
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light C–H amidation of heteroarenes with benzoyl azides†
E.
Brachet
,
T.
Ghosh
,
I.
Ghosh
and
B.
König
*
University of Regensburg Faculty of Chemistry and Pharmacy, Institute of Organic Chemistry, Universitätsstraße 31, 93053 Regensburg, Germany. E-mail: Burkhard.Koenig@chemie.uni-regensburg.de
First published on 30th October 2014
Abstract
Benzoyl azides were used for the direct and atom economic C–H amidation of electron rich heteroarenes in the presence of phosphoric acid, a photocatalyst and visible light. Hetero-aromatic amides are obtained in good yields at very mild reaction conditions with dinitrogen as the only by-product. The reaction allows the use of aryl-, heteroaryl- or alkenyl acyl azides and has a wide scope for heteroarenes, including pyrroles, indole, furan, benzofuran and thiophene giving good regio-selectivities and yields.
Introduction
Introducing amide moieties into organic molecules is a key transformation in organic chemistry due to the ubiquitous presence of this functional group in natural products, pharmaceuticals or functional materials.1 Amidation methods have been improved constantly in terms of functional group tolerance and conditions. Using transition metal catalysis, e.g. in Buchwald–Hartwig amidations, proved to be particularly advantageous.2 However, some limitations still exist: catalytic systems can be expensive, and often high reaction temperatures and pre-functionalized substrates, e.g. halides or pseudo-halides (Scheme 1a) are required. More recently, different groups have developed C–H amidations for heteroarenes or arenes with or without the use of a directing group (Scheme 1b).3 A drawback of this attractive strategy are the often required harsh reaction conditions. With the aim to improve direct C–H amidations towards milder conditions and a broader reaction scope we have developed a photocatalytic4 variant of the reaction (Scheme 1c).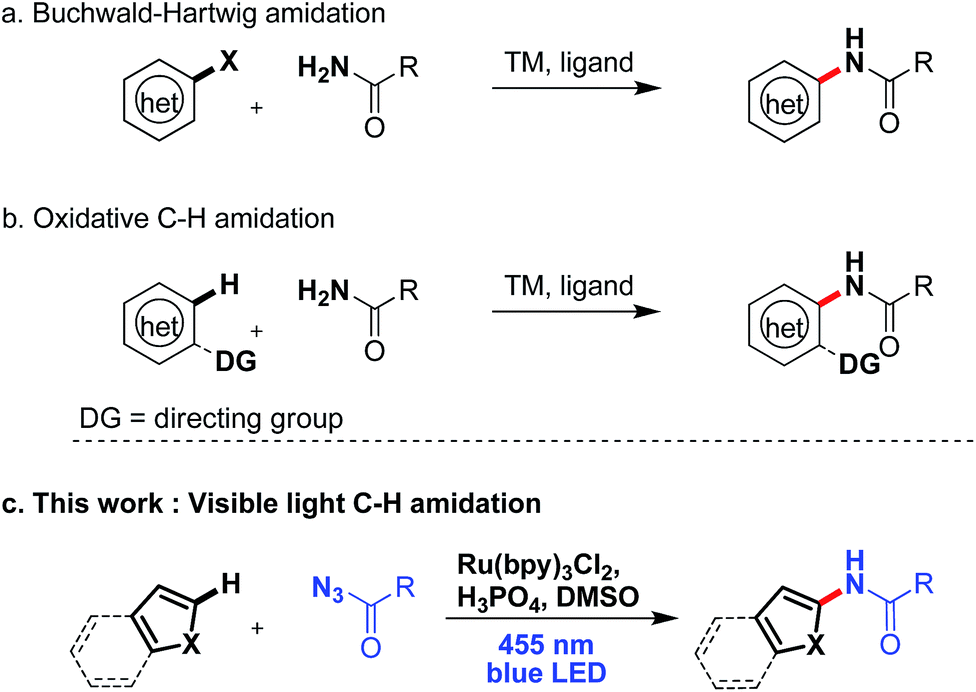 | ||
| Scheme 1 Catalytic aromatic amidation reactions. | ||
Very few examples of photocatalyzed C–H amidations have been reported in the literature. MacMillan and coworkers5 described in 2013 the amidation of enamines yielding α-amino aldehydes with good yields and excellent stereocontrol. A second example was reported by Sanford et al.6 exploring the reactivity of N-acyloxyphtalimides under visible light to generate phthalimide radicals that add to arenes and heteroarenes with good to excellent yields. This methods has been recently extended to N-chlorophthalimide as aminating agent under photocatalytic conditions.7 Yu et al. used activated hydroxylamines introducing amine groups into heteroarenes.8 The reported methods require a functionalized aminating reagent5,8 or yield phthalimide moieties.6,7
Inspired by this work we developed an alternative photocatalytic method for the functionalization of heteroarenes with aromatic amides, which may be particularly useful in synthesis. Many bioactive compounds show this structural motif and typical examples are found among analgestic and anti-parasitic compounds or hepatitis C drugs.9 A key advantage of our method is the introduction of the amide at a late stage of a synthesis of a more complex structure.
As aminating agents we choose benzoyl azides, which are easily accessible and allow for the introduction of simple benzamides into a substrate, which was so far not possible using visible light photocatalysis. Dinitrogen is the only stoichiometric by-product. Benzoyl azides have been used previously in transition metal catalysis, but the reaction requires the presence of a directing group.10 Exposed to UV irradiation11 benzoyl azides give oxazolines, dioxazoles or aziridines, but no conversion of heterocycles or formal amidation was observed under these conditions.12 We activate benzoyl azides using visible light and a photocatalyst allowing a direct C–H amidation of heteroarenes.
Results and discussion
Optimization of the reaction conditions
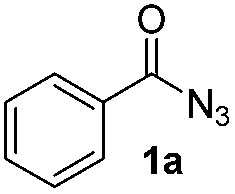
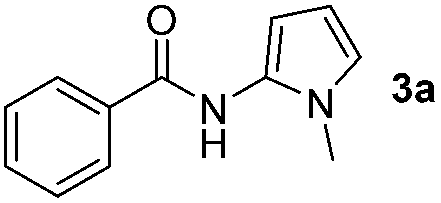
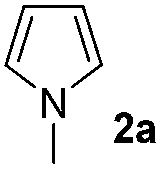
The reaction of benzoyl azide 1a with N-methylpyrrole 2a was used to establish and optimize the conditions. The results of the optimization are summarized in Table 1. Using ruthenium(II)trisbipyridine dichloride (A) as the photosensitizer in DMSO with blue light irradiation did not yield any amidation product. Part of the benzoyl azide is converted into the corresponding isocyanate by Curtius rearrangement and benzamide, as the nitrene hydrogen abstraction product (see ESI† for GC-MS analyses). The addition of an acid as additive is essential for the formation of the desired amidation product. Isocyanates are not stable under these conditions and only their decomposition products are observed. Initial low amidation product yields of 10% were improved to 40% after 12 h of irradiation using phosphoric acid, while changing the photocatalyst or the solvent had no significant effect on the product yield. Working at lower concentrations gave 65% product yield already after 4 h of blue light irradiation. Control experiments showed that the photocatalyst, irradiation, and acid are required to get full conversion of the starting material (entries 1, 15 and 16). The amount of ruthenium catalyst can be reduced to 1 mol% to provide an acceptable product yield of 51%.|
|
|||||
|---|---|---|---|---|---|
|
|
|||||
| Entry | Cata | Additiveb | Solvent | Conv. [%] | Yieldc |
| a 2.5 mol% of catalyst. b 2 equiv. of additive with respect to 1a. c Isolated yields. d Phenylisocyanate and benzamide are obtained. e Reaction conditions: 1a (0.34 mmol, 1 equiv.), 2a (1.7 mmol, 5 equiv.), additives (0.68 mmol, 2 equiv.) and photocatalyst (8.5 μmol, 2.5 mol%) in dry solvent (0.09 M) under N2 was irradiated with blue light for 4 h. f 1 mol% of the catalyst used. g 1 equiv. of N-methylpyrrole. h Without light. | |||||
| 1 | A | — | DMSO (0.34 M) | ∼35% | —d |
| 2 | A | (BnO)2P(O)OH | DMSO (0.34 M) | 100 | 10a% |
| 3 | A | (BnO)2P(O)OH | DMSO (0.34 M) | 100 | 30% |
| 4 | A | Benzoic acid | DMSO (0.34 M) | 100 | 8% |
| 5 | A | Acetic acid | DMSO (0.34 M) | 100 | 10% |
| 6 | A | PTSA | DMSO (0.34 M) | 100 | 0% |
| 7 | A | H3PO4 | DMSO (0.34 M) | 100 | 40% |
| 8 | B | H3PO4 | DMSO (0.34 M) | 60 | — |
| 9 | C | H3PO4 | DMSO (0.34 M) | 75 | — |
| 10 | A | H3PO4 | DMF (0.34 M) | 60 | — |
| 11 | A | H3PO4 | ACN (0.34 M) | 20 | — |
| 12 | A | H3PO4 | DMSO (0.09 M) | 100 | 65e% |
| 13 | A | H3PO4 | DMSO (0.09 M) | 100 | 51f% |
| 14 | A | H3PO4 | DMSO (0.09 M) | 95 | 30g% |
| 15 | — | H3PO4 | DMSO (0.09 M) | 0 | — |
| 16 | A | H3PO4 | DMSO (0.09 M) | 0 | —h |

Scope of the reaction
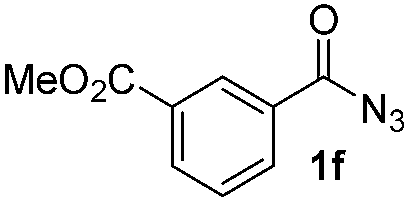
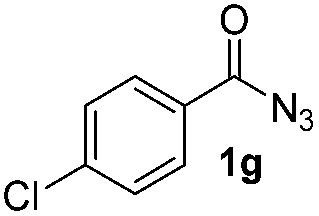
Having established the reaction conditions we explored the scope of benzoyl azides (Table 2). Benzoyl azides (1a–i) are readily obtained from their corresponding benzoyl chlorides and sodium azide. Gratifyingly, differently substituted benzoyl azides react with N-methylpyrrole 2a in good yields and a perfect selectivity for position 2 as confirmed by the crystal structure analysis of compound 3b (Table 2, entry 2). Benzoyl azides bearing electron donating groups or electron withdrawing groups are tolerated. The sterically hindered 1-naphthoyl azide 1c reacts, albeit with a lower yield of 47%. Sensitive groups like cyano or ester moieties are well tolerated (compound 3e and 3f respectively). It was reported that benzoyl azides react under light irradiation with cyanide groups to give oxadiazole,11,12 but in our conditions we only obtained the desired product in 63% yield. Product 3g bearing a chlorine atom allows for subsequent functionalization by transition metal mediated cross-coupling,13 and the use of heteroaryl acyl azide 1h yields diheteroaryl amide 3h in 49%. However, alkyl, phenyl, diphenylphosphoryl or benzyl acyl azides do not react under the photocatalytic conditions (entry 9). We explain this selectivity and limitation in substrate scope by the available energy of the long-lived [Ru(bpy)32+]* triplet state (46 kcal mol−1), which is sufficient for energy transfer to acyl azides (first electronically excited triplet state ∼41 kcal mol−1), but not for e.g. phenyl azide (68 kcal mol−1) (vide infra for mechanistic proposal and ESI† for data and references).|
|
|||
|---|---|---|---|
| Entry | Benzoyl azide | Product | Yieldb [%] |
| a Reaction conditions: 1a–i (0.34 mmol, 1 equiv.), 2a–i (1.7 mmol, 5 equiv.), H3PO4 (0.68 mmol, 2 equiv.) and Ru(bpy)3Cl2·6H2O (8.5 μmol, 2.5 mol%) in dry DMSO (0.09 M) were irradiated with blue light under N2. b Isolated yield, average of two reactions. | |||
| 1 |
|
|
65% |
| 2 |
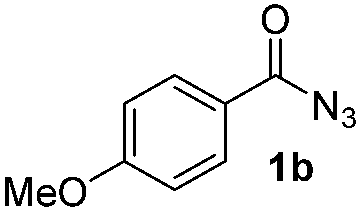
|
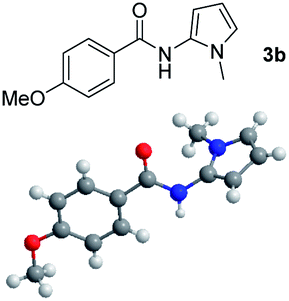
|
71% |
| 3 |
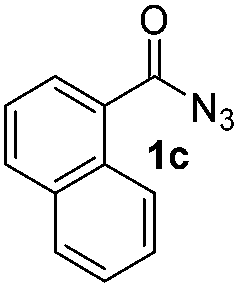
|
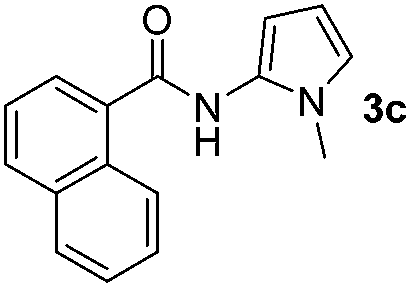
|
47% |
| 4 |
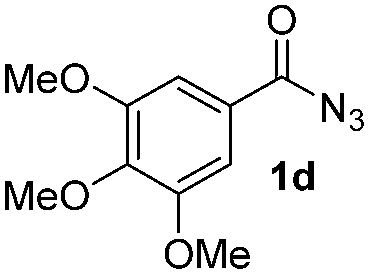
|
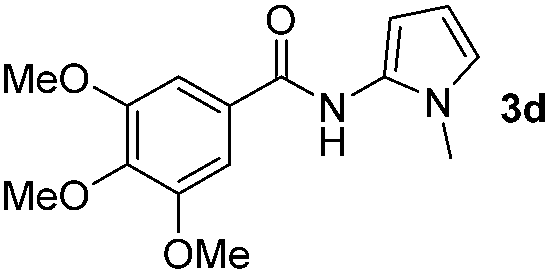
|
54% |
| 5 |
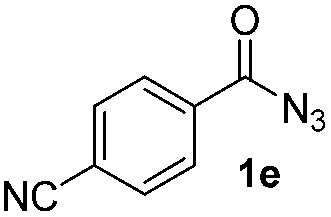
|
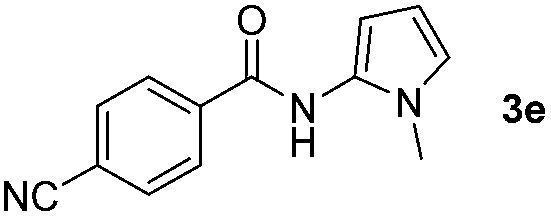
|
63% |
| 6 |
|
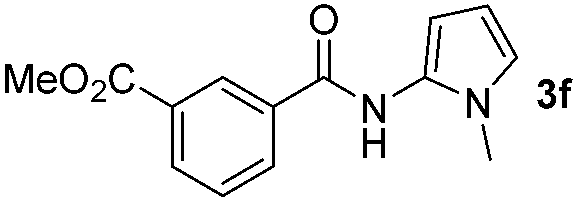
|
46% |
| 7 |
|
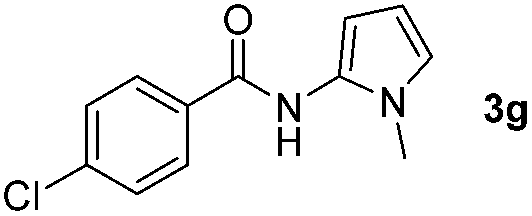
|
61% |
| 8 |

|
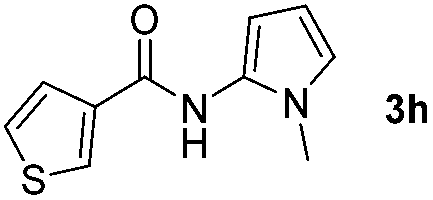
|
49% |
| 9 |
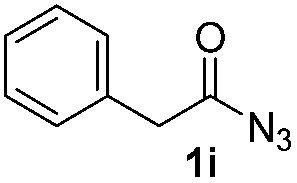
|

|
0% |
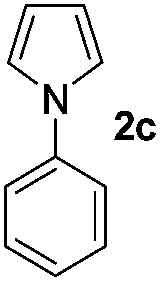
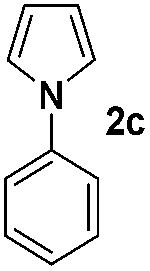
We then explored the scope of the reaction towards substituted pyrroles and other heterocycles like indole, furan, thiophene, and benzofuran (Table 3). Substituted pyrroles; either with electron withdrawing or electron donating groups gave the corresponding products in moderate to good yields of up to 88%.
|
|
||||
|---|---|---|---|---|
| Entry | Benzoyl azide | Heteroarene | Product | Yieldb [%] |
| a Reaction conditions: 1a–k (0.34 mmol, 1 equiv.), 2a–k (1.7 mmol, 5 equiv.), H3PO4 (0.68 mmol, 2 equiv.) and Ru(bpy)3Cl2·6H2O (8.5 μmol, 2.5 mol%) in dry DMSO (0.09 M) were irradiated with blue light under N2. b Isolated yield, average of two reactions. | ||||
| 1 |
|
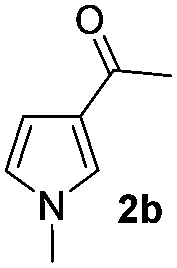
|
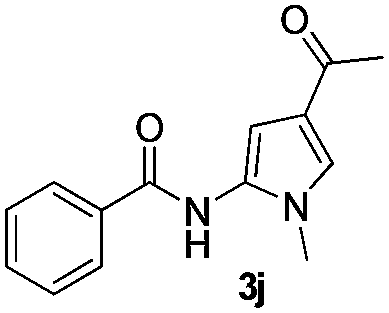
|
65% |
| 2 |
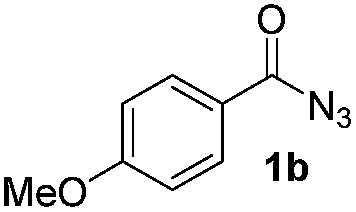
|
|
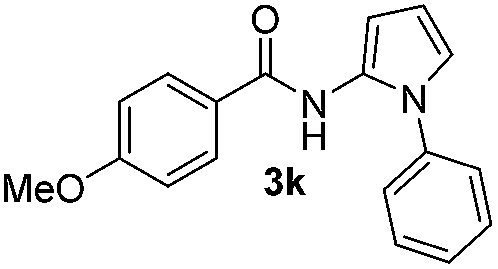
|
72% |
| 3 |
|
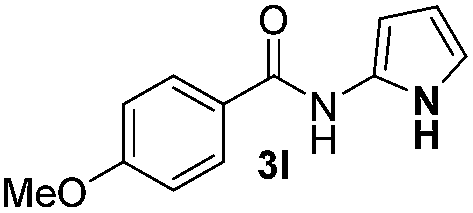
|
69% | |
| 4 |

|
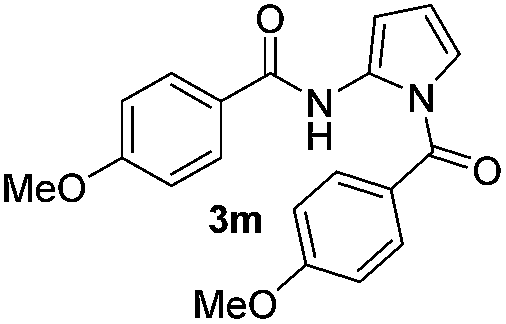
|
64% | |
| 5 |
|
|
49% | |
| 6 |
|
|
15% | |
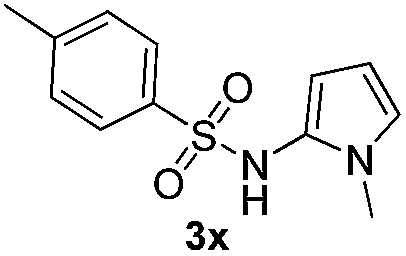
| 7 |
|
|
59% | |
| 8 |
|
|
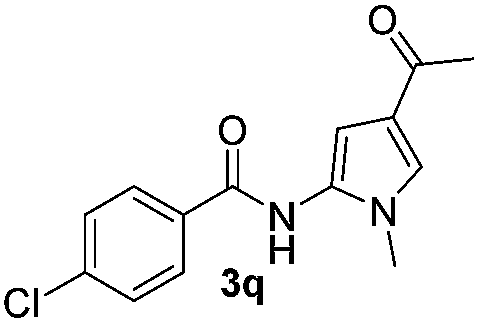
|
46% |
| 9 |
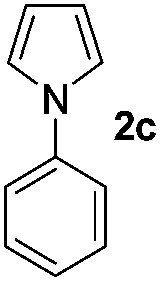
|
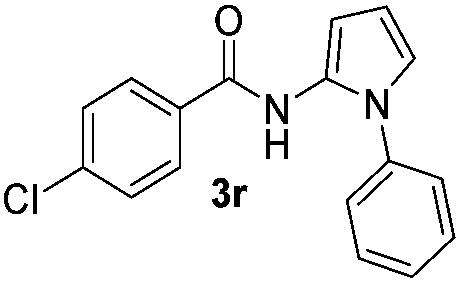
|
88% | |
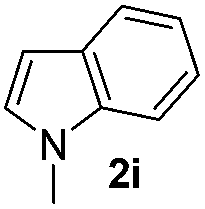
| 10 |
|
|
59% | |
| 11 |
|
|
35% | |
| 12 |
|
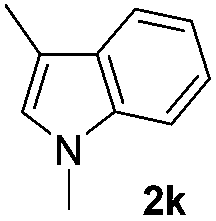
|

|
61% |
| 13 |
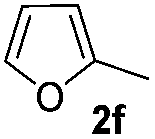
|
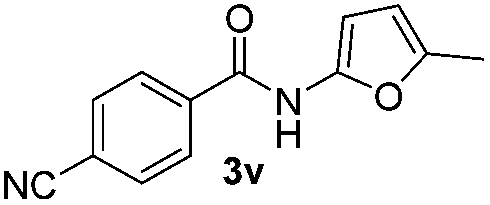
|
44% | |
| 14 |
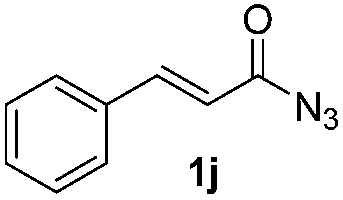
|
|
|
55% |
| 15 |
|
|
|
52% |
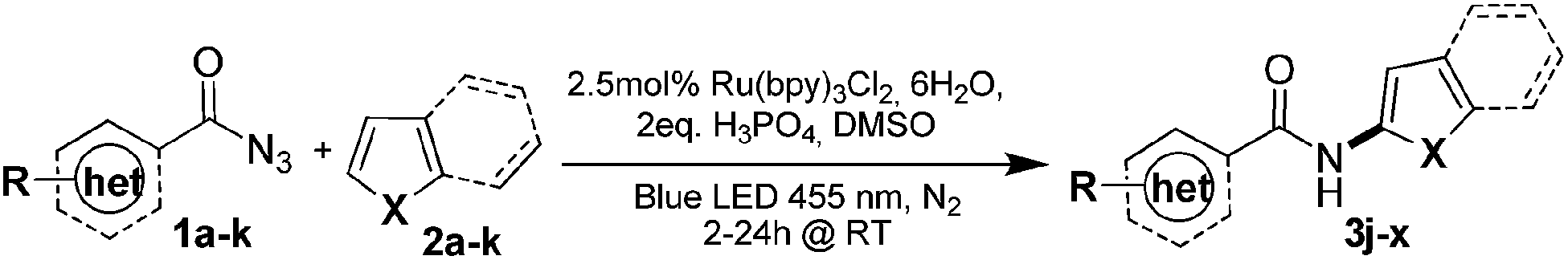
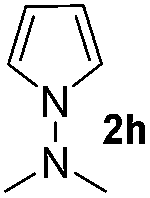
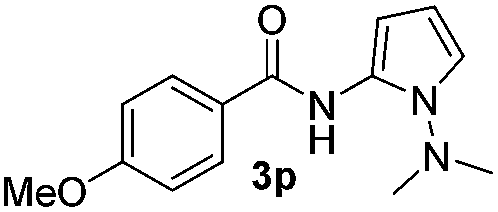
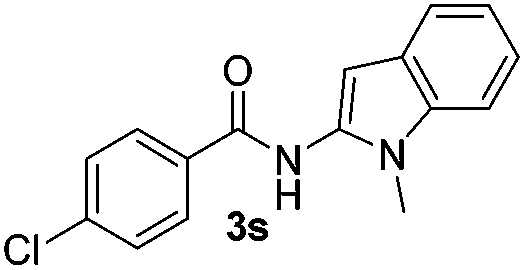
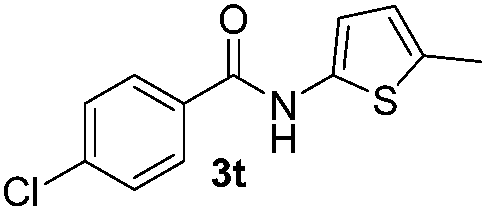
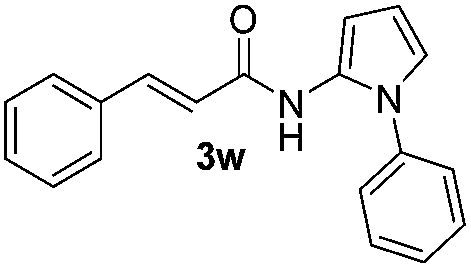
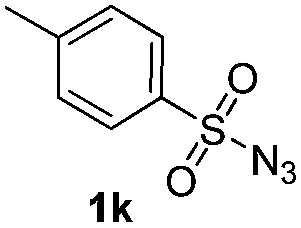
Protection of the N–H moiety of pyrrole is not necessary, which is advantageous for the functionalization of such heterocycles and their late stage amidation.14 (Table 3, entry 3). N-Aminoazoles are found in bioactive compounds,15 but their sensitive hydrazine group limits a direct functionalization in synthesis.16 Visible light photocatalysis allows the conversion with benzoyl azide 1b yielding the substituted N-aminopyrrole 3p in 59%. Indoles, furan and thiophene gave the expected amidation products in 35–61% yield. Analogous to benzoyl azides, we investigated the reaction with alkenyl acyl azide and sulfonylazide, which are cleanly converted to the corresponding products in more than 50% yield (Table 3, entries 14 and 15).
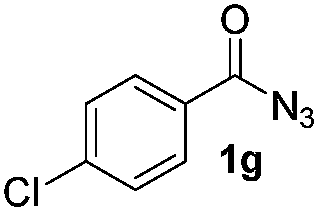
Only benzofuran reacts with a significantly lower yield of 15% (Table 3, entry 6), due to the formation of a side product (see mechanistic proposal). To illustrate the potential of the new method for synthesis, it was applied to the preparation of amide 6 having analgesic and anti-inflammatory properties (Scheme 2).7 The previously reported route used trifold substituted hydrazine 4, which is cyclized to the indole core and acylated to give the desired product 6. This route is limited to symmetrical diarylhydrazines to avoid product mixtures in the cyclization step. The photocatalytic C–H-amidation gave the target compound in one step with a slightly improved yield of 65% (Scheme 2).
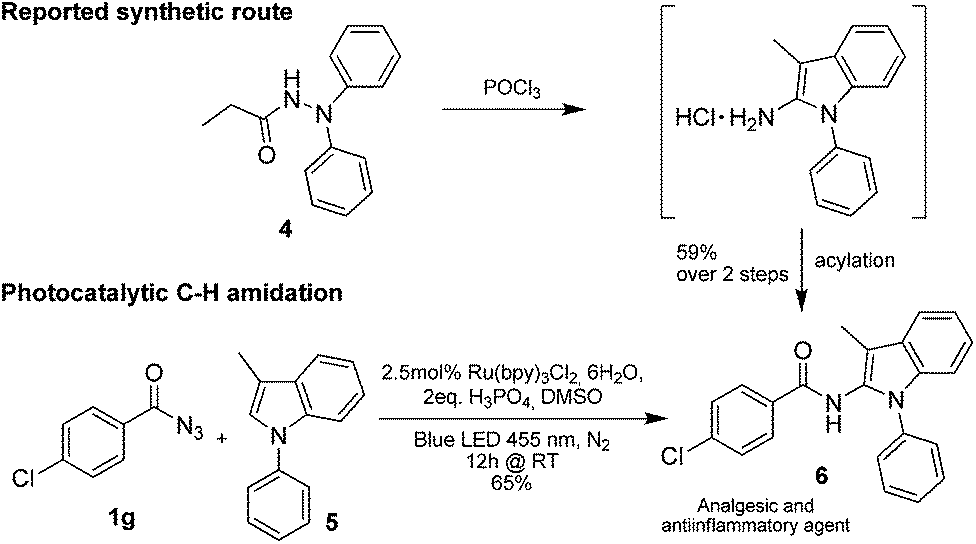 | ||
| Scheme 2 Synthesis of compound 6 using photocatalytic C–H amidation with benzoyl azide 1g. | ||
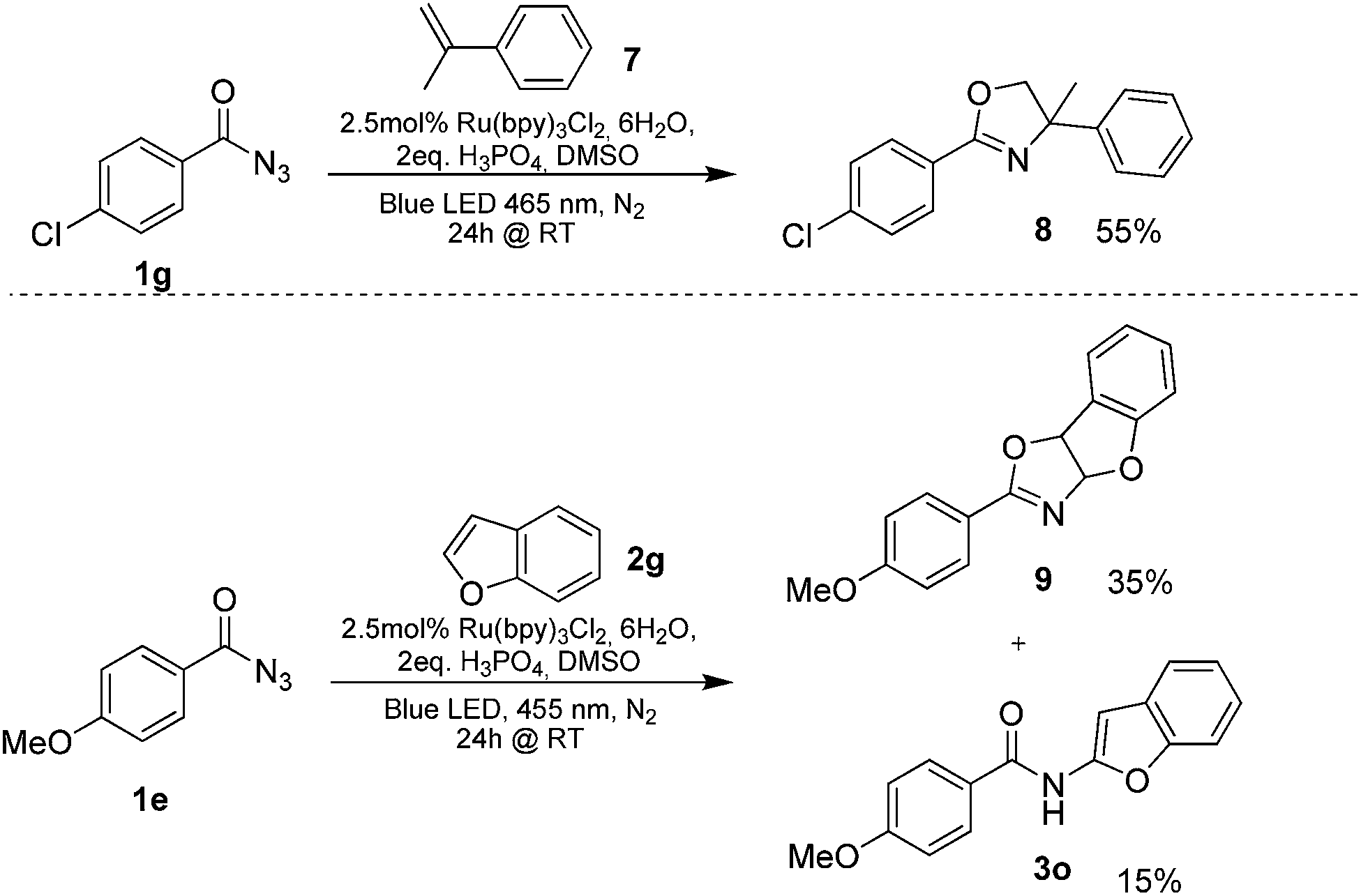 | ||
| Scheme 3 Isolated oxazolines from the photoreaction. | ||
Mechanistic investigations
Benzoyl azides are known to loose dinitrogen upon sensitization yielding nitrenes, which convert into isocyanates by the Curtius rearrangement.17 In the presence of hydrogen donors benzamides are obtained and the reaction with double bonds yields aziridines or oxazolines.11,12,18 The ratio between isocyanate and nitrene formation from the excited benzoyl azide is reported to be independent of the presence and the amount of alkenes as trapping reagent;19 the photophysical mechanism of sensitized and direct benzoyl azide decomposition has been investigated in detail.20 Some recent examples using azides in photocatalyzed reactions suggest a nitrene intermediate from single electron transfer or by sensitization.21 Based on these findings and a series of control experiments, we propose the following mechanism for the described amidation reaction: Ru(bpy)3Cl2 acts upon blue light irradiation as a triplet sensitizer for benzoyl azides. The interaction of the excited ruthenium complex and the azide was confirmed by a Stern–Volmer experiment (see ESI† for data). An electron transfer pathway is excluded, as benzoyl azides are insufficiently electron deficient to be easily reduced by ruthenium photocatalysis: the reduction potentials of [Ru(bpy)3Cl2]* and benzoyl azide 1b, respectively, are −0.89 and −1.49 V versus the saturated calomel electrode (SCE). Addition of TEMPO does not interfere with the reaction, which also indicates the absence of radical intermediates in this reaction. The energy transfer triggers the loss of dinitrogen yielding the benzoyl nitrene, which converts in part via the Curtius rearrangement to the corresponding isocyanate. GC-MS analysis of the reaction mixture in the absence of acid clearly identified the isocyanate. However, amidation products are only observed in the presence of an acid, e.g. H3PO4, in the reaction mixture. As neither the absorption of the ruthenium complex and the benzoyl azide, nor the emission of the ruthenium complex and the reduction potential of the benzoyl azide (from cyclic voltammetry) change by the addition of acid (see ESI† for data), the sensitization step is not affected.22 The benzoyl nitrene may be protonated under the strongly acidic conditions, as analogously reported for substituted aryl nitrenes,23 giving electrophilic nitrenium ions, which react with the electron rich heteroarene. The carbenium ion intermediate reacts under a loss of a proton and rearomatization to the amidation product.24 In the case of benzofurane and α-methylstyrene 7 the oxazoline formation25 is an alternative reaction pathway. Benzoyl azide 1g reacts with α-methylstyrene 7 under blue light irradiation yielding oxazoline 8 in 55% yield (Scheme 3), while in the reaction with benzofurane, oxazoline 9 was obtained as the major product accompanied by benzofuran amide 3o in 15%. Isolated oxazoline 9 does not convert into benzofuran 3o when exposed to the reaction conditions, which indicates that its formation is irreversible and it is not a reaction intermediate (Scheme 4).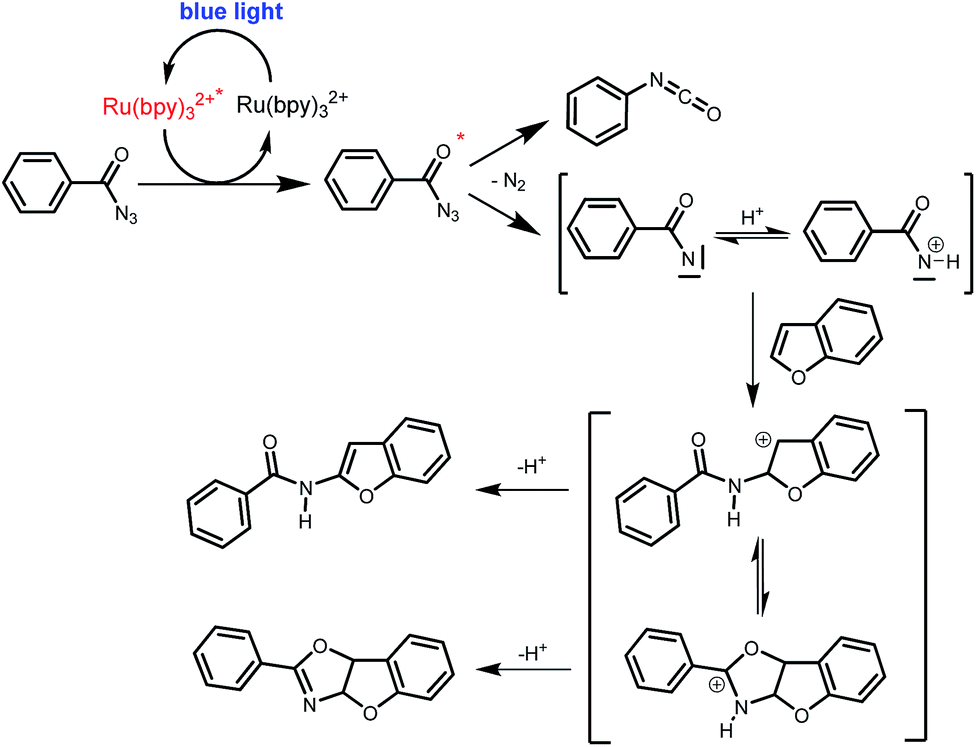 | ||
| Scheme 4 Proposed mechanism of the photo CH-amidation of benzofuran with benzoyl azide in the presence of acid, Ru(bpy)3Cl2 and blue light. | ||
Conclusions
We have reported the C–H amidation of heterocycles with benzoyl azides under very mild and practical conditions using visible light. The only byproduct of this atom economic process is dinitrogen. Heterocycles, such as pyrroles, indoles, furan, benzofuran or thiophene and substituted benzoyl azides give the corresponding amide coupling products in moderate to good yields in a single step. This first application of benzoyl azides in photocatalysis provides C–H amidation products of classic benzamides, which are typical structural motifs of many bioactive compounds. The photoreaction may therefore be a valuable alternative in the synthesis of bioactive heterocycles to the previously reported use of phthalimides or functionalized amines. A particular advantage of the new method is the amidation under mild conditions allowing for a late stage functionalization of complex heterocyclic molecules.Experimental section
Standard procedure for the photocatalyzed CH amidation reaction
In a 5 mL snap vial equipped with magnetic stirring bar Ru(bpy)3Cl2·6H2O, (0.025 equiv.), benzoyl azide (1 equiv.), o-H3PO4 (2 equiv.) and the heteroarene (5 equiv.) were dissolved in dry DMSO (0.09 mmol mL−1) and the resulting mixture was degassed by “pump–freeze–thaw” cycles (×2) via a syringe needle and filled with nitrogen. The vial was irradiated through the vial's plane bottom side using blue LEDs. After complete conversion of the starting material, as monitored by TLC, the pressure in the vial was released by a needle and the reaction mixture was transferred into a separating funnel, diluted with ethyl acetate and washed with 15 mL of water. The aqueous layer was washed three times with ethyl acetate. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuum. Purification of the crude product was achieved by flash column chromatography using petrol ether/ethyl acetate as eluent.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (GRK 1626, Chemical Photocatalysis) for financial support.Notes and references
- (a) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284 CrossRef CAS PubMed; (b) A. Ricci, Amino Group Chemistry: From Synthesis to the Life Science, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) E. C. Taylor and R. A. Jones, Pyrroles, Wiley, New York, 1990 Search PubMed; (d) R. J. Sundberg, Indoles, Academic, New York, 1996 Search PubMed.
- (a) D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13 RSC; (b) D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27 RSC; (c) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS PubMed; (d) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed.
- (a) M. L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901 RSC; (b) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092 RSC; (c) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (d) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926 RSC.
- Recent reviews on photocatalysis: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (b) J. Xuan and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (d) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (e) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem.–Eur. J., 2014, 20, 3874 CrossRef CAS PubMed; (f) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355 CrossRef CAS; (g) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688 RSC; (h) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS PubMed; (i) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef CAS PubMed; (j) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785 CrossRef CAS PubMed.
- G. Cecere, C. M. König, J. L. Alleva and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521 CrossRef CAS PubMed.
- L. J. Allen, P. J. Cabrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 5607 CrossRef CAS PubMed.
- H. Kim, T. Kim, D. G. Lee, S. W. Roh and C. Lee, Chem. Commun., 2014, 50, 9273 RSC.
- Q. Qin and S. Yu, Org. Lett., 2014, 16, 3504 CrossRef CAS PubMed.
- (a) L. Zhou, G. Stewart, E. Rideau, N. J. Westwood and T. K. Smith, J. Med. Chem., 2013, 56, 796 CrossRef CAS PubMed; (b) V. N. Barinova, V. P. Zhestkov, Yu. N. Portnov, S. E. Metkalova and Yu. V. Burov, Pharm. Chem. J., 1991, 25, 18 CAS; (c) R. Singh, U. Ramesh, J. Huang, S. D. Issakani, L. Tsvetkov, M. D. Petroski, PCT Int. Appl., WO 2008115259 A2 20080925, 2008.
- Selected references : (a) J. Ryu, J. Kwak, K. Shin, D. Lee and S. Chang, J. Am. Chem. Soc., 2013, 135, 12861 CrossRef CAS PubMed; (b) D. Lee, Y. Kim and S. Chang, J. Org. Chem., 2013, 78, 11102 CrossRef CAS PubMed; (c) J. Kim and S. Chang, Angew. Chem., Int. Ed., 2014, 53, 2203 CrossRef CAS PubMed; (d) K. Shin, J. Ryu and S. Chang, Org. Lett., 2014, 16, 2022 CrossRef CAS PubMed.
- In a single example the decomposition was achieved with visible light: K.-U. Clauss, K. Buck and W. Abraham, Tetrahedron, 1995, 51, 7181 CrossRef CAS.
- (a) K. Buck, D. Jacobi, Y. Plögert and W. Abraham, J. Prakt. Chem., 1994, 678 CrossRef CAS; (b) Y. Roeske and W. Abraham, Synthesis, 2001, 1125 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS PubMed.
- M. Su, N. Hoshiya and S. L. Buchwald, Org. Lett., 2014, 16, 832 CrossRef CAS PubMed.
- (a) R. C. Effland, J. T. Klein, L. Davis, G. E. Olson, Eur.Pat., EP0402752, 1990; (b) A. S. Gurkan, A. Karabay, Z. Buyukbingol, A. Adejare and E. Buyukbingol, Arch. Pharm., 2005, 338, 67 CrossRef CAS PubMed; (c) T. Itoh, M. Miyazaki, H. Maeta, Y. Matsuya, K. Nagata and A. Ohsawa, Bioorg. Med. Chem., 2000, 8, 1983 CrossRef CAS.
- (a) B. A. Frontana-Uribe, C. Moinet and L. Toupet, Eur. J. Org. Chem., 1999, 419 CrossRef CAS; (b) E. Brachet, S. Messaoudi, J.-F. Peyrat, J.-D. Brion and M. Alami, Adv. Synth. Catal., 2012, 354, 2829 CrossRef CAS.
- T. Curtius, Ber. Dtsch. Chem. Ges., 1890, 23, 3023 CrossRef.
- Review on products obtained from azides: (a) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef PubMed , For triazole formation, see; (b) G. Broggini, L. Garanti, G. Molteni and G. Zecchi, J. Chem. Res., 1998, 688 RSC; (c) J. Kubicki, Y. Zhang, J. Xue, H. L. Luk and M. Platz, Phys. Chem. Chem. Phys., 2012, 14, 10377 RSC , N-centered radicals; (d) M. Minozzi, D. Nanni and P. Spagnolo, Chem.–Eur. J., 2009, 15, 7830 CrossRef CAS PubMed.
- N. Gritsan and M. Platz, Photochemistry of Azides: The Azide/Nitrene Interface, in Organic Azides: Syntheses and Applications, ed. S. Bräse and K. Banert, Wiley, Chichester, 2010 Search PubMed.
- (a) J. Kubicki, Y. Zhang, J. Wang, H. L. Luk, H.-L. Peng, S. Vyas and M. S. Platz, J. Am. Chem. Soc., 2009, 131, 4212 CrossRef CAS PubMed; (b) K.-U. Clauss, K. Buck and W. Abraham, Tetrahedron, 1995, 51, 7181 CrossRef CAS.
- (a) Y. Chen, A. S. Kamlet, J. B. Steinman and D. R. Liu, Nat. Chem., 2011, 3, 146 CrossRef CAS PubMed; (b) E. P. Farney and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 793 CrossRef CAS PubMed; (c) J. Xuan, X.-D. Xia, T.-T. Zeng, Z.-J. Feng, J. –R. Chen, L.-Q. Lu and W. –J. Xiao, Angew. Chem., Int. Ed., 2014, 53, 5653 CrossRef CAS PubMed.
- Isocyanates are not stable under the reaction conditions containing acid and traces of water; GC-MS analysis of the crude reaction mixture shows decomposition products, e.g. the corresponding aniline. Benzamides as hydrogen abstraction products of the nitrene are identified. E. Eibler and J. Sauer, Tetrahedron Lett., 1974, 30, 2565 CrossRef.
- (a) V. Voskresenska, R. M. Wilson, M. Panov, A. N. Tarnovsky, J. A. Krause, S. Vyas, A. H. Winter and C. M. Hadad, J. Am. Chem. Soc., 2009, 131, 11535 CrossRef CAS PubMed; (b) J. Wang, G. Burdzinski, Z. Zhu, M. S. Platz, C. Carra and T. Bally, J. Am. Chem. Soc., 2007, 129, 8380 CrossRef CAS PubMed.
- A. Makino and S. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1251 CrossRef CAS.
- (a) J. Du Bois, C. S. Tomooka, J. Hong and E. M. Carreira, J. Am. Chem. Soc., 1997, 119, 3179 CrossRef CAS; (b) D. Ferrarris, W. J. Drury III, C. Cox and T. Lectka, J. Org. Chem., 1998, 63, 4568 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization and synthesis of new compounds, general procedures, proton and carbon NMR spectra, crystal structure analysis, gas chromatographic analyses. CCDC 1017661. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02365j |
| This journal is © The Royal Society of Chemistry 2015 |