
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpecific methionine oxidation of cytochrome c in complexes with zwitterionic lipids by hydrogen peroxide: potential implications for apoptosis†
Daiana A.
Capdevila‡
a,
Waldemar A.
Marmisollé‡
a,
Florencia
Tomasina
b,
Verónica
Demicheli
b,
Magdalena
Portela
c,
Rafael
Radi
b and
Daniel H.
Murgida
*a
aDepartamento de Química Inorgánica, Analítica y Química Física and INQUIMAE (CONICET-UBA), Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Pab. 2, piso 1, C1428EHA-Buenos Aires, Argentina. E-mail: dhmurgida@qi.fcen.uba.ar
bDepartamento de Bioquímica and Center for Free Radical and Biomedical Research, Facultad de Medicina, Universidad de la República, Montevideo, Uruguay
cUnidad de Bioquímica y Proteómica Analíticas, Institut Pasteur de Montevideo, Montevideo, Uruguay
First published on 1st September 2014
Abstract
Cytochrome c (Cyt-c) has been previously shown to participate in cardiolipin (CL) oxidation and, therefore, in mitochondrial membrane permeabilization during the early events of apoptosis. The gain in this function has been ascribed to specific CL/Cyt-c interactions. Here we report that the cationic protein Cyt-c is also able to interact electrostatically with the main lipid components of the mitochondrial membranes, the zwitterionic lipids phosphatidylcholine (PC) and phosphatidylethanolamine (PE), through the mediation of phosphate anions that bind specifically to amino groups in the surfaces of protein and model membranes. In these complexes, Cyt-c reacts efficiently with H2O2 at submillimolar levels, which oxidizes the sulfur atom of the axial ligand Met80. The modified protein is stable and presents significantly enhanced peroxidatic activity. Based on these results, we postulate that the rise of H2O2 concentrations to the submillimolar levels registered during initiation of the apoptotic program may represent one signaling event that triggers the gain in peroxidatic function of the Cyt-c molecules bound to the abundant PE and PC membrane components. As the activated protein is a chemically stable species, it can potentially bind and oxidize important targets, such as CL.
1. Introduction
Cytochrome c (Cyt-c) is a highly conserved monohemic protein that serves as an electron shuttle between the respiratory complexes III and IV in the inner mitochondrial membrane (IMM), where it is also implicated in the generation, oxidation and trapping of reactive oxygen species (ROs).1,2 The release of Cyt-c from the mitochondrial intermembrane space to the cytosol, on the other hand, plays a pivotal role in apoptosis, thereby highlighting the importance of this small multifunctional protein in sustaining and terminating cellular life.3 Once in the cytosol, Cyt-c binds Apaf-1, ATP and pro-caspase-9 to form a multimeric complex called apoptosome that triggers a cascade of events leading to cellular suicide.2,4–7 The Cyt-c mediated caspase activation apoptotic pathway is essential for a large variety of biological events, including brain development, immune system homeostasis, and stress-induced and genotoxic-induced cell death and, therefore, is involved in protecting living organisms from diseases such as cancer. It may also amplify other apoptotic pathways.2,4–7Several mechanisms have been proposed to explain the permeabilization of the outer mitochondrial membrane (OMM) that precedes Cyt-c release and are extensively reviewed elsewhere.2,4–9 In this context, different groups have paid particular attention to the interactions between cationic Cyt-c and the anionic phospholipid cardiolipin (CL),2,10–24 which represents ca. 10–20% of the total lipid content of the IMM and a much smaller proportion of the OMM.25–28 In the IMM, CL plays a crucial role in stabilizing the respiratory complexes and in mediating electron transport by Cyt-c. Indeed, it has been estimated that about 15% of the available Cyt-c is bound to CL.29–32 A number of biophysical studies on model systems suggest that, in addition to nonspecific electrostatic interactions, CL forms a specific association with Cyt-c that involves the insertion of one or two hydrocarbon chains into the hydrophobic channels of the protein.11,14–20,24 The interplay of electrostatic and hydrophobic interactions triggers conformational changes that include the detachment of the iron axial ligand Met80 and augmented accessibility of potential substrates to the metal center. The result is a gain in peroxidase activity of Cyt-c that has been implicated in the selective oxidation of CL observed during the early events of apotosis.10,11,23 Good arguments in favor of this proposal are the findings that (a) during apoptosis, the amount of CL in the OMM increases significantly and (b) the oxidation of CL appears to be a requisite for the release of proapoptotic factors, such as the release of Cyt-c to the cytosol across the OMM.10,11 Yet, the mechanistic details and, more specifically, the regulation of these processes remain elusive.
In addition to CL, mitochondrial membranes are rich in zwitterionic phospholipids such as phosphatidylcholine (PC) and phophatidylethanolamine (PE), which comprise more than 70% of the total lipid content and have a quaternary ammonium and a primary amine bound to the phosphate group, respectively.25–27 As previously shown, Cyt-c is not expected to present significant electrostatic affinity for such lipids.14,33,34 Very recent studies, however, have demonstrated high affinity binding of Cyt-c to model systems containing amino and ammonium functional groups, that is specifically mediated by inorganic and organic phosphate anions at millimolar levels.35,36 In the present work, we have investigated the chemical reactivity of Cyt-c associated to similar model systems and in complexes with PE and PC liposomes. We found that the phosphate mediated binding of Cyt-c to these model lipids results in efficient oxidation of the axial ligand Met80 in the presence of H2O2 at submillimolar levels, thus yielding a stable modified protein with high peroxidatic activity. Interestingly, the early stages of the apoptotic program are characterized by a rise in H2O2 intracellular levels from submicromolar to submillimolar.37,38 Therefore, we propose that such a mechanism might be involved in the induction of peroxidatic activity of Cyt-c that precedes the liberation of proapoptotic molecules from the mitochondria.
2. Experimental
Chemicals
6-mercapto-1-hexanol, 6-mercaptohexanoic acid, 11-mercapto-1-undecanol, 11-mercaptoundecanoic acid, 11-mercaptoundecylphosphoric acid, (11-mercaptoundecyl)-N,N,N-trimethylammonium bromide and horse heart cytochrome c (Cyt-c) were purchased from Sigma-Aldrich. 11-amino-1-undecanethiol hydrochloride was purchased from Dojindo. 1,2-dioleoyl-sn-glycero-3-phosphocholine (PC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (PE) were purchased from Avanti Polar Lipids. Hydrogen peroxide was purchased from Mallinckrodt Baker, Inc. Amplex Red Ultra was purchased from Invitrogen. The M80A mutant of Cyt-c was prepared according to published procedures.39All chemicals were of the highest available purity and were used without further purification. The water used in all experiments was purified by a Millipore system and its resistance was 18.2 MΩ.
Electrochemistry
Cyclic voltammetry (CV) experiments were performed using a Gamry REF600 potentiostat. The jacketed three-electrode electrochemical cell was placed inside a Faraday cage (Gamry Vista Shield) and was equipped with a polycrystalline Au bead working electrode, a Pt wire counter electrode and a Ag/AgCl (3 M KCl) reference electrode to which all potentials in this work are referenced. Unless stated otherwise, the electrolyte solution was a 10 mM phosphate buffer, pH = 7.0, which was thoroughly deoxygenated by Ar bubbling. All experiments were carried out at room temperature (22–25 °C).Raman spectroscopy
Resonance Raman (RR) and surface-enhanced resonance Raman (SERR) spectra were acquired with a confocal Raman microscope (Dilor XY) equipped with a liquid-nitrogen cooled CCD detector. The excitation source was the 413 nm line of a krypton ion laser (Spectra-Physics BeamLok 2060). For SERR determinations, the laser beam was focused onto the surface of a home-made rotating Ag electrode by means of a long-working-distance objective (20×, numerical aperture of 0.35). The SERR spectroelectrochemical cell has been described in detail elsewhere.40 RR measurements were performed using a cylindrical quartz cell mounted on a home-made rotating device to avoid laser-induced sample degradation. Typically, experiments were performed with laser powers of 3.5 mW measured at the samples.Electrode treatment and self-assembled monolayer preparation
The Au electrodes used for the CV experiments were first oxidized in 10% HClO4 by applying a potential of 2.5 V for 2 minutes, sonicated in 10% HCl for 15 minutes and rinsed with water. The electrodes were then cleaned with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v H2O2:H2SO4 mixture at 120 °C. Finally, they were cycled between −0.2 and 1.6 V in 10% HClO4 and thoroughly washed with water and ethanol.
:1 v/v H2O2:H2SO4 mixture at 120 °C. Finally, they were cycled between −0.2 and 1.6 V in 10% HClO4 and thoroughly washed with water and ethanol.
The area of these electrodes was determined by CV from the integration of the reduction peak of the surface oxide. For these determinations, cyclic voltammograms (CVs) were recorded at 0.1 V s−1 between −0.25 and 1.6 V in 0.5 M H2SO4, and the conversion factor was 0.44 mC cm−2 for an oxide monolayer. The average surface area of the electrodes used in this work was calculated to be 0.18 ± 0.08 cm2 with a roughness factor of 3.4 ± 1.1.
The Ag ring electrodes employed for the SERR measurements were mechanically polished and then treated with repetitive electrochemical oxidation/reduction cycles in 0.1 M KCl to create a SER-active nanostructured surface.40
After the treatment described above, the Au and Ag electrodes were immersed in 2 mM ethanolic solutions of the desired thiols for self-assembly. Unless stated otherwise, the incubation solutions were 2 mM 1:1 mixtures of 11-mercapto-1-undecanol, which is added as a diluting component for the self-assembled monolayer (SAM), and a second ω-functionalized undecanethiol with negatively or positively charged ω-groups: carboxylate, phosphate, amino and trimethylammonium. The obtained SAMs are designated as COO-SAM, PO4-SAM, NH2-SAM and TA-SAM, respectively.
In the specific case of NH2-SAM, the incubation solvent was a 4:1 v/v mixture of C2H5OH–HClO4, 10−4 M, pH = 4, to prevent adsorption via the amino group. In all cases, the incubations were carried out overnight and the SAM-modified electrodes were kept in darkness until use.41 Prior to their use, the electrodes were rinsed thoroughly with ethanol and water and subjected to electrochemical cycling in the phosphate buffer as a conditioning treatment.36
Preparation of the lipid vesicles
Large unilamellar vesicles were prepared by the extrusion of equimolar PC/PE mixtures. Appropriate amounts of the lipid stock solutions were mixed in chloroform, evaporated to dryness under a gentle nitrogen stream and then left under reduced pressure for 1.5 h to remove any residual solvent. The dry lipids were subsequently hydrated with 20 mM HEPES, pH = 7, at room temperature to yield a lipid concentration of 0.5 mM. Afterwards, the samples were passed 20 times through polycarbonate filters with a pore size of 100 nm (Nuclepore, Whatman), yielding liposomes of the desired composition. The vesicle size, distribution and stability were checked by dynamic light scattering (SLS 90 Plus/BI-MAS equipped with He–Ne laser operating at 632.8 nm and 15 mW).Synthesis and purification of the SO-Cyt protein variant
Chloramine-T was employed as a selective reagent for oxidizing the Met80 residue of Cyt-c, according to previous reports.42,43 The product obtained was designated as ex situ generated SO-Cyt. For this purpose, 100 μL of 1 mM Cyt-c were mixed with 100 μL of 5 mM chloramine-T and the pH was adjusted to 8.4. After 3 hours at room temperature, the mixture was washed 6 times with 10 mL of 10 mM phosphate buffer, pH = 7, using centrifugal filters (Amicon 10 kDa filters). The progress of the reaction was monitored by the disappearance of the 695 nm band from the UV-visible spectrum of Cyt-c (Thermo Scientific Evolution Array spectrophotometer).The reaction mixture was passed through a cation exchange sulfopropyl-TSK preparative column (21.5 mm × 15.0 cm; Tosoh Biosep) at a flow rate of 3 mL min−1. The column was equilibrated with a 5 mM ammonium acetate buffer (pH = 9.0), kept for 5 min in this buffer and then eluted using linear gradients of the same salt from 5 mM to 150 mM (5 to 30 min), from 150 mM to 400 mM (30 to 75 min) and, finally, 500 mM ammonium acetate from 76 to 90 min (see ESI†).
The desired product was characterized by mass spectrometry (MS) employing a 4800 MALDI TOF/TOF instrument (Applied Biosystems) in a positive ion reflector mode. The mass spectra were externally calibrated using a mixture of peptide standards (Applied Biosystems). MS/MS spectra were recorded to confirm the site of oxidation in the analyzed peptides (see ESI†). The mass spectra were analyzed using DATA explorer and GPMAW4 software.
Peroxidatic activity determination
The assessment of the peroxidatic activity was performed using an Amplex UltraRed Reagent (Invitrogen). The fluorescence of the oxidation product, resorufin, was monitored at λem = 585 nm with λex = 570 nm. Briefly, 0.5 μM Cyt-c was incubated in HEPES (10 mM plus 100 μM DTPA), pH = 7. Then, 50 μM Amplex Red and 25 μM H2O2 were added to HEPES (100 mM plus 100 μM DTPA), pH = 7, and the fluorescence was registered using a FLUOstar Optima plate fluorimeter (BMG Labtech) or a Varioskan Flash Multimode Reader (Thermo). The reaction rate was determined by a linear fit of the fluorescence intensity profile.3. Results and discussion
Reactivity of Cyt-c in biomimetic complexes
Hydrogen peroxide is a well-documented bleaching agent of Cyt-c that produces oxidative damage of the porphyrin ring.44 Recent studies, however, suggest that the formation of electrostatic complexes with negatively charged surfaces has a protective effect on Cyt-c, thus preventing its degradation in the presence of H2O2 up to concentrations of ca. 20 mM.45 In this context, we investigated the reactivity of Cyt-c towards H2O2 in electrostatic complexes with different model systems that mimic some essential features of the natural protein/membrane interactions. Specifically, we employed liposomes of different compositions and metal electrodes coated with SAMs of ω-functionalized alkanethiols. In the last case, the ω functional groups explored were carboxylate, phosphate, amino and trimethylammonium (COO-SAM, PO4-SAM, NH2-SAM and TA-SAM, respectively). The adsorption of Cyt-c on COO- and PO4-SAMs involves direct electrostatic interactions through the ring of positively charged lysine residues that surround the partially exposed heme edge of the protein.46 Roughly the same surface residues are implicated in the adsorption of Cyt-c onto NH2- and TA-SAMs, although in these cases the interactions are mediated by phosphate ions from the buffer solution.35,36 In all cases, the adsorbed protein exhibits quasi-reversible cyclic voltammetric (CV) responses (Fig. S1†) typical of one-electron redox couples. The formal reduction potentials obtained for Cyt-c adsorption on COO-, PO4- and NH2-SAMs are very close to the value for the native protein in solution while, in excellent agreement with previous observations, the value for adsorption on TA-SAMs is somewhat more positive.35,47 Therefore, the detected electrochemical signals can be ascribed to adsorbed Cyt-c which largely preserves its native structure, particularly at the level of the redox site. It is important to point out that the absence of other electrochemical signals does not rule out the presence of alternative Cyt-c conformations in the adsorbed state that might be kinetically impaired. Indeed, the larger peak separations observed for PO4-SAM compared to the other SAMs is indicative of a much slower interfacial electron transfer reaction of Cyt-c in this case, which is compatible with a less favorable and more rigid orientation of the protein in such complexes.48Upon addition of H2O2 to Cyt-c adsorbed on NH2-SAMs, we observe the appearance of a second monoelectronic and quasi-reversible voltammetric signal with an apparent reduction potential of −119 mV (Fig. 1). The anodic and cathodic peak currents of the new redox active species rise at the expense of the corresponding signals of native Cyt-c, with a complete transformation at H2O2 concentrations slightly above 1 mM. Interestingly, the new redox couple presents enhanced cathodic currents in the presence of H2O2, thus indicating the electrocatalytic reduction of H2O2 by the modified protein (pseudoperoxidase activity).
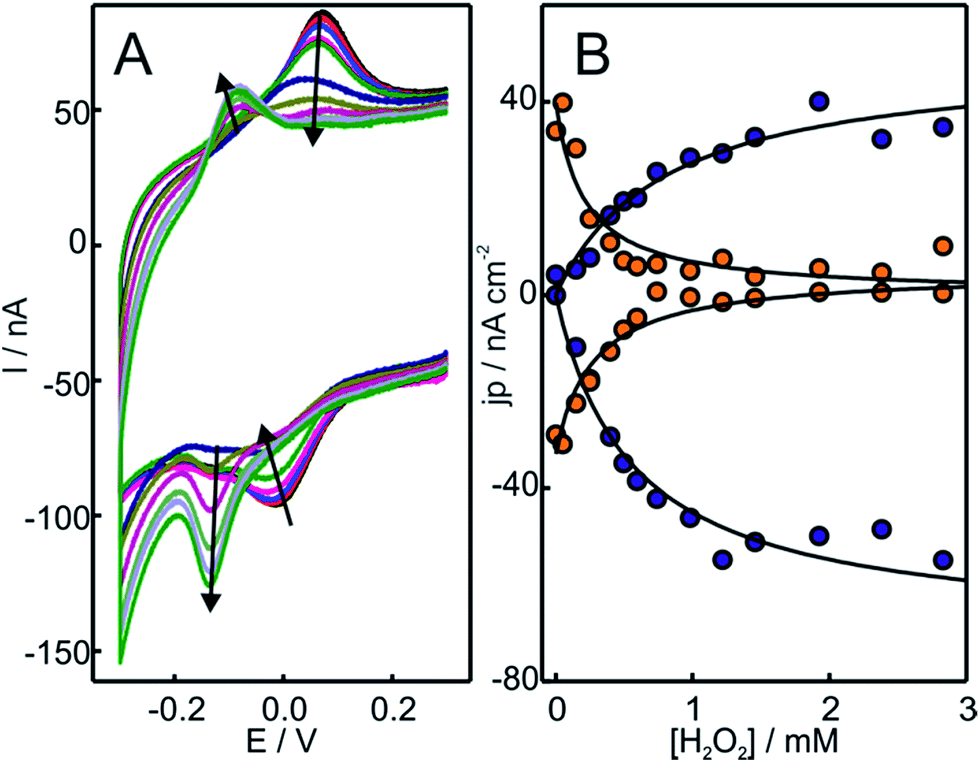 | ||
| Fig. 1 (A) Voltammetric response of Cyt-c adsorbed on electrodes coated with NH2-SAMs. The measurements were performed at 0.05 V s−1 in 10 mM phosphate buffer, pH = 7.0, in the presence of increasing concentrations of H2O2 from 0 to 500 μM. (B) Background corrected peak current densities for the native Cyt-c redox couple (orange circles) and the newly generated couple (blue circles) as a function of H2O2 concentration. The solid lines are only intended as guides. | ||
The downshift of the reduction potential of Cyt-c after treatment with H2O2 may in principle indicate a more polar heme environment as a consequence of increased solvent accessibility.49 However, the addition of several complexing agents such as imidazole, azide, DTPA and methionine has no effect on the electrochemical response (Fig. S2†), suggesting that the solvent exposure of the heme crevice is not significantly altered.
The replacement of the electrolyte by fresh phosphate buffer once the H2O2-induced transformation is complete results in stable voltammograms that retain the electrochemical features of the newly generated redox couple (Fig. S3†). These results suggest a permanent chemical modification of the adsorbed Cyt-c into a stable species that presents pseudoperoxidase activity and, potentially, peroxidase-like characteristics. Remarkably, this behavior is only observed when Cyt-c is adsorbed on the NH2- and TA-SAMs in the presence of the phosphate buffer (Fig. 1A and S5A†). In contrast, when the protein is directly adsorbed on negatively charged COO- and PO4-SAMs, the addition of H2O2 produces a slight decrease of the voltammetric peaks of native Cyt-c, but this partial bleaching is not accompanied by the appearance of new CV signals (Fig. S6†). Moreover, pretreatment of the Cyt-c solutions with different concentrations of H2O2 (between 1 and 10 mM) prior to incubation with the electrodes coated with NH2-SAMs leads exclusively to weak voltammetric signals at the potentials expected for the native Cyt-c, but there is no indication of other redox active species (Fig. 2B).
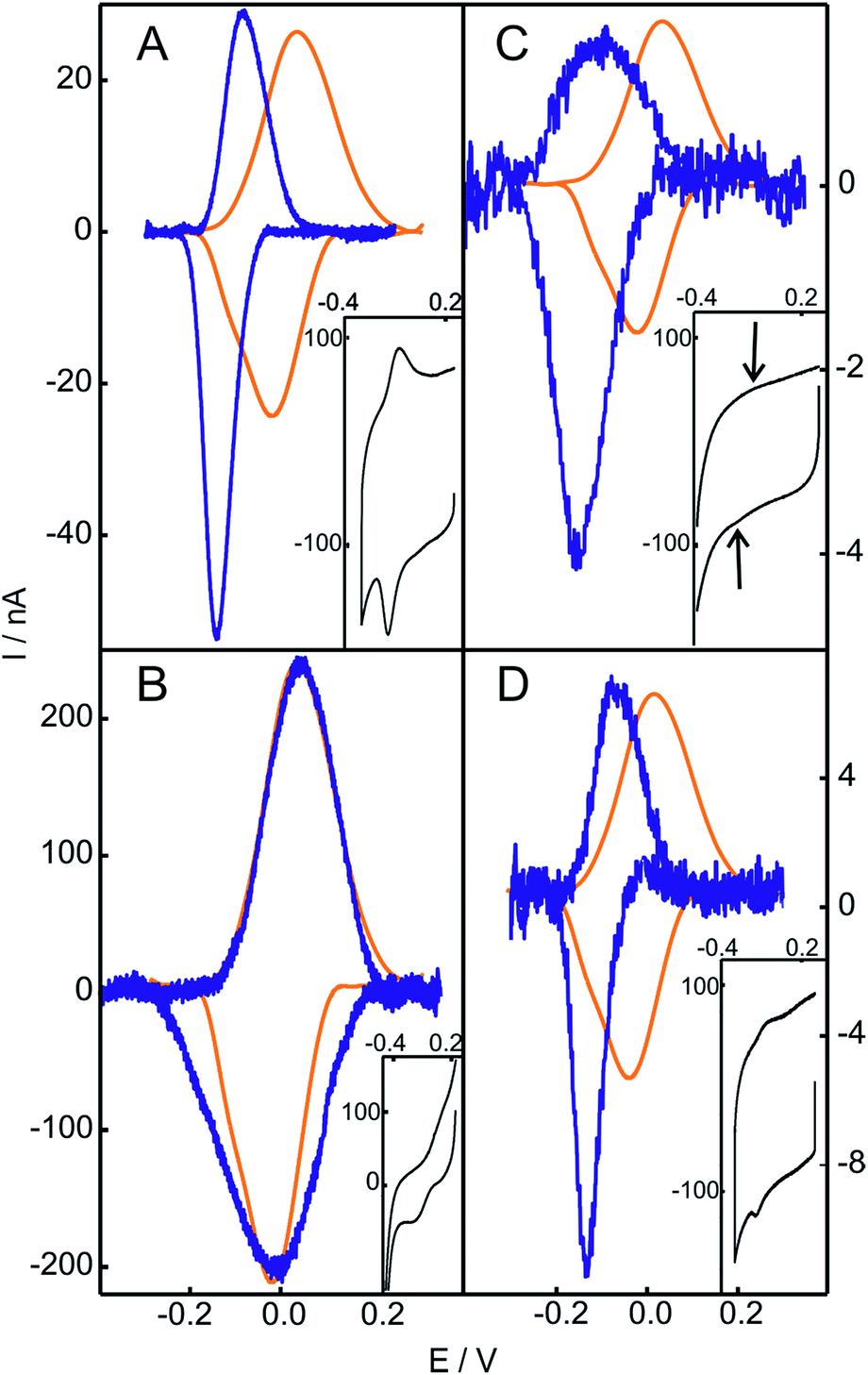 | ||
| Fig. 2 Baseline subtracted cyclic voltammetries of Cyt-c before (orange) and after (blue) chemical treatment. (A) Cyt-c adsorbed on an NH2-SAM electrode treated with 2 mM H2O2. (B) Cyt-c pretreated in solution with 2 mM H2O2 and subsequently adsorbed on an NH2-SAM electrode. (C) Cyt-c pretreated in solution with 2 mM H2O2 in the presence of PE/PC liposomes, and subsequently adsorbed on an NH2-SAM electrode. (D) Ex situ generated and purified SO-Cyt adsorbed on an NH2-SAM electrode. All measurements were performed at 0.1 V s−1 in 10 mM phosphate buffer, pH = 7.0. The insets are the voltammetric signals before background correction. | ||
Based on these results, we investigated the effect of unilamellar vesicles of PC and mixtures of PC/PE. Typically, up to 1 mM concentrations of H2O2 were added to solutions containing 200 μM Cyt-c and 0.5 mM PC (or PC/PE). After this treatment, the Cyt-c solutions were thoroughly washed using centrifugal filters and the supernatant was incubated with electrodes coated with NH2-SAMs in the phosphate buffer. Similar to the in situ treatment of Cyt-c adsorbed on NH2-SAMs, the modified electrodes exhibit a CV signal with E1/2 = −119 mV that also presents enhanced cathodic currents in the presence of H2O2 (Fig. 2C and S7†). Therefore, the H2O2-induced chemical modification that leads to a gain in pseudoperoxidatic (and possibly peroxidatic) activity appears to be a general feature of Cyt-c molecules adsorbed on positively charged and zwitterionic membrane models via mediating phosphate anions.
Characterization of the catalytically competent species
The structure of Cyt-c in the different biomimetic complexes was investigated by RR and SERR, as these spectroscopic techniques are able to monitor the redox state, spin and coordination pattern of the heme iron, as well as heme–protein interactions.40,48,50 In agreement with previous reports, the SERR spectra of Cyt-c adsorbed on COOH-SAMs are identical to the corresponding RR spectra of the native protein in solution (Fig. S8†).50 Moreover, in agreement with the electrochemical results, the spectra remain unchanged upon the addition of moderate amounts of H2O2 (ca. 4 mM). The SERR spectra of Cyt-c adsorbed on PO4-SAMs are also largely insensitive to the addition of similar amounts of H2O2 but in this case they reveal, in addition to the native protein, a ca. 30% spectral contribution of a second species consistent with the replacement of the heme axial ligand Met80 by a histidine residue (Fig. S9A†).50 A similar contribution of bis-His species is also observed for Cyt-c adsorbed on NH2-SAMs (Fig. S9B†). The assignment of the bis-His species is based on the spectral parameters of the high frequency region shown in Table S2† and on the appearance of a band at 405 cm−1 assigned to the asymmetric stretching of the heme Fe bound to two imidazolic axial ligands. Upon treatment of the Cyt-c/NH2-SAM complexes with 1 mM H2O2, we detect the appearance of a new spectroscopic component that resembles the RR spectrum of the ferric M80A mutant and, therefore, is compatible with a OH−/His coordination pattern (Fig. 3 and S10†).51 The rise of the OH−/His spectral component is concomitant with the appearance of the pseudoperoxidatic redox couple and occurs at the expense of the native spectral component, without significant alteration of the bis-His contribution (Fig. S10†). Nevertheless, within the error of the quantification, we cannot discard the possibility that the OH−/His species actually originates from the reaction of H2O2 with the bis-His species, which is in equilibrium with the main native component.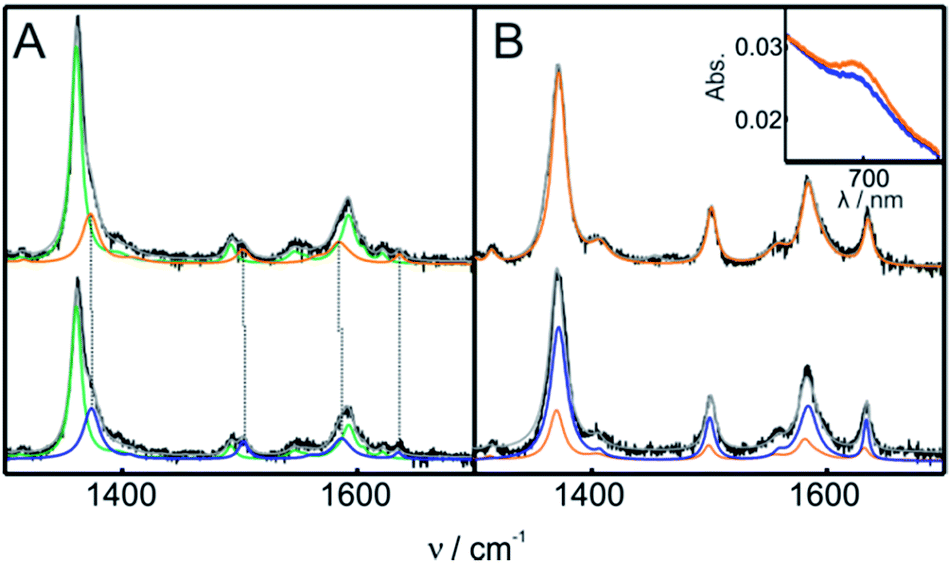 | ||
| Fig. 3 (A) SERR spectra of Cyt-c adsorbed on an Ag electrode coated with NH2-SAMs recorded before (top) and after (bottom) the addition of 1 mM H2O2 (final concentration). The spectra were acquired while cycling the electrode potential between 0.1 V and −0.3 V at 0.05 V s−1 in 10 mM phosphate buffer, pH = 7. The colors indicate the different spectral components. Green: ferrous native Cyt-c. Orange: ferric native Cyt-c. Blue: OH−/His ferric component. (B) RR spectra of 0.2 mM ferric Cyt-c in 10 mM phosphate buffer, pH = 7, containing 0.5 mM PC/PE liposomes. The spectra were recorded before (top) and after (bottom) the addition of 1 mM H2O2 (final concentration). The color code of the RR spectral components is the same as that for the SERR spectra in panel (A). The inset shows the UV-vis charge transfer band before (orange) and after (blue) the addition of H2O2. | ||
The fact that the adsorbed Cyt-c is sensitive to H2O2 only on amino-terminated SAMs suggests the involvement of hydroxyl radical species (˙OH) that may be generated by the reaction of the amine groups of the SAMs with H2O2.52 To test this possibility, we performed a H2O2 activation of Cyt-c/NH2-SAM complexes in the presence of DMSO, a very efficient scavenger of ˙OH.53–55 The results were identical to those obtained in the absence of DMSO, thereby indicating that, most likely, ˙OH is not involved in the formation of the pseudoperoxidatic OH−/His species.
In contrast to the native Cyt-c, the reduction potential of the OH−/His species adsorbed on NH2- and QA-SAMs exhibits a strong linear pH dependence (Fig. 4A and S5B†), with a slope of −0.057 V per pH unit, which is consistent with the uptake of one proton per incoming electron. For the M80A mutant of Cyt-c cross-linked to electrodes coated with COOH-terminated SAMs, we obtain an almost identical pH-dependence (Fig. 4B). Moreover, the slope is also very similar to the one reported for horseradish peroxidase,56 thus further suggesting close similarities of this species to the “professional” peroxidases.
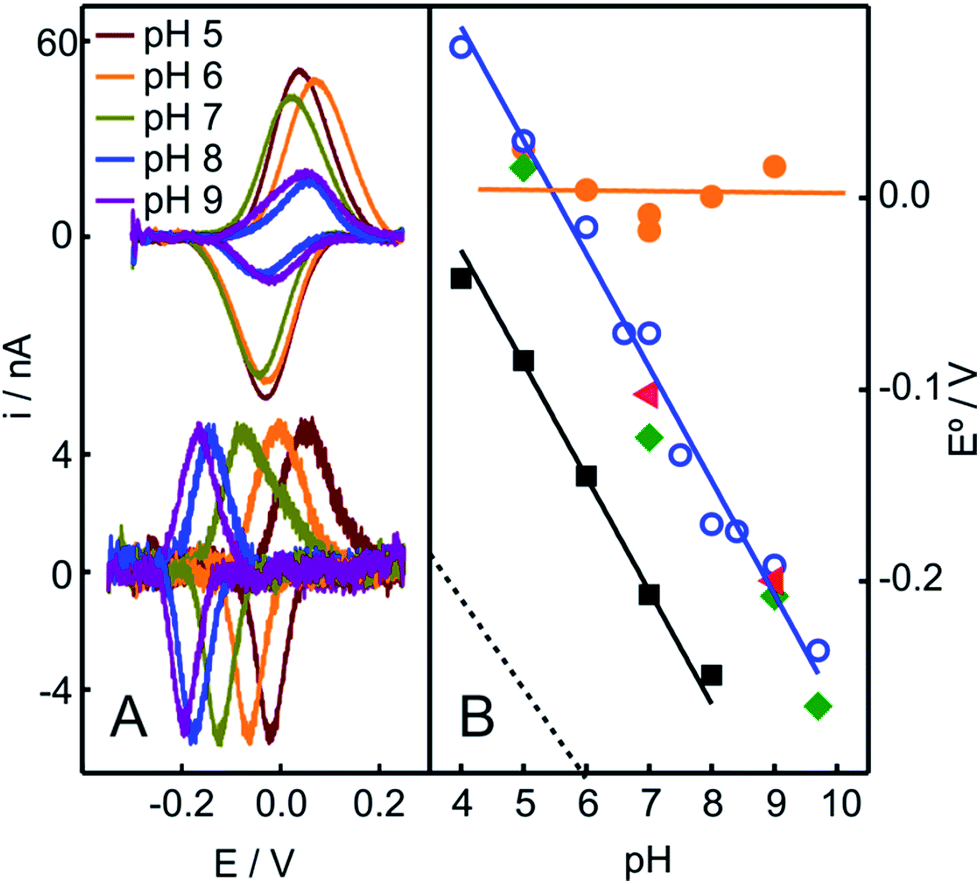 | ||
| Fig. 4 (A) Voltammetric signals of Cyt-c adsorbed on NH2-SAMs recorded at different pH values before (top) and after (bottom) the addition of 2 mM H2O2 (final concentration). (B) pH-dependencies of the reduction potentials of different protein species adsorbed on NH2-SAM electrodes. Orange circles: native Cyt-c. Blue circles: Cyt-c adsorbed on the same electrode after in situ pretreatment with 1 mM H2O2. Green diamonds: Cyt-c pretreated ex situ with 1 mM H2O2 in the presence of 5 mM PE liposomes. Red triangles: ex situ synthesized and purified SO-Cyt. Additionally, the black squares correspond to the M80A Cyt-c mutant cross-linked to a COO-SAM (this work) and the dotted line corresponds to horseradish peroxidase on glassy carbon electrodes, adopted from ref. 56. | ||
The treatment of Cyt-c/liposome complexes with H2O2 yields results very similar to the Cyt-c/NH2-SAM complexes. On the one hand, the RR spectra of the treated Cyt-c/liposome systems are largely dominated by the same spectral component assigned to a ferric OH−/His species and, moreover, this spectral change is complemented by a decrease of the characteristic MetS-Fe charge transfer absorption band at 695 nm (Fig. 3B). On the other, the reduction potentials of the Cyt-c products generated by treatment with H2O2 in both types of complexes exhibit identical values and pH dependencies (Fig. 4B).
Hence, the spectroscopic and electrochemical results point to the same chemical modification of Cyt-c in the different phosphate-mediated biomimetic complexes that lead to the detachment of the native axial ligand Met80 and the concomitant gain in catalytic activity.
A potential target for the reaction with H2O2 is the sulfur atom of the Met80 residue, as shown for methionines in general with a variety of oxidant species.42,57–59 Therefore, we treated Cyt-c with chloramine-T, which has been described as a selective reagent to oxidize Met80 and generate the corresponding sulfoxide species (SO-Cyt).42,43 The product was purified by high-performance liquid chromatography and characterized by mass spectrometry-based peptide mapping (see ESI† for further details).
The purified SO-Cyt exhibits an identical RR spectrum to that of the H2O2-generated OH−/His species obtained in the biomimetic complexes (Fig. S13†). Moreover, as shown in Fig. 2D and 4B, when the ex situ generated SO-Cyt is adsorbed on electrodes coated with NH2-SAMs, it exhibits nearly identical voltammetric responses and pH dependencies as the in situ generated OH−/His species.
The CV experiments reveal very similar heterogeneous electron transfer rate constants for native Cyt-c and SO-Cyt at all pH values (Table S1†), but a large downshift of the reduction potential of SO-Cyt at physiological pH. Thus, the loss of electron shuttling ability of Cyt-c upon Met80 oxidation is essentially a thermodynamic effect.
In summary, the presented results provide convincing evidence that submillimolar concentrations of H2O2 are able to efficiently and selectively oxidize the axial M80 ligand of Cyt-c in phosphate-mediated complexes with the main lipid components of the mitochondrial membrane. The oxidized product SO-Cyt is stable and undergoes a coordination and conformational transition that leads to a gain in pseudoperoxidatic activity. The remaining question of whether this transformation also results in a significant gain in truly peroxidatic activity is addressed next.
Peroxidatic and pseudoperoxidatic activity
The peroxidase-like activity of in situ generated SO-Cyt was evaluated following different strategies depending on whether it was obtained on NH2-SAMs or on PC/PE liposomes. Both peroxidase and pseudoperoxidase activities refer to the ability to catalyze the reduction of H2O2 to water. In the first case, electrons are supplied by a molecule (substrate) in solution, which is then oxidized, and homogeneous reaction rates are measured. In contrast, for pseudoperoxidase activity we refer to the electrocatalytic reduction of H2O2 by the immobilized protein using the heterogeneous electron transfer reaction from the metal electrode to the protein as the electron source. The resulting currents measured by CV in the presence of increasing concentrations of H2O2 are a measure of the reaction rate. As shown in Fig. 5, the addition of H2O2 results in enhanced cathodic currents, without a significant alteration of other parameters such as peak potentials or FWHMs. This type of electrocatalytic behavior has been previously reported for the M80A mutant of yeast iso-cytochrome c, and was interpreted by a mechanism that involves O2 generation and its subsequent reduction.60 On the other hand, for the same mutant of the mammalian protein, Águila and coworkers showed some evidence of the formation of Fe(III)OOH (compound 0) in the reaction with H2O2, which then would evolve to ferryl species (compounds I and II).61 Assuming that this mechanism is correct, and considering that the reduction potential of the Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O intermediate is significantly more positive than that of the other species,62 we can quantitatively analyze the cathodic peak currents in Fig. 5 in terms of a Michaelis–Menten formalism (see ESI† for further details).
O intermediate is significantly more positive than that of the other species,62 we can quantitatively analyze the cathodic peak currents in Fig. 5 in terms of a Michaelis–Menten formalism (see ESI† for further details).
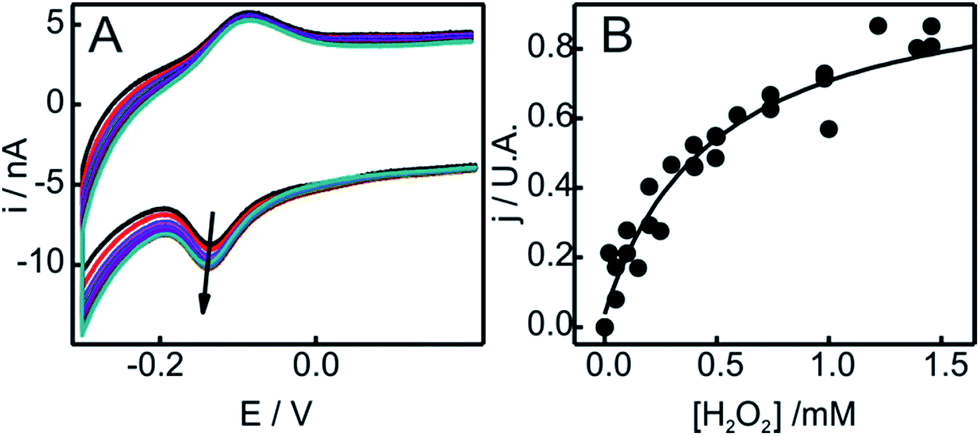 | ||
| Fig. 5 (A) Voltammetric response as a function of H2O2 concentration of SO-Cyt generated in situ upon pretreatment with H2O2 of Cyt-c adsorbed on an NH2-SAM electrode. All measurements were performed in 10 mM phosphate buffer, pH = 7, at 0.05 V s−1. The arrow indicates increasing concentrations of H2O2 from 0 to 2 mM. (B) Normalized cathodic peak current densities obtained from panel (A) as a function of H2O2 concentration. The line corresponds to the fit to the Michaelis–Menten-like equation (see ESI†). | ||
The catalytic parameters obtained through this analysis are summarized in Table 1. While the kcat values obtained for in situ and ex situ generated SO-Cyt are similar to native Cyt-c, the oxidation of M80 results in a ca. 300–600 times increase in affinity towards H2O2. As a reference, the value of Km reported for horseradish peroxidase on glassy carbon electrodes is ca. 53 μM,56 while for the M80A mutant of iso-cytochrome c it is only 3.25 μM, although this unusually high affinity is accompanied by protein bleaching at H2O2 concentrations of about 4 μM.60
| Activity | K m/mM−1 | k cat/s−1 | % of control | |
|---|---|---|---|---|
| a Standard deviations are indicated in brackets. b Determined for Cyt-c adsorbed on NH2-SAMs, this work. c Cyt-c adsorbed on COO-SAMs, from ref. 45. d Cyt-c in solution with ABTS, from ref. 63. e SO-Cyt in solution with ABTS, from ref. 64. f SO-Cyt obtained in situ on NH2-SAMs, this work. g From ref. 60. h Iso-cyt M80A mutant, from ref. 65. i SO-Cyt obtained by pretreatment with H2O2 in the presence of PC/PE liposomes. | ||||
| WT | Pseudoperoxidase | 144.3c | 8c | — |
| Peroxidase | 65d | 5.5d | 100 | |
| SO-Cyt ex situ | Pseudoperoxidaseb | 0.24 (0.08) | 2.6 (0.8) | — |
| Peroxidase | 19e | — | 719 | |
| SO-Cyt in situ | Pseudoperoxidaseb | 0.44f (0.07) | 1.8f (0.6) | — |
| Peroxidase | — | — | 644i | |
| M80A | Pseudoperoxidase | 0.00325g | — | — |
| Peroxidase | 23.2h | 72h | 780 | |
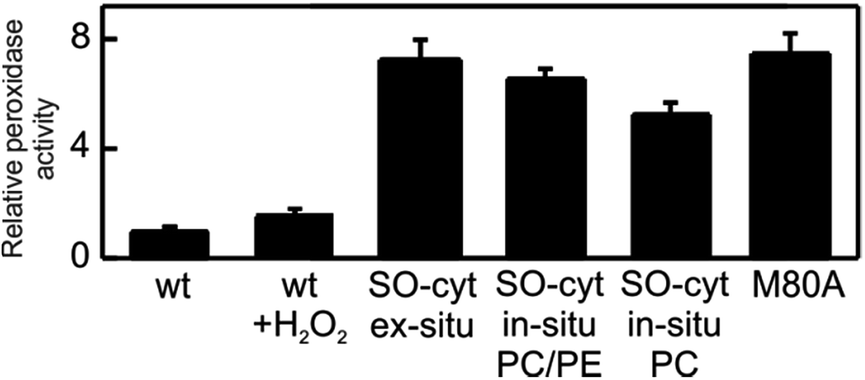 | ||
| Fig. 6 Peroxidase activities of the different Cyt-c variants relative WT Cyt-c, as determined fluorometrically using Amplex UltraRed reagent. | ||
The peroxidatic activity of the different species in solution was assessed fluorometrically using Amplex UltraRed reagent. As summarized in Table 1 and Fig. 6, both the oxidation of Met80 (SO-Cyt) as well as its replacement by non-coordinating alanine (M80A) result in increased peroxidatic activity with respect to native Cyt-c by almost one order of magnitude, in agreement with previous observations.39,65 The effect is quantitatively similar for purified SO-Cyt generated ex situ and for Cyt-c treated with H2O2 in the presence of PE/PC liposomes. In spite of the bleaching effect, the treatment of a Cyt-c solution with only H2O2 has no significant effect on the measured peroxidatic activity (Fig. 6). Similarly, the mere presence of PE/PC liposomes has no effect unless H2O2 is added, thus confirming that the H2O2-induced gain in peroxidatic activity is selective for Cyt-c molecules adsorbed on zwitterionic lipids via mediating phosphate anions.
4. Conclusions
Cyt-c is able to interact electrostatically with the main lipid components of the mitochondrial membranes, the zwitterionic lipids PE and PC, in the presence of biologically relevant amounts of mediating phosphate anions. Under these conditions, the adsorbed protein reacts efficiently with H2O2. The reaction does not lead to the bleaching of the heme group, but is restricted to the oxidation of the sulfur in the iron axial ligand Met80, leading to its detachment and to a concomitant increase of the peroxidatic activity by one order of magnitude.There is no evidence, though, that the PE and PC lipids are peroxidated by Cyt-c to significant amounts.10,23 Cardiolipin, in contrast, is effectively peroxidated by Cyt-c during the early events of apoptosis, thus leading to membrane permeabilization. The gain in peroxidatic activity has been ascribed to specific Cyt-c/CL interactions. The present findings, on the other hand, suggest a possible additional pathway for membrane permeabilization. Specifically, the rise of H2O2 concentrations to submillimolar levels that characterizes the initiation of apoptosis37,38 appears to be sufficient to chemically modify a fraction of the Cyt-c molecules that interact (via phosphate anions) with the PE and PC membrane components. The modified protein is a stable peroxidase that could later bind and catalyze the oxidation of membrane components such as CL, thus facilitating the liberation of pro-apoptotic factors, including unmodified Cyt-c.
Acknowledgements
Financial support from ANPCyT (PICT 2010-070 and 2011-1249) and UBACyT (20020130100206BA) to D.H.M. and from ANII (FCE 2486) and CSIC-UdelaR to R.R is gratefully acknowledged. D.A.C. thanks CONICET for a fellowship. W.A.M and D.H.M. are staff members of CONICET. F.T. was partially supported by a fellowship of Universidad de la República. F.T., V.D. and R.R. acknowledge financial support from Programa de Desarrollo de Ciencias Básicas.Notes and references
- R. A. Scott and A. G. Mauk, Cytochrome c: a multidisciplinary approach, Univ Science Books, Sausalito, California, 1995 Search PubMed.
- M. Hüttemann, P. Pecina, M. Rainbolt, T. H. Sanderson, V. E. Kagan, L. Samavati, J. W. Doan and I. Lee, Mitochondrion, 2011, 11, 369 CrossRef PubMed.
- X. Liu, C. N. Kim, J. Yang, R. Jemmerson and X. Wang, Cell, 1996, 86, 147 CrossRef CAS.
- Y. Kushnareva and D. Newmeyer, Ann. N. Y. Acad. Sci., 2010, 50, 1201 Search PubMed.
- R. Caroppi, P. Sinibaldi, F. Fiorucci and L. Santucci, Curr. Med. Chem., 2009, 16, 4058 CrossRef.
- X. Jiang and X. Wang, Annu. Rev. Biochem., 2004, 73, 87 CrossRef CAS PubMed.
- A. V. Kulikov, E. S. Shilov, I. A. Mufazalov, V. Gogvadze, S. A. Nedospasov and B. Zhivotovsky, Cell. Mol. Life Sci., 2012, 69, 1787 CrossRef CAS PubMed.
- C. Garrido, L. Galluzzi, M. Brunet, P. E. Puig, C. Didelot and G. Kroemer, Cell Death Differ., 2006, 13, 1423 CrossRef CAS PubMed.
- V. Gogvadze, S. Orrenius and B. Zhivotovsky, Biochim. Biophys. Acta, 2006, 1757, 639 CrossRef CAS PubMed.
- V. E. Kagan, V. A. Tyurin, J. Jiang, Y. Y. Tyurina, V. B. Ritov, A. A. Amoscato, A. N. Osipov, N. A. Belikova, A. A. Kapralov, V. Kini, I. I. Vlasova, Q. Zhao, M. Zou, P. Di, D. A. Svistunenko, I. V. Kurnikov and G. G. Borisenko, Nat. Chem. Biol., 2005, 1, 223 CrossRef CAS PubMed.
- V. E. Kagan, H. A. Bayır, N. A. Belikova, O. Kapralov, Y. Y. Tyurina, V. A. Tyurin, J. Jiang, D. A. Stoyanovsky, P. Wipf, P. M. Kochanek, J. S. Greenberger, B. Pitt, A. A. Shvedova and G. Borisenko, Free Radicals Biol. Med., 2009, 46, 1439 CrossRef CAS PubMed.
- P. Ascenzi, C. Ciaccio, F. Sinibaldi, R. Santucci and M. Coletta, Biochem. Biophys. Res. Commun., 2011, 404, 190 CrossRef CAS PubMed.
- P. Ascenzi, R. Santucci, M. Coletta and F. Polticelli, Biophys. Chem., 2010, 152, 21 CrossRef CAS PubMed.
- V. M. Trusova, G. P. Gorbenko, J. G. Molotkovsky and P. K. J. Kinnunen, Biophys. J., 2010, 99, 1754 CrossRef CAS PubMed.
- L. Macchioni, T. Corazzi, M. Davidescu, E. Francescangeli, R. Roberti and L. Corazzi, Mol. Cell. Biochem., 2010, 341, 149 CrossRef CAS PubMed.
- F. Sinibaldi, B. D. Howes, M. C. Piro, F. Polticelli, C. Bombelli, T. Ferri, M. Coletta, G. Smulevich and R. Santucci, JBIC, J. Biol. Inorg. Chem., 2010, 15, 689 CrossRef CAS PubMed.
- J. Hanske, J. R. Toffey, A. M. Morenz, A. J. Bonilla, K. H. Schiavoni and E. V. Pletneva, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 125 CrossRef CAS PubMed.
- C. L. Bergstrom, P. A. Beales, Y. Lv, T. K. Vanderlick and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6269 CrossRef PubMed.
- Y. A. Vladimirov, E. V. Proskurnina and A. V. Alekseev, Biokhim., Biochem., 2013, 78, 1086 CrossRef CAS PubMed.
- J. M. Bradley, G. Silkstone, M. T. Wilson, M. R. Cheesman and J. N. Butt, J. Am. Chem. Soc., 2011, 133, 19676 CrossRef CAS PubMed.
- S. L. Iverson and S. Orrenius, Arch. Biochem. Biophys., 2004, 423, 37 CrossRef CAS PubMed.
- Y.-L. P. Ow, D. R. Green, Z. Hao and T. W. Mak, Nat. Rev. Mol. Cell Biol., 2008, 9, 532 CrossRef CAS PubMed.
- N. Belikova and Y. Vladimirov, Biochemistry, 2006, 45, 4998 CrossRef CAS PubMed.
- E. Kalanxhi and C. J. A. Wallace, Biochem. J., 2007, 407, 179 CrossRef CAS PubMed.
- C. Osman, D. R. Voelker and T. Langer, J. Cell Biol., 2011, 192, 7 CrossRef CAS PubMed.
- D. Ardail, J. Privat and M. Egret-Charlier, J. Biol. Chem., 1990, 265, 18797 CAS.
- N. Gebert, A. S. Joshi, S. Kutik, T. Becker, M. McKenzie, X. L. Guan, V. P. Mooga, D. A. Stroud, G. Kulkarni, M. R. Wenk, P. Rehling, C. Meisinger, M. T. Ryan, N. Wiedemann, M. L. Greenberg and N. Pfanner, Curr. Biol., 2009, 19, 2133 CrossRef CAS PubMed.
- M. Garcia Fernandez, L. Troiano, L. Moretti, M. Nasi, M. Pinti, S. Salvioli, J. Dobrucki and A. Cossarizza, Cell Growth Differ., 2002, 13, 449 Search PubMed.
- Z. T. Schug and E. Gottlieb, Biochim. Biophys. Acta, 2009, 1788, 2022 CrossRef CAS PubMed.
- J. Montero, M. Mari, A. Colell, A. Morales, G. Basañez, C. Garcia-Ruiz and J. C. Fernández-Checa, Biochim. Biophys. Acta, 2010, 1797, 1217 CrossRef CAS PubMed.
- M. Schlame, D. Rua and M. L. Greenberg, Prog. Lipid Res., 2000, 39, 257 CrossRef CAS.
- V. E. Kagan, G. G. Borisenko, Y. Y. Tyurina, V. A. Tyurin, J. Jiang, A. I. Potapovich, V. Kini, A. A. Amoscato and Y. Fujii, Free Radicals Biol. Med., 2004, 37, 1963 CrossRef CAS PubMed.
- Z. Salamon and G. Tollin, Biophys. J., 1996, 71, 848 CrossRef CAS.
- T. Ahn, D. B. Oh, B. C. Lee and C. H. Yun, Biochemistry, 2000, 39, 10147 CrossRef CAS PubMed.
- D. A. Capdevila, W. A. Marmisollé, F. J. Williams and D. H. Murgida, Phys. Chem. Chem. Phys., 2013, 15, 5386 RSC.
- W. A. Marmisollé, D. A. Capdevila, E. de la Llave, F. J. Williams and D. H. Murgida, Langmuir, 2013, 29, 5351 CrossRef PubMed.
- M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722 CrossRef CAS PubMed.
- C. Cerella, S. Coppola, V. Maresca, M. De Nicola, F. Radogna and L. Ghibelli, Ann. N. Y. Acad. Sci., 2009, 1171, 559 CrossRef CAS PubMed.
- L. C. Godoy, C. Muñoz-Pinedo, L. Castro, S. Cardaci, C. M. Schonhoff, M. King, V. Tórtora, M. Marín, Q. Miao, J. F. Jiang, A. Kapralov, R. Jemmerson, G. G. Silkstone, J. N. Patel, J. E. Evans, M. T. Wilson, D. R. Green, V. E. Kagan, R. Radi and J. B. Mannick, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2653 CrossRef CAS PubMed.
- D. H. Murgida and P. Hildebrandt, J. Phys. Chem. B, 2001, 105, 1578 CrossRef CAS.
- S.-H. Lee, W.-C. Lin, C.-H. Kuo, M. Karakachian, Y.-C. Lin, B.-Y. Yu and J.-J. Shyue, J. Phys. Chem. C, 2010, 114, 10512 CAS.
- Y.-R. Chen, L. J. Deterding, B. E. Sturgeon, K. B. Tomer and R. P. Mason, J. Biol. Chem., 2002, 277, 29781 CrossRef CAS PubMed.
- J. Pande, K. Kinnally, K. K. Thallum, B. Verma, Y. Myer, L. Rechsteiner and H. Bosshard, J. Protein Chem., 1987, 6, 295 CrossRef CAS.
- B. Valderrama, M. Ayala and R. Vazquez-Duhalt, Chem. Biol., 2002, 9, 555 CrossRef CAS.
- L. Wang and D. H. Waldeck, J. Phys. Chem. C, 2008, 112, 1351 CAS.
- D. Alvarez-Paggi, D. F. Martín, P. M. DeBiase, P. Hildebrandt, M. A. Martí and D. H. Murgida, J. Am. Chem. Soc., 2010, 132, 5769 CrossRef CAS PubMed.
- X. Chen, R. Ferrigno, J. Yang and G. M. Whitesides, Langmuir, 2002, 18, 7009 CrossRef CAS.
- D. H. Murgida and P. Hildebrandt, Acc. Chem. Res., 2004, 37, 854 CrossRef CAS PubMed.
- G. Battistuzzi, M. Borsari, J. A. Cowan, A. Ranieri and M. Sola, J. Am. Chem. Soc., 2002, 124, 5315 CrossRef CAS PubMed.
- D. H. Murgida and P. Hildebrandt, Chem. Soc. Rev., 2008, 37, 937 RSC.
- G. Battistuzzi, C. A. Bortolotti, M. Bellei, G. Di Rocco, J. Salewski, P. Hildebrandt and M. Sola, Biochemistry, 2012, 51, 5967 CrossRef CAS PubMed.
- G. Cohen and R. Heikkila, J. Biol. Chem., 1974, 249, 2447 CAS.
- R. C. Scaduto, Free Radicals Biol. Med., 1995, 18, 271 CrossRef CAS.
- M. G. Steiner and C. F. Babbs, Arch. Biochem. Biophys., 1990, 278, 478 CrossRef CAS.
- C. Tai, J.-F. Peng, J.-F. Liu, G.-B. Jiang and H. Zou, Anal. Chim. Acta, 2004, 527, 73 CrossRef CAS PubMed.
- B. Wang, J.-J. Zhang, Z.-Y. Pan, X.-Q. Tao and H.-S. Wang, Biosens. Bioelectron., 2009, 24, 1141 CrossRef CAS PubMed.
- W. Vogt, Free Radicals Biol. Med., 1995, 18, 93–105 CrossRef CAS.
- B. Valderrama and R. Vazquez-Duhalt, J. Mol. Catal. B: Enzym., 2005, 35, 41 CrossRef CAS PubMed.
- J. Thariat, F. Collin, C. Marchetti, N. S. Ahmed-Adrar, H. Vitrac, D. Jore and M. Gardes-Albert, Biochimie, 2008, 90, 1442 CrossRef CAS PubMed.
- S. Casalini, G. Battistuzzi, M. Borsari, C. A. Bortolotti, G. Di Rocco, A. Ranieri and M. Sola, J. Phys. Chem. B, 2010, 114, 1698 CrossRef CAS PubMed.
- S. Águila, A. M. Vidal-Limón, J. B. Alderete, M. Sosa-Torres and R. Vázquez-Duhalt, J. Mol. Catal. B: Enzym., 2013, 85–86, 187 CrossRef PubMed.
- G. Battistuzzi, M. Bellei, C. A. Bortolotti and M. Sola, Arch. Biochem. Biophys., 2010, 500, 21 CrossRef CAS PubMed.
- R. Radi, L. Thomson, H. Rubbo and E. Prodanov, Arch. Biochem. Biophys., 1991, 288, 112 CrossRef CAS.
- X. Chen, R. Ferrigno, J. Yang and G. Whitesides, Langmuir, 2002, 7009 CrossRef CAS.
- A. Ranieri, F. Bernini, C. A. Bortolotti and E. Castellini, Catal. Sci. Technol., 2012, 2, 2206 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: CV, RR, SERR, UV-vis, HPLC and MS data. See DOI: 10.1039/c4sc02181a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |