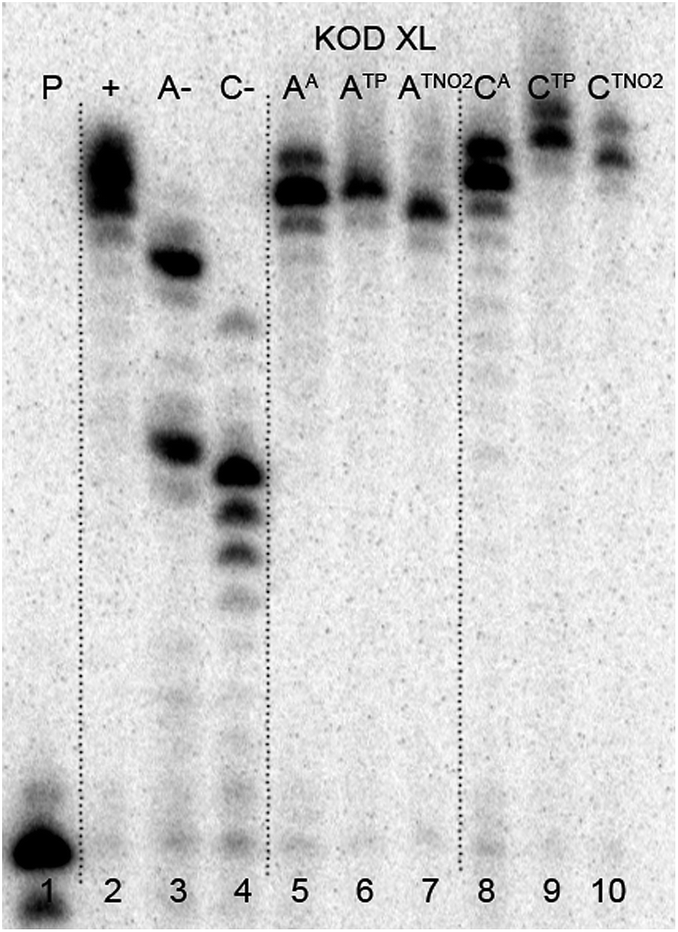 | ||
| Fig. 3 PAGE analysis of PEX incorporations into 31 nt DNA using dNATP, template temprnd16 and KOD XL polymerase, followed by click reaction with 1-ethynyl-4-nitrobenzene and phenylacetylene. | ||
In order to study the CuAAC click modification, the azidophenyl-labelled dsDNA was synthesized on a larger scale (increased 10 times) and the PEX products were purified using a QIAquick Nucleotide Removal Kit to remove the dNTPs. Then the Cu(I)-catalyzed CuAAC reaction of the azidophenyl-labelled dsDNA with phenylacetylene or 1-ethynyl-4-nitrobenzene was performed under standard conditions in the presence of CuBr, TBTA (tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine) ligand and sodium ascorbate, in aqueous tBuOH–DMSO (1:3) at 37 °C for 2 hours. The products were purified once again and analyzed by PAGE (Fig. 3, lane 6, 7, 9, 10) to show no apparent degradation of DNA and MALDI-TOF (see ESI, Fig. S21–S24†) to confirm the conversion.
The direct PEX incorporation of dNTP and dNTNO2 nucleotides into DNA was also studied using either a 19 nt or 31 nt template, KOD XL (Fig. 4 and 5) or Pwo polymerase and dNTPTP or dNTNO2TP as the substrate (Fig. S2 in ESI†). In single incorporations all dNTx nucleotides were successfully incorporated into DNA (Fig. 4, lane 4, 5, 8, 9). In multiple incorporations, dCTxTPs gave fully extended products (Fig. 5, lane 5, 7, 8), whereas for the PEX using dATNO2TP (Fig. 5, lane 6) the product stopped in the same line as the negative control A-, probably due to steric hindrance of the bulky nitrophenyltriazolylphenyl group.
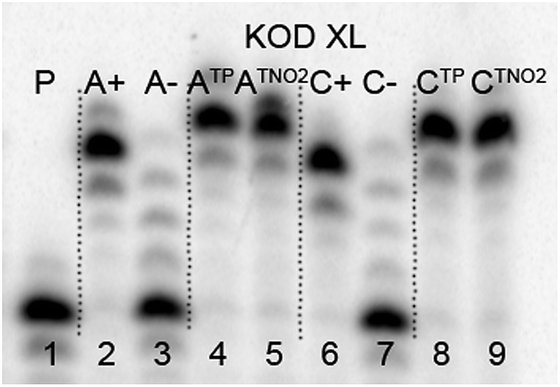 | ||
| Fig. 4 PAGE analysis of PEX single-incorporations into 19 nt DNA using dNTNO2TP or dNTPTP, tempA or tempC template and KOD XL polymerase. | ||
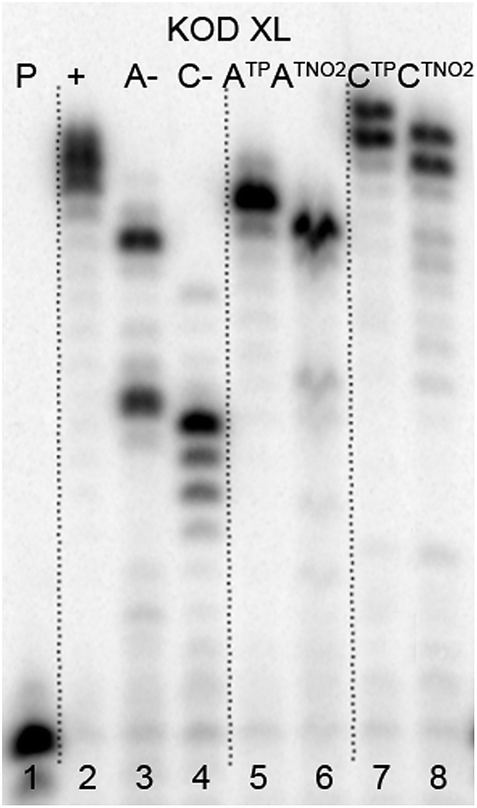 | ||
| Fig. 5 PAGE analysis of PEX reactions with dNTPTP or dNTNO2TP using template temprnd16 and KOD XL polymerase leading to 31 nt DNA. | ||
Electrochemistry of the modified DNA
The voltammetric properties of modified DNA (the PEX products) containing AA or CA were studied by using cyclic voltammetry (CV) at a HMDE. Similarly to the electrochemistry of modified dNxTPs, cyclic voltammograms of PEX products prepared with the temprnd16 template containing azidophenyl-modified nucleobases (AA or CA) show an irreversible cathodic peak at around −0.9 V due to reduction of the azido group (peak Nred3, Fig. 6). A detailed study of the electrode reaction mechanism will be published elsewhere. In a negative control experiment of PEX reactions with no polymerase added to the mixture, no signal corresponding to the azido group was detected, which excludes the presence of unremoved dNATPs in the mixture. PEX products containing azido groups AA or CA were transformed by click reaction to PEX products containing phenyltriazole groups ATP or CTP, with no redox signal from the label being observed (blue curves). On the other hand, the CuAAC click reaction of NA-modified DNA with nitrophenylacetylene provides DNA products containing nitrophenyltriazole groups ATNO2 and CTNO2, which produce the irreversible cathodic peak at around −0.4 V due to the reduction of the nitro group (peak NOred2, Fig. 6).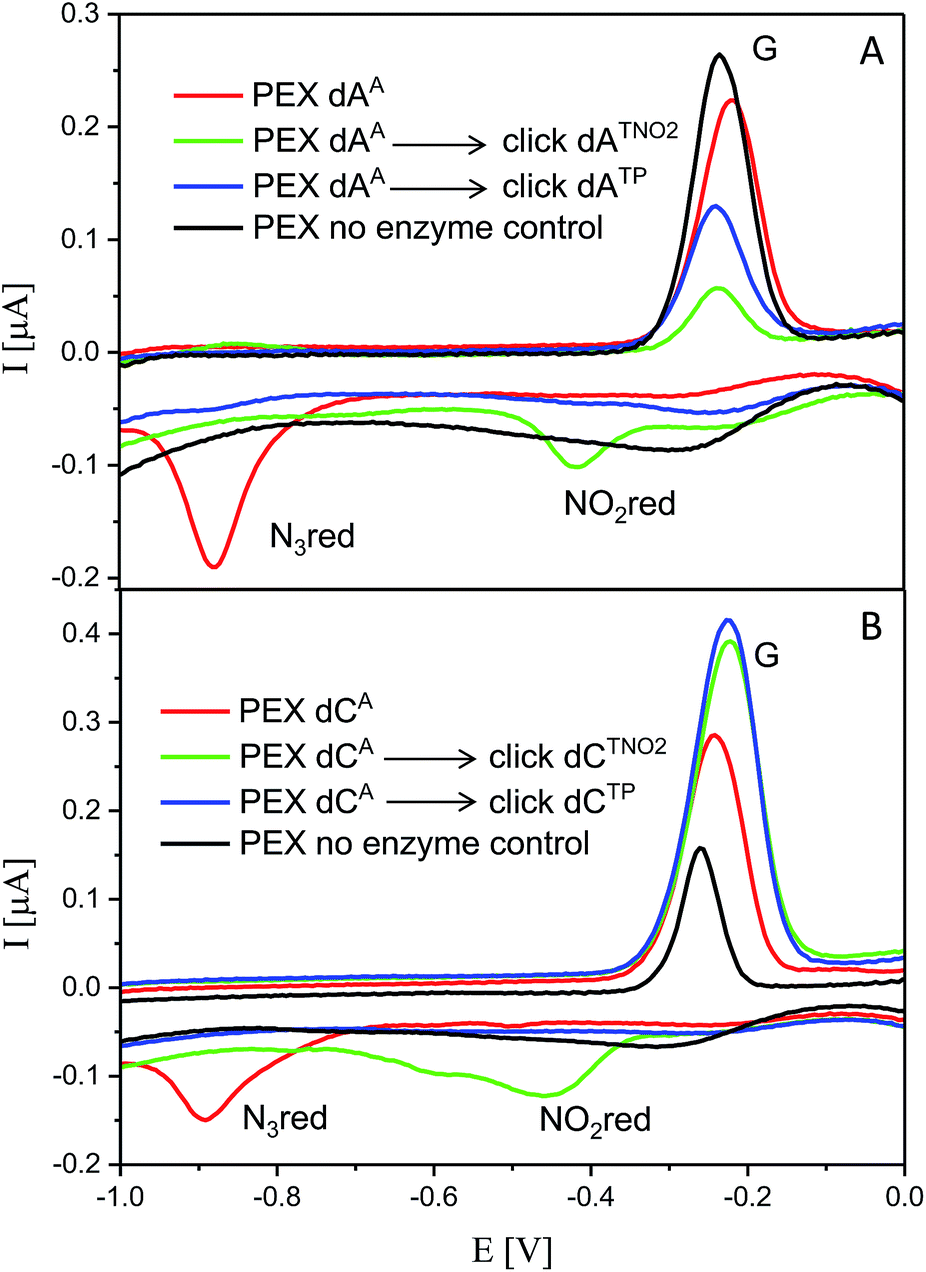 | ||
| Fig. 6 CV responses at a HMDE of PEX products synthesized with a temprnd16 template and dNTP mixes containing a dNATP conjugate (as specified in the legend) complemented with three respective unmodified dNTPs and PEX products after click reaction with (nitro)phenyltriazole. Peak G corresponds to oxidation of a reduction product of guanine generated at the electrode.1a For full CV scans and other details see Fig. S14–S15† and Experimental section. | ||
For comparison, we also prepared PEX products containing the NTP and NTNO2 modifications by polymerase-catalyzed incorporation of the corresponding triazole-modified dNTxTPs. Voltammetric responses of the PEX products were again measured at a HMDE and Fig. 7 confirms the irreversible cathodic peak NOred2 at around −0.4 V corresponds to PEX products containing the nitrophenyltriazole label whereas PEX products containing the phenyltriazole label did not produce any redox signals from the label.
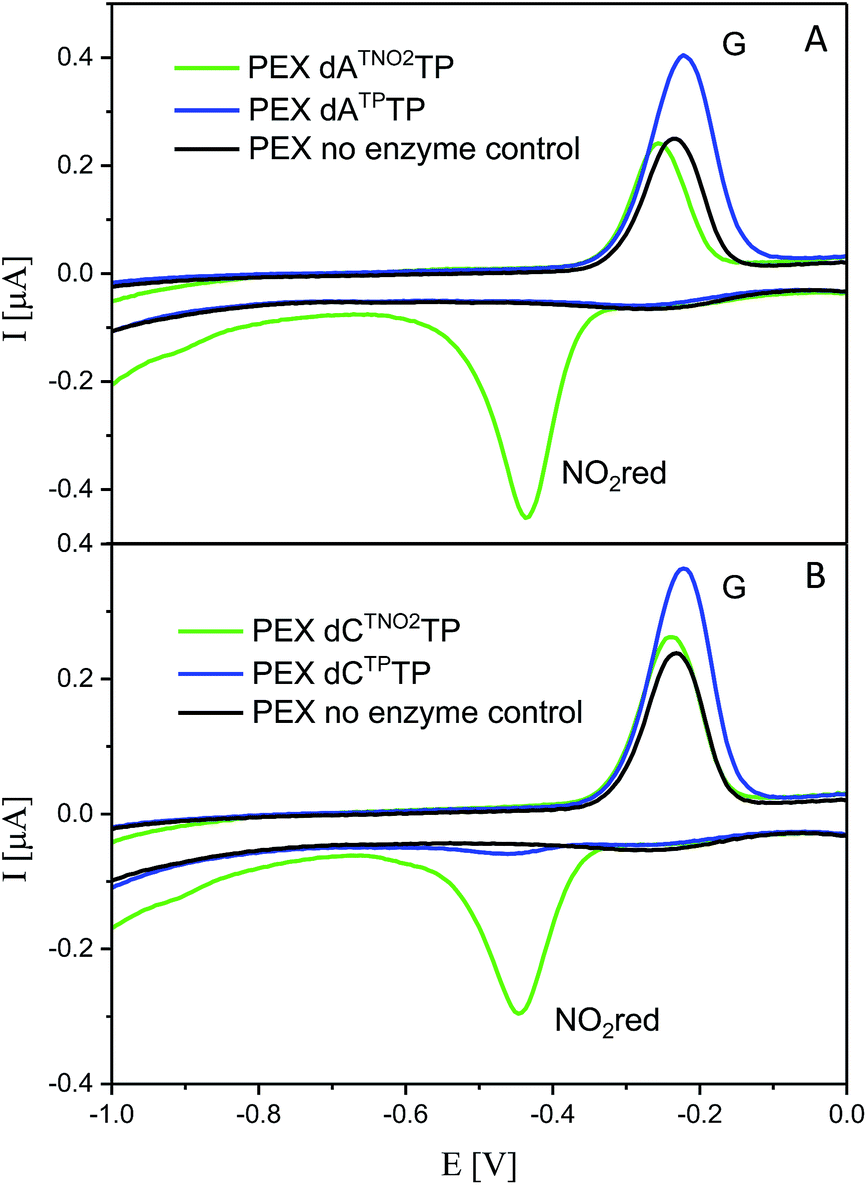 | ||
| Fig. 7 CV responses at a HMDE of PEX products synthesized with a temprnd16 template and dNTP mixes containing a dNTxTP conjugate (as specified in the legend) complemented with three respective unmodified dNTPs. For full CV scans and other details see Fig. S16–S17† and Experimental section. | ||
Application of the click transformations of the redox labels in detection of DNA–protein interactions
DNA–protein interactions are of paramount importance in DNA recombination, transcription, methylation and repair. The current techniques available for footprinting of these interactions are mostly based on specific DNA cleavage.6,7 Based on our encouraging results in the transformation of an azido redox label to nitrophenyltriazole, we envisaged that this approach could be used as a new method for the detection of protein–DNA interactions (Scheme 2). We assumed that if we incorporate several azidophenyl-modifications into a DNA probe, incubate the probe with a protein and then perform the CuAAC click reaction with nitrophenylacetylene, only the freely accessible azido-groups (not shielded by the protein) should be transformed to nitrophenyltriazoles and the ratio of azido/nitro redox signals should indicate whether the protein was bound to the DNA and how large was the sequence of contact. | ||
| Scheme 2 The principle of electrochemical detection of protein–DNA interactions. | ||
To test this idea, we chose a GST-tagged DNA binding (core) domain of tumor-suppressor protein p53 (ref. 17 and 18) (p53CD_GST) as a biologically relevant example of a sequence-specific19 binder to DNA. We have previously shown that p53 retained binding to a specific DNA sequence containing vinylsulfonamide modifications in the major groove, which efficiently cross-linked with a cysteine of p53 through Michael addition.20 We prepared two different sequences of 50-bp DNA by PEX (using KOD XL polymerase and template tempp53_1a2G or tempp53_2CON_4) in which 6 azido-groups are inside and 6 azido-groups are outside of the sequence specifically recognized by p53. Both azido-modified dNATPs gave full length ON-products which were characterized by PAGE (Fig. 8, lane 5 and 6 and Fig. S7 in the ESI†).
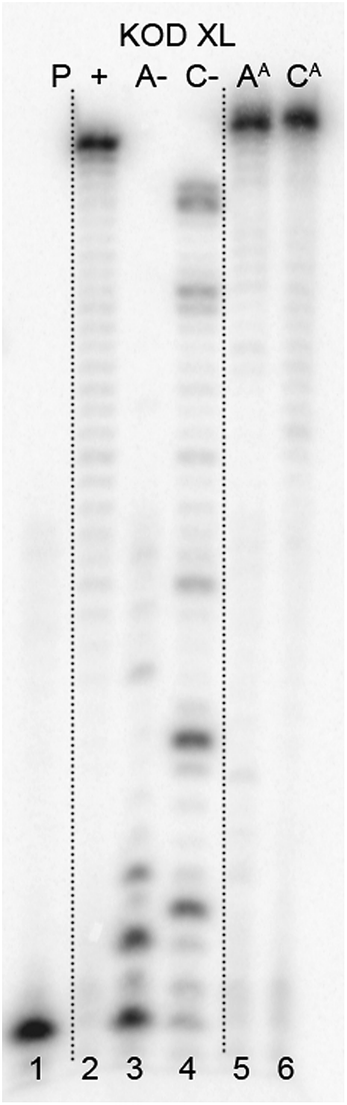 | ||
| Fig. 8 PAGE analysis of PEX reactions with dNATP using template tempp53-1a2G and KOD XL DNA polymerase, giving 50 nt DNA products. | ||
After successful synthesis of the azido-modified DNA, it was incubated with different ratios of p53CD_GST protein to test its binding activity. The recognition of the binding sequence by p53CD_GST was monitored by 6% native PAGE (Fig. 9a, lane 1–10, and Fig. S8 in the ESI†). For further experiments we decided to use 1.2 equiv. of protein p53CD_GST (under these conditions the modified DNA was quantitatively bound by the protein, forming predominantly a 1:1 p53–DNA complex, Fig. 9). Then it was necessary to test the thermal stability of the DNA–protein complex (DNA_p53CD_GST complex), by incubating the created DNA_p53CD_GST complexes at the stated temperatures for 1 h. The DNA_p53CD_GST complex was found to be stable at 20 °C for 1 hour (Fig. S10 and S11,† lane 3). At higher temperatures, the binding of p53 to DNA is inefficient (Fig. S10 and S11,† lane 5).
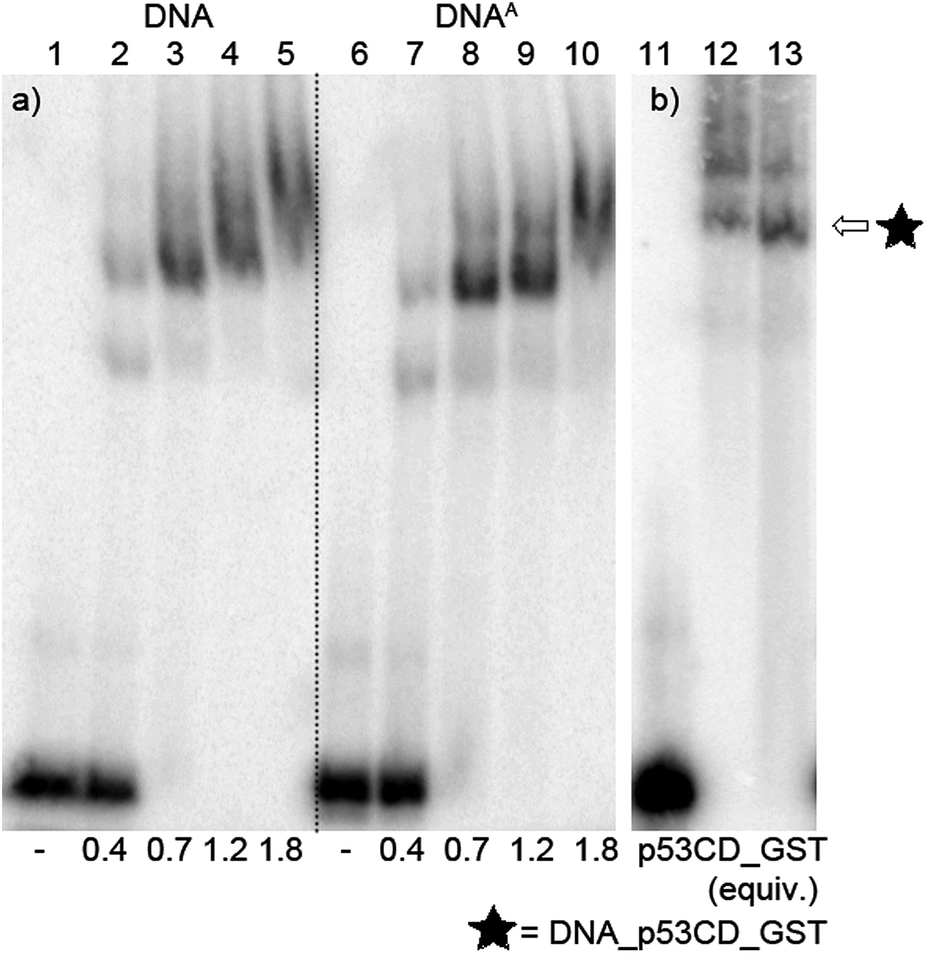 | ||
| Fig. 9 (a) Native PAGE analysis of the 50-mer DNA1a2G_p53CD_GST complex. Lane 1: natural DNA; 2:0.4 equiv.; 3:0.7 equiv.; 4:1.2 equiv.; 5:1.8 equiv. of protein p53CD_GST to DNA; lane 6: DNAA; 7:0.4 equiv.; 8:0.7 equiv.; 9:1.2 equiv.; 10:1.8 equiv. of protein p53CD_GST to DNA. (b) Native PAGE analysis of the stability of the DNA_p53CD_GST complex after click reaction of the DNA. Templatep53_1a2G:lane 11: DNAA; lane 12: protein–DNA complex; lane 13: protein–DNA complex, 0.5 mM 4-nitrophenylacetylene, 5 μM CuBr; 25 μM TBTA ligand, 65 μM Na ascorbate, 20 °C, 1 h. | ||
Transition metals, such as nickel, copper, cobalt and zinc, in high concentrations may also cause the inhibition or disruption of DNA–protein binding.21 Therefore the stability of the DNA_p53CD_GST complex under different CuI concentrations was studied. DNA_p53CD_GST complexes were mixed with different concentrations of CuBr solution in the presence or absence of a TBTA ligand at 20 °C for 1 h. Relatively low concentrations of CuBr (10 μM) prevent the inhibitory effect due to copper from occurring on binding of p53 to DNA (Fig. S12 and S13,† lane 6 and 7). At higher concentrations of CuBr (20 μM), the binding of p53 to DNA is completely inhibited (Fig. S13 and S14,† lane 8 and 9). For the next experiments we decided to use 5 μM CuBr. In the last control experiment, we tested the stability of the DNA_p53CD_GST complex during the CuAAC click reaction. The DNA_p53CD_GST complex was mixed with 0.5 mM 4-nitrophenylacetylene, 5 μM CuBr, 25 μM TBTA ligand, 65 μM sodium ascorbate at 20 °C for 1 h. Fig. 9b, lane 13 (and Fig. S9,† lane 3 in ESI) shows that the DNA–protein complex was stable during the reaction under these conditions.
Then we proceeded to the experiments involving electrochemical detection of DNA–protein interactions. The 50-bp dsDNA containing 12 azidophenyl groups was prepared by PEX with tempp53-1a2G template, and the CV showed the characteristic peak for N3 reduction at −0.9 V (Fig. 10, red curve). This AA-linked DNA was then reacted with nitrophenylacetylene (3) under the previously optimized conditions (suitable for survival of DNA–protein complexes), in the presence of CuBr, TBTA and sodium ascorbate and in the absence of p53CD_GST, to show that full conversion of all the azido-groups to nitrophenyltriazoles occurred, which was confirmed by the disappearance of the signal at −0.9 V and appearance of a new signal at −0.4 V due to reduction of the NO2 group (Fig. 10, green curve). In a further experiment, the AA-linked DNA was incubated with 1.2 equiv. of p53CD_GST (for 45 min on ice) to form a complex and then treated with nitrophenylacetylene (3) under the above mentioned conditions, followed by denaturation. The voltammetric analysis of the product (Fig. 10, violet curve) revealed a ca. 50% decrease in intensity of the peak Nred3 for reduction of azido-group and an increase of the peak NOred2 corresponding to the reduction of the nitro group (to ca. 50% intensity compared to the reaction in the absence of p53). This confirms that only those azido-groups which are not shielded by protein binding can undergo the click transformation to nitrophenyltriazole, whereas the N3 groups covered by the protein remain intact. As a control, we performed the CuAAC reaction of AA-linked DNA in the presence of bovine serum albumin (BSA), which does not bind DNA, and obtained the same results as for the experiment conducted in the absence of any protein (Fig. 10, black curve, all azido groups were converted to nitrophenyltriazoles). Almost identical results were obtained with AA-modified DNA synthesized using the tempp53-2CON4 template (Fig. S19 and S20†).
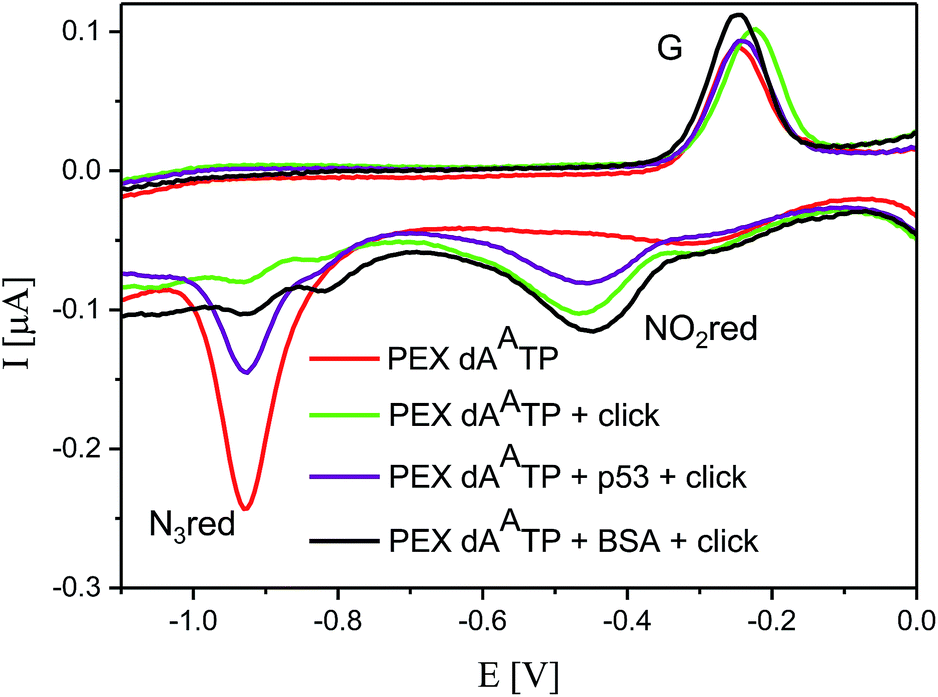 | ||
| Fig. 10 CV responses at a HMDE of PEX products synthesized with tempp53_1a2G template and a dAATP conjugate complemented with three respective unmodified dNTPs (red curve); PEX products after click reaction with nitrophenylacetylene (green curve); DNA–p53 complex after click reaction followed by denaturation (violet curve); and the control with BSA (black curve). For full CV scans and other details see Fig. S18† and Experimental section. | ||
Conclusions
We have designed and prepared nucleosides and dNTPs bearing azidophenyl groups and developed a polymerase mediated incorporation of azido-modified nucleotides into DNA by primer extension using these dNATPs as substrates. Both nucleotides and DNA bearing the azidophenyl modifications exert a strong reduction peak around −0.9 V under voltammetry. The azido-group readily undergoes a CuAAC click reaction with phenyl- or nitrophenylacetylene under conditions compatible to working with dsDNA and DNA–protein complexes. The phenyltriazole-modified dNTPs do not produce any reduction signals in the voltammetric scans, and so the transformation of the azido-modification to a triazole results in silencing of the voltammetric signal. On the other hand, the nitrophenyltriazoles (resulting from CuAAC transformation of the azides with nitrophenylacetylene) give a strong reduction signal at −0.4 V. Thus the azidophenyl modified nucleotides are useful redox labels for DNA, which can be easily either silenced or transformed to a different redox label with a distinctly different redox potential. This transformation was utilized in the detection and mapping of DNA–protein interactions. When an AA-containing DNA is incubated with a protein, binding to a part of its sequence, and then undergoes the CuAAC reaction with nitrophenylacetylene, only the free azido-groups not covered by the protein would react whereas the azides in close contact with the protein remain intact. The electrochemical readout is used for the analysis of the outcome and one can not only distinguish whether or not the protein formed a stable complex with the DNA but also directly deduce the length of the sequence in contact with the particular protein (from the ratio of transformed versus not transformed labels resulting from analysis of changes in intensity of the corresponding redox peaks in voltammetry). Apparently, by the proper design of a series of differently labelled probes, one could even determine the binding sequences for DNA–protein footprinting. Moreover, the method has potential for being conducted in parallel and applied to high-throughput screening of ligands that interfere with protein–DNA binding. Since the azido-group can be photolyzed to reactive nitrenes,22 the azido-modified DNA could also be applied to cross-linking with proteins. Studies along these lines and toward practical applications of this approach will continue in our laboratories.Experimental section
NMR spectra were measured at 500 MHz for 1H and 125.7 MHz for 13C, or at 600 MHz for 1H and 150.9 MHz for 13C when using D2O (reference to dioxane as internal standard, δH = 3.75 ppm, δC = 67.19 ppm) or [D6]DMSO (reference to the residual solvent signal) as the solvent. Chemical shifts are given in ppm (δ scale) and coupling constants (J) in Hz. Complete assignment of all NMR signals was achieved using a combination of H,H-COSY, H,C-HSQC, and H,C-HMBC experiments. Mass spectra were measured with a LCQ classic (Thermo-Finnigan) spectrometer using ESI or a Q-Tof Micro spectrometer (Waters, ESI source, internal calibration with lockspray). Semipreparative separation of nucleoside triphosphates was performed using HPLC on a column packed with 10 μm C18 reversed phase (Phenomenex, Luna C18 (2)). IR spectra were measured using the ATR technique or by using KBr discs. High-resolution mass spectra were measured using an ESI ionization technique. Mass spectra of the functionalized DNA were measured using Maldi-TOF, Reflex IV (Bruker) with a nitrogen laser. Melting points were determined on a Kofler block. Known starting compounds were prepared by literature procedures (compound potassium 4-azidophenyltrifluoroborate14).Method A: Suzuki–Miyaura cross-coupling reaction
Method B: triphosphorylation – synthesis of the modified nucleoside triphosphates
:1) as the eluent. Several co-distillations with water and conversion to the sodium salt form (Dowex 50WX8 in Na+ cycle) followed by freeze-drying from water gave the solid product.
Method C: general procedure for the CuAAC reactions15
:1). The appropriate alkyne (2 equiv.) was subsequently added, and the mixture was stirred overnight at room temperature. The 1,4-disubstituted 1,2,3-triazole derivatives (generally) precipitated from this reaction medium and were isolated by filtration with water.
Method D: triphosphorylation – synthesis of modified nucleoside triphosphates
:1) as the eluent. Several co-distillations with water and conversion to the sodium salt form (Dowex 50WX8 in Na+ cycle) followed by freeze-drying from water gave the solid product.
:1), TBTA ligand (4 μL, 100 mM in DMSO–tBuOH 3:1) and DMSO–tBuOH 3:1 (3 μL). To the DNA solution (50 μL, 50 ng μL−1), a solution of acetylene (phenylacetylene or 1-ethynyl-4-nitrobenzene) (30 μL, 10 mM in DMSO), sodium ascorbate (2 μL, 5 mM in water), pre-complexed Cu(I) and 10 μL DMSO–tBuOH 3:1 were added. The mixture was incubated for 2 h at 37 °C and with 500 rpm stirring. After the reaction, the crude mixture was purified once again and then was desalted using dialysis membranes (Millipore).
:1; 4 °C, 400 V/2.5 hours). Visualization was performed using phosphoimaging (Fig. 9a, Fig. S8†).
:1; 4 °C, 400 V/2.5 hours). Visualization was performed using phosphoimaging.
:1; 4 °C, 400 V/2.5 hours). Visualization was performed using phosphoimaging.
:1, 0.75 μL), TBTA (1 mM in DMSO–tBuOH 3:1, 0.4 μL), sodium ascorbate (5 mM in water, 0.2 μL), KCl (50 mM, 1.5 μL) and tris (5 mM, pH 7.6, 1.5 μL) were added and the reaction mixture was incubated at 20 °C for 1 h. Then glycerol was added (60%, 2 μL) and a part of the reaction mixture (3 μL) was separated by use of a 6% native PAGE (acrylamide/bisacrylamide 37.5:1; 4 °C, 400 V/2.5 hours). Visualization was performed using phosphoimaging. For the electrochemical measurements we applied the same conditions described above but in higher scale (increased 5 times), using three parallel samples for exact comparison (DNA mixed with binding protein, control sample – DNA mixed with BSA). BSA was used as control protein.
Acknowledgements
This work was supported by institutional support from the Academy of Sciences of the Czech Republic (RVO 61388963 and 68081707), by the Czech Science Foundation (P206/12/G151) and by Gilead Sciences, Inc.Notes and references
- (a) E. Paleček, F. Jelen in Electrochemistry of nucleic acids and proteins: Towards electrochemical sensors for genomics and proteomics, ed. E. Paleček, F. Scheller, J. Wang, Elsevier, Amsterdam, 2005, pp. 74–174 Search PubMed; (b) J. Wang, in Electrochemistry of nucleic acids and proteins: Towards electrochemical sensors for genomics and proteomics, ed. E. Paleček, F. Scheller, J. Wang, Elsevier, Amsterdam, 2005, pp. 175–194 Search PubMed; (c) E. Palecek and M. Bartosik, Chem. Rev., 2012, 112, 3427–3481 CrossRef CAS PubMed.
- (a) M. Hocek and M. Fojta, Chem. Soc. Rev., 2011, 40, 5802–5814 RSC; (b) P. Brázdilová, M. Vrábel, R. Pohl, H. Pivoňková, L. Havran, M. Hocek and M. Fojta, Chem.–Eur. J., 2007, 13, 9527–9533 CrossRef PubMed; (c) H. Cahová, L. Havran, P. Brázdilová, H. Pivoňková, R. Pohl, M. Fojta and M. Hocek, Angew. Chem., Int. Ed., 2008, 47, 2059–2062 CrossRef PubMed; (d) J. Balintová, R. Pohl, P. Horáková, P. Vidláková, L. Havran, M. Fojta and M. Hocek, Chem.–Eur. J., 2011, 17, 14063–14073 CrossRef PubMed; (e) H. Macíčková-Cahová, R. Pohl, P. Horáková, L. Havran, J. Špaček, M. Fojta and M. Hocek, Chem.–Eur. J., 2011, 17, 5833–5841 CrossRef PubMed; (f) V. Raindlová, R. Pohl, B. Klepetářová, L. Havran, E. Šimková, P. Horáková, H. Pivoňková, M. Fojta and M. Hocek, ChemPlusChem, 2012, 77, 652–662 CrossRef.
- (a) M. Vrábel, P. Horáková, H. Pivoňková, L. Kalachova, H. Černocká, H. Cahová, R. Pohl, P. Šebest, L. Havran, M. Hocek and M. Fojta, Chem.–Eur. J., 2009, 15, 1144–1154 CrossRef PubMed; (b) J. Balintová, M. Plucnara, P. Vidláková, R. Pohl, L. Havran, M. Fojta and M. Hocek, Chem.–Eur. J., 2013, 19, 12720–12731 CrossRef PubMed.
- (a) A. A. Gorodetsky, M. C. Buzzeo and J. K. Barton, Bioconjugate Chem., 2008, 19, 2285–2296 CrossRef CAS PubMed; (b) H. F. Wang, N. B. Muren, D. Ordinario, A. A. Gorodetsky, J. K. Barton and C. Nuckolls, Chem. Sci., 2012, 3, 62–65 RSC.
- (a) P. Horakova, H. Macickova-Cahova, H. Pivonkova, J. Spacek, L. Havran, M. Hocek and M. Fojta, Org. Biomol. Chem., 2011, 9, 1366–1371 RSC; (b) K. Nemcova, P. Sebest, L. Havran, P. Orsag, M. Fojta and H. Pivonkova, Anal. Bioanal. Chem., 2014, 406, 5843–5852 CrossRef CAS PubMed.
- Review: B. Dey, S. Thukral, S. Krishnan, M. Chakrobarty, S. Gupta, C. Manghani and V. Rani, Mol. Cell. Biochem., 2012, 365, 279–299 CrossRef CAS PubMed.
- (a) D. J. Galas and A. Schmitz, Nucleic Acids Res., 1978, 5, 3157–3170 CrossRef CAS PubMed; (b) R. Sandaltzopoulos and P. B. Becker, Nucleic Acids Res., 1994, 22, 1511–1512 CrossRef CAS PubMed; (c) M. J. Storek, A. Suciu and G. L. Verdine, Org. Lett., 2002, 4, 3867–3869 CrossRef CAS; (d) M. J. Storek, A. Ernst and G. L. Verdine, Nat. Biotechnol., 2002, 20, 183–186 CrossRef CAS PubMed.
- (a) Review: H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) D. K. Scrafton, J. E. Taylor, M. F. Mahon, J. S. Fossey and T. D. James, J. Org. Chem., 2008, 73, 2871–2874 CrossRef CAS PubMed; (c) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
- (a) Review: A. H. El-Sagheer and T. Brown, Chem. Soc. Rev., 2010, 39, 1388–1405 RSC; (b) J. Gierlich, G. A. Burley, P. M. E. Gramlich, D. M. Hammond and T. Carell, Org. Lett., 2006, 8, 3639–3642 CrossRef CAS PubMed; (c) A. H. El-Sagheer and T. Brown, Chem. Commun., 2011, 47, 12057–12058 RSC.
- (a) P. M. E. Gramlich, C. T. Wirges, A. Manetto and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 8350–8358 CrossRef CAS PubMed; (b) P. M. E. Gramlich, S. Warncke, J. Gierlich and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 3442–3444 CrossRef CAS PubMed; (c) F. Seela, V. R. Sirivolu and P. Chittepu, Bioconjugate Chem., 2008, 19, 211–224 CrossRef CAS PubMed; (d) F. Seela and V. R. Sirivolu, Org. Biomol. Chem., 2008, 6, 1674–1687 RSC; (e) K. Gutschmiedl, D. Fazio and T. Carell, Chem.–Eur. J., 2010, 16, 6877–6883 CrossRef PubMed; (f) S. A. Ingale and F. Seela, J. Org. Chem., 2013, 78, 3394–3399 CrossRef CAS PubMed.
- A. B. Neef and N. W. Luedtke, ChemBioChem, 2014, 15, 789–793 CrossRef CAS PubMed.
- Review on chemistry of azides: S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- Selected examples on polymerase incorporations of base-modified dNTPs: (a) S. Jäger, G. Rasched, H. Kornreich-Leshem, M. Engeser, O. Thum and M. Famulok, J. Am. Chem. Soc., 2005, 127, 15071–15082 CrossRef PubMed; (b) S. Obeid, M. Yulikow, G. Jeschke and A. Marx, Angew. Chem., Int. Ed., 2008, 47, 6782–6785 CrossRef CAS PubMed; (c) N. Ramsay, A.-S. Jemth, A. Brown, N. Crampton, P. Dear and P. Holliger, J. Am. Chem. Soc., 2010, 132, 5096–5104 CrossRef CAS PubMed; (d) A. Baccaro and A. Marx, Chem.–Eur. J., 2010, 16, 218–226 CrossRef CAS PubMed; (e) P. Kielkowski, H. Macíčková-Cahová, R. Pohl and M. Hocek, Angew. Chem., Int. Ed., 2011, 50, 8727–8730 CrossRef CAS PubMed; (f) P. Ménová and M. Hocek, Chem. Commun., 2012, 48, 6921–6923 RSC; (g) A. Baccaro, A.-L. Steck and A. Marx, Angew. Chem., Int. Ed., 2012, 51, 254–257 CrossRef CAS PubMed; (h) M. Hollenstein, Chem.–Eur. J., 2012, 18, 13320–13330 CrossRef CAS PubMed; (i) J. Riedl, R. Pohl, N. P. Ernsting, P. Orsag, M. Fojta and M. Hocek, Chem. Sci., 2012, 3, 2797–2806 RSC; (j) S. Obeid, H. Busskamp, W. Welte, K. Diederichs and A. Marx, J. Am. Chem. Soc., 2013, 135, 15667–15669 CrossRef CAS PubMed; (k) Z. Vaníková and M. Hocek, Angew. Chem., Int. Ed., 2014, 53, 6734–6737 CrossRef PubMed; (l) P. Kielkowski, J. Fanfrlík and M. Hocek, Angew. Chem., Int. Ed., 2014, 53, 7552–7555 CrossRef CAS PubMed.
- Y. A. Cho, D. Kim, H. R. Ahn, B. Canturk, G. A. Molander and J. Ham, Org. Lett., 2009, 11, 4330–4333 CrossRef CAS PubMed.
- For related CuAAC reactions of 2-azidopurines, see: L. Cosyn, K. K. Palaniappan, S.-K. Kim, H. T. Duong, Z.-G. Gao, K. A. Jacobson and S. van Calenbergh, J. Med. Chem., 2006, 49, 7373–7383 CrossRef CAS PubMed.
- T. Kovacs and L. Ötvös, Tetrahedron Lett., 1988, 29, 4525–4528 CrossRef CAS.
- (a) G. Matlashewski, P. Lamb, D. Pim, J. Peacock, L. Crawford and S. Benchimol, EMBO J., 1984, 3, 3257–3262 CAS; (b) M. Isobe, B. S. Emanuel, D. Givol, M. Oren and C. M. Croce, Nature, 1986, 320, 84–85 CrossRef CAS PubMed; (c) S. E. Kern, K. W. Kinzler, A. Bruskin, D. Jarosz, P. Friedman, C. Prives and B. Vogelstein, Science, 1991, 252, 1708–1711 CrossRef CAS; (d) O. W. McBride, D. Merry and D. Givol, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 130–134 CrossRef CAS.
- For expression of p53, see: (a) M. Brazdova, J. Palecek, D. I. Cherny, S. Billova, M. Fojta, P. Pecinka, B. Vojtesek, T. M. Jovin and E. Palecek, Nucleic Acids Res., 2002, 30, 4966–4974 CrossRef CAS PubMed; (b) M. Fojta, H. Pivonkova, M. Brazdova, K. Nemcova, J. Palecek and B. Vojtesek, Eur. J. Biochem., 2004, 271, 3865–3876 CrossRef CAS PubMed; (c) C. Klein, G. Georges, K.-P. Künkele, R. Huber, R. A. Engh and S. Hansen, J. Biol. Chem., 2001, 276, 37390–37401 CrossRef CAS PubMed; (d) J. Buzek, L. Latonen, S. Kurki, K. Peltonen and M. Laiho, Nucleic Acids Res., 2002, 30, 2340–2348 CrossRef CAS PubMed; (e) V. Tichy, L. Navratilova, M. Adamik, M. Fojta and M. Brazdova, Biochem. Biophys. Res. Commun., 2013, 433, 445–449 CrossRef CAS PubMed; (f) M. Brazdova, L. Navratilova, V. Tichy, K. Nemcova, M. Lexa, R. Hrstka, P. Pecinka, M. Adamik, B. Vojtesek, E. Palecek, W. Deppert and M. Fojta, PLoS One, 2013, 8, e59567 CAS.
- (a) W. S. El-Deiry, S. E. Kern, J. A. Pietenpol, K. W. Kinzler and B. Vogelstein, Nat. Genet., 1992, 1, 45–49 CrossRef CAS PubMed; (b) Y. Wang, J. F. Schwedes, D. Parks, K. Mann and P. Tegtmeyer, Mol. Cell. Biol., 1995, 15, 2157–2165 CAS; (c) J. L. Kaar, N. Basse, A. C. Joerger, E. Stephens, T. J. Rutherford and A. R. Fersht, Protein Sci., 2010, 19, 2267–2278 CrossRef CAS PubMed.
- J. Dadová, P. Orság, R. Pohl, M. Brázdová, M. Fojta and M. Hocek, Angew. Chem., Int. Ed., 2013, 52, 10515–10518 CrossRef PubMed.
- (a) E. Paleček, M. Brázdová, H. Černocká, D. Vlk, V. Brázda and B. Vojtešek, Oncogene, 1999, 18, 3617–3625 CrossRef PubMed; (b) G. W. Verhaegh, M. J. Richard and P. Hainaut, Mol. Cell. Biol., 1997, 17, 5699–5706 CAS.
- (a) J. Sumranjit and S. J. Chung, Molecules, 2013, 18, 10425–10451 CrossRef CAS PubMed; (b) S.-W. Yang and H. A. Nash, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 12183–12187 CrossRef CAS; (c) R. K. Evans and B. E. Haley, Biochemistry, 1987, 26, 269–276 CrossRef CAS.
- P. Ménová, V. Raindlová and M. Hocek, Bioconjugate Chem., 2013, 24, 1081–1093 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures of PAGE analyses of PEX experiments, additional electrochemistry figures, copies of MALDI spectra, copies of NMR spectra. See DOI: 10.1039/c4sc01906g |
| This journal is © The Royal Society of Chemistry 2015 |