
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extremely strong tubular stacking of aromatic oligoamide macrocycles†
Mark A.
Kline‡
a,
Xiaoxi
Wei‡
a,
Ian J.
Horner
a,
Rui
Liu
a,
Shuang
Chen
b,
Si
Chen
c,
Ka Yi
Yung
a,
Kazuhiro
Yamato
a,
Zhonghou
Cai
c,
Frank V.
Bright
a,
Xiao Cheng
Zeng
b and
Bing
Gong
*ad
aDepartment of Chemistry, the State University of New York at Buffalo, Buffalo, New York, USA 14260. E-mail: bgong@buffalo.edu; Web: http://www.chemistry.buffalo.edu/people/faculty/gong/
bDepartment of Chemistry, University of Nebraska-Lincoln, Lincoln, Nebraska 68588, USA
cX-ray Science Division, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, IL 60439, USA
dCollege of Chemistry, Beijing Normal University, Beijing 100875, China
First published on 16th September 2014
Abstract
As the third-generation rigid macrocycles evolved from progenitor 1, cyclic aromatic oligoamides 3, with a backbone of reduced constraint, exhibit extremely strong stacking with an astoundingly high affinity (estimated lower limit of Kdimer > 1013 M−1 in CHCl3), which leads to dispersed tubular stacks that undergo further assembly in solution. Computational study reveals a very large binding energy (−49.77 kcal mol−1) and indicates highly cooperative local dipole interactions that account for the observed strength and directionality for the stacking of 3. In the solid-state, X-ray diffraction (XRD) confirms that the aggregation of 3 results in well-aligned tubular stacks. The persistent tubular assemblies of 3, with their non-deformable sub-nm pore, are expected to possess many interesting functions. One such function, transmembrane ion transport, is observed for 3.
Introduction
Tubular structures, with their cylindrical shapes, defined outer and inner surfaces, and internal pores, provide versatile structural modules for constructing functional structures.1,2 Nanopores with precisely defined diameters of less than 2 nm, such as those of carbon nanotubes (CNTs), exhibit many fascinating properties.3,4 Compared with carbon nanotubes, organic nanotubes5 have unique advantages such as ready functionalization, versatile compatibility, and modular assembly. Among known strategies,2,5,6 the superposition of cyclic building blocks5a,e–g,7 represents an approach that combines the ready modifiability of small molecules and the power of self-assembly, leading to nanotubes with structural and functional tunability. However, the alignment of cyclic molecules based on non-covalent forces is often impeded by limitations such as the poor directionality of π–π stacking and/or the limited strength of hydrogen-bonding, especially in polar media, which frequently lead to undesired outcomes upon even a slight structural modification on an otherwise promising building block.Given the many remarkable functions exhibited by or expected of non-deformable nanopores,3,4 organic nanotubes resulted from the stacking of rigid macrocycles, which contain non-collapsible inner pores, is especially attractive.8 While many rigid macrocycles such as those with π-conjugated9,10 and other backbones,11–15 along with tubular stacks of some of these macrocycles in the solid and liquid crystalline phases,5d–f,7 are known, self-assembling nanotubes that stably exist in solution are rare. The availability of stable nanotubular assemblies should greatly advance the development of systems with properties typically associated with biological structures. Achieving this objective requires the strong, directional stacking of cyclic building blocks.
Over the years, we have developed several different classes of rigid macrocycles containing non-deformable internal cavities.16 The first generation of such molecules are aromatic oligoamide macrocycles 1,14a which were found to form efficiently in one pot while we attempted to prepare folding aromatic oligoamides17,18 and polyamides19 having similar backbones. The one-pot macrocyclization we found has led to rigid macrocycles containing internal cavities of 5 to 30 Å across.16,20 The benzene residues of macrocycles 1 are connected via amide groups engaging in highly favourable three-centre intramolecular hydrogen-bonding interaction21 that constrains the macrocyclic backbones. With fully constrained, non-deformable backbones, macrocycles 1 were observed to strongly aggregate in solution and form tubular stacks in the solid state.22
To better control the alignment of these molecules, amide side chains are attached to 1, which led to the second-generation macrocycles 2. Being flanked by alkoxy side chains, the amide side chains of 2 are perpendicular to the benzene rings to which they are attached and should thus be predisposed to intermolecular H-bonding that enforces the macrocycles to stack on top of one another into a tubular stack. Surprisingly, studies using multiple analytical techniques suggested that macrocycles 2 underwent insignificant aggregation.23 It seemed that the amide side chains of 2 not only failed to engage in intermolecular H-bonding, but also abolished the otherwise strong aggregation of 1.
The unexpected behavior of macrocycles 2 may be due to steric crowdedness that hampers side-chain H-bonding and backbone π–π stacking. Such a besetment could be evaded by removing the alkoxy groups flanking the amide side chains of 2, which leads to 3, our third-generation aromatic oligoamide macrocycles.24 Herein, we report the unusually strong tubular stacking of 3. It was found that, in solution, macrocycles 3 underwent aggregation that was mediated by their backbones and weakened by polar solvents. The self-association of 3 is extremely strong, with a strength that is, to the best of our knowledge, unprecedentedly high. The strong association of 3 results in individually dissolved columnar stacks that dominate at low concentrations and further pack at elevated concentrations. X-ray diffraction (XRD) revealed the columnar stacks of 3 and their hexagonal packing in the solid state. Consistent with their reliable tubular self-assembly, macrocycles 3 were found to mediate efficient transmembrane transport of ions.
Results and discussion
Backbone-mediated aggregation
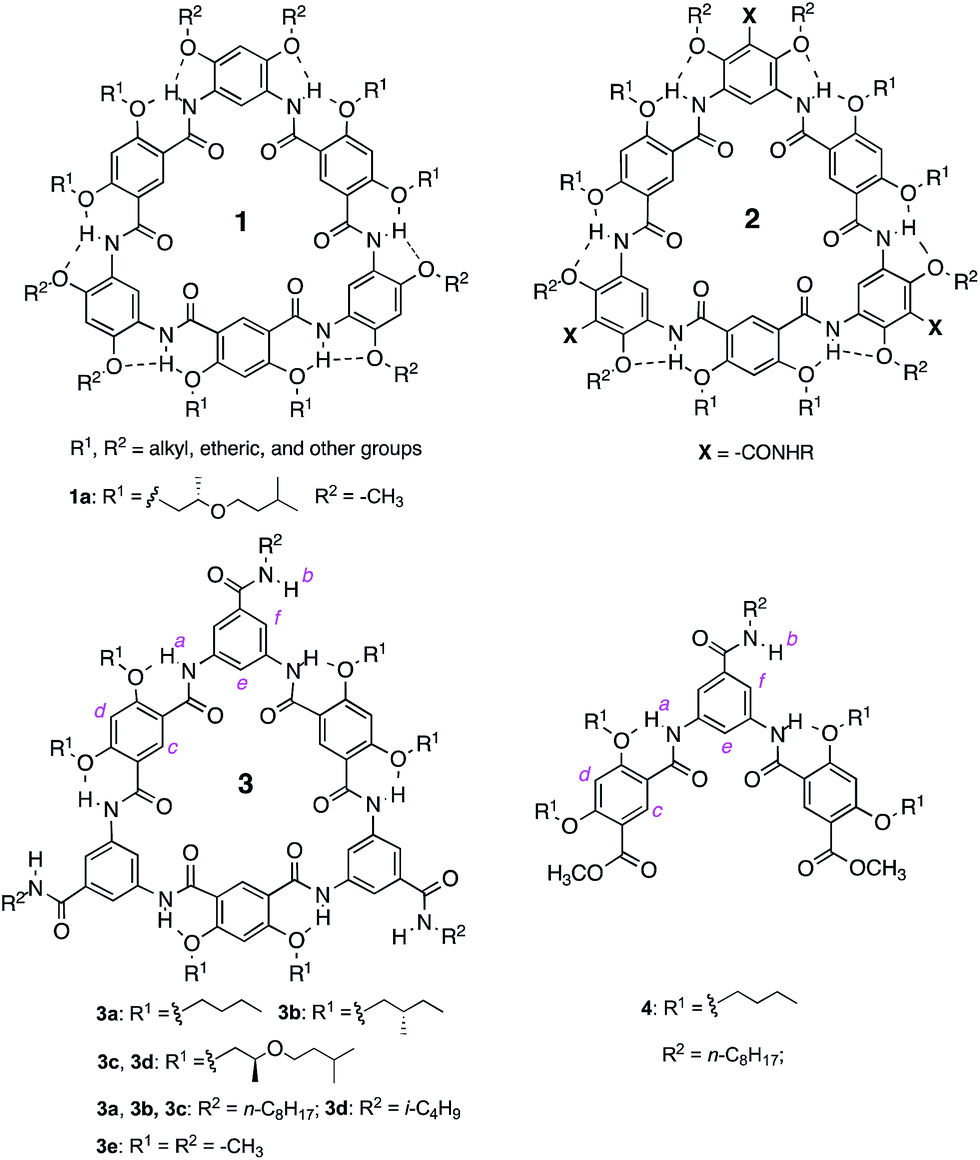
The aggregation of macrocycles 3a–d was first indicated by their 1H NMR spectra. In CDCl3, no signals could be found in the amide and aromatic region. The only peaks observed are those from 0.5 ppm to 1.8 ppm, which belong to the terminal alkyl groups of the side chains (Fig. S1 in the ESI†). This observation suggests that 3a–d undergo decreased molecular motion due to aggregation involving their oligoamide backbones. In DMF-d7 or DMSO-d6, the 1H NMR spectra of 3a–d contain well dispersed signals (Fig. S2†), suggesting that the aggregation of these molecules is interrupted in polar solvents.To gain additional insights, the 1H NMR spectra of 3a were recorded in CDCl3 (1 mM) containing incremental proportions of DMSO-d6. The signals of amide and aromatic protons only become obvious in solvents with 30% or more DMSO-d6 (Fig. S3†). The same trend was observed with increasing ratios of DMF-d7 (Fig. S4†). In comparison to macrocycles 1 that gave well dispersed 1H NMR signals with as few as 5% DMSO-d6 or DMF-d7 in CDCl3,14a the aggregation of 3a, as indicated by the effect of DMF or DMSO, is much stronger. Plotting the chemical shifts of the amide protons of 3a and those of 4 against DMSO-d6 contents indicates that amide protons b of 3a and 4 follow the same trend with changing solvent polarity (Fig. 1a). This observation suggests that the side-chain NH groups of 3a, like that of the molecularly dissolved 4 (Fig. S5†), are exposed to solvent. In contrast, with increasing ratios of DMSO-d6, the downfield shifts of amide protons a are non-linear for 3a, and linear for trimer 4 (Fig. 1b). The different behavior of protons a of 3a and 4 can be explained by the stacking of 3a in CDCl3, which shields the oligoamide backbone from solvent molecules. Increasing solvent polarity weakens and eventually breaks up the aggregates, which exposes individual molecules, and hence protons a, of 3a to solvent molecules.
 | ||
| Fig. 1 Plots of the chemical shifts of (a) protons b and (b) protons a of 3a (1 mM, blue) and 4 (1 mM, red), against volume percent DMSO-d6 in CDCl3. | ||
Experiments based on diffusion-ordered spectroscopy (DOSY) in CDCl3 containing 40% DMF-d7 clearly demonstrated the aggregation of 3a and the lack of aggregation in pure DMF-d7 (Fig. S6†). Dynamic light scattering (DLS) was also used to compare the aggregation of 3a and 1a. In DMF, neither 3a nor 1a formed any noticeable aggregate. In contrast, aggregates of 3a (1 mM), with an average hydrodynamic diameter [(2.6 ± 0.6) × 104 nm] that is much larger than that [(250 ± 18) nm] of the aggregates formed by 1a were observed in chloroform. The DLS results corroborate those from DOSY and 1H NMR, confirming that the aggregation of 3a is much stronger than that of 1a.
Ground-state aggregation
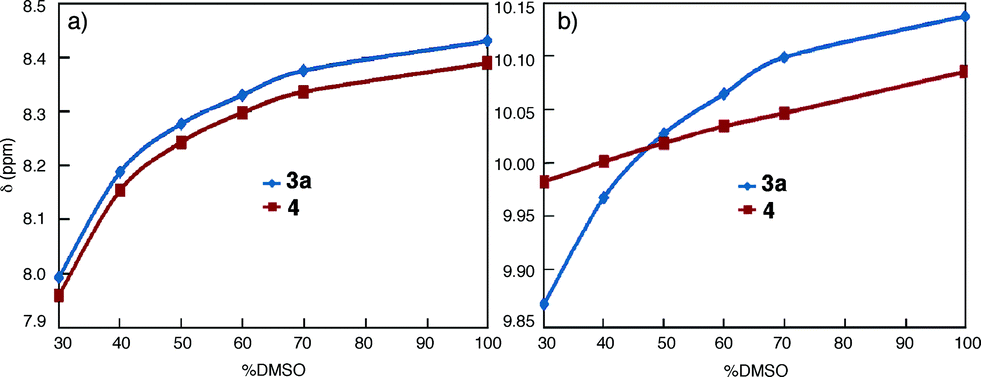
Macrocycles 1a and 3a were then examined at 1 μM, a concentration that is three orders of magnitude lower than those used for NMR and DLS studies, with fluorescence spectroscopy. In DMF, emission bands centred at 350 nm, which can be ascribed to molecularly dissolved monomers, are observed (Fig. 2a). In CHCl3, macrocycles 1a and 3a display broad, red-shifted bands at 450 nm (Fig. 2b). The 450 nm bands are reminiscent of excimer fluorescence typical of π-stacked aromatic rings25 that exist in the ground state (i.e., due to aggregation) and give “excimer-like” emission.26 Consistent with the ground-state association of 3a, monitoring the emission bands of 3a (125 nM and 0.1 pM in CHCl3) at 350 nm and 450 nm revealed two different peaks at 260 nm and 280 nm, respectively, in the excitation spectra (Fig. S7†).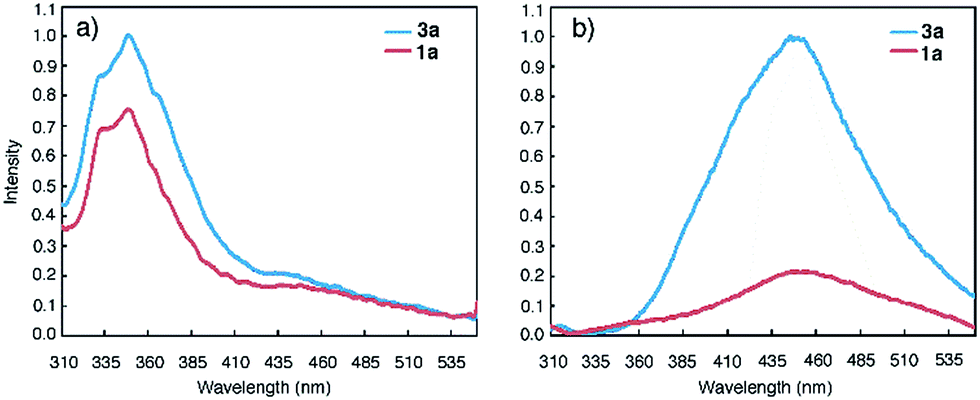 | ||
| Fig. 2 Fluorescence spectra of 1a (red) and 3a (blue) in (a) DMF (1 μM) and, (b) CHCl3 (1 μM) using an excitation wavelength of 282 nm. | ||
Emission spectra collected at reduced concentrations in CHCl3 indicate that 3a remains aggregated down to 1 pM and exists as monomers only at 0.1 pM (Fig. 3a). Assuming that, at 1 pM, macrocycle 3a is involved in a monomer-dimer equilibrium10b with a 10% dissociation, a lower limit of 4.5 × 1013 M−1 for the dimerization constant can be estimated, which suggests that 3a engages in remarkably strong association. In contrast, the fluorescence spectra of 1a recorded below 100 nM contain emission bands at both 350 nm and 450 nm; at 10 nM, the emission band at 450 nm greatly weakens while the one around 350 nm becomes dominant (Fig. 3b). These observations demonstrate that the aggregation of 3a is several orders of magnitude stronger than that of 1a.
 | ||
| Fig. 3 Fluorescence spectra of (a) 3a and (b) 1a recorded at different concentrations in CHCl3 using an excitation wavelength of 282 nm. | ||
The effect of solvent polarity on the aggregation of 3a and 1a (1 μM) was revealed by plotting E450/E350, the ratios of normalized fluorescence emissions at 450 nm and 350 nm, as a function of volume percent CHCl3 in DMF (Fig. S8 and Table S1†). The E450/E350 ratio rises with increasing volume percent CHCl3 in DMF. In contrast, the intensity of the 450 nm band of 1a (1 μM) is much less prominent than that of 3a. These observations confirm the high sensitivity of the aggregation of 3a, even at a very low concentration, to solvent polarity, which implies the involvement of a strong dipole–dipole factor in the self-association of this compound.
Insights from computational study
To gain insights into the strong self-association of 3, ab initio computation was performed on a dimer consisting of two stacked molecules of model macrocycle 3e. The potential energy as a function of the relative stacking angle between two such macrocyclic units was calculated at the density-functional theory (DFT) level of M06-2X/6-31G(d), with the molecular structure being optimized at the DFT BLYP-D3/GTH level with inclusion of dispersion correction (see the ESI†). It was found that the dimer with a stacking angle of 60.5° gave the strongest binding, with a binding energy of −49.77 kcal mol−1. In contrast, the dimer involving two “eclipsed” molecules, i.e., with a stacking angle of 0°, had a binding energy of −24.42 kcal mol−1. The drastically enhanced stability of the most stable dimer may be explained by the highly cooperative interaction of local dipoles. With a stacking angle of 60°, the two different types of benzene residues, one derived from the diacid monomer and the other derived from the diamine monomer, stack directly on top of each other and undergo favourable dipole–dipole attraction. The highly cooperative action of six such pairs of benzene residues is most likely responsible for the observed strong association of 3a.Time course of aggregation: a two-stage process
The progress of the aggregation of 1a and 3a was monitored by following the intensity of the 450 nm band upon adding a solution of 3a or 1a in DMF into CHCl3. It was found that the rate of aggregation increased with increasing proportion of CHCl3 (Fig. S9†). Fig. 4 shows the time courses for the 450 nm band of 1a or 3a (1 μM) in CHCl3 and DMF (1/1, v/v). The aggregation of 3a involves two stages: an initial rapid growth phase that lasts for about 37 min, followed by a much slower growth phase (Fig. 4a, red). In the same solvent, the aggregation of 1a is negligible, with no obvious increase being observed for its 450 nm band (Fig. 4a, blue). Lowering the concentration of 3a decreased the rates of aggregation considerably (Fig. S10†) and, below certain concentration, resulted in the disappearance of the second growth phase, even at greatly increased CHCl3 content. For example, at 10 nM, macrocycle 3a, being aggregated as shown by its 450 nm band (Fig. S11†), gives one growth phase (Fig. 4b). At 1 pM in the same solvent, macrocycle 3a exists mainly as monomers (Fig. S12†) and, consistent with the lack of aggregation, shows insignificant increase of emission at 450 nm (Fig. S13†).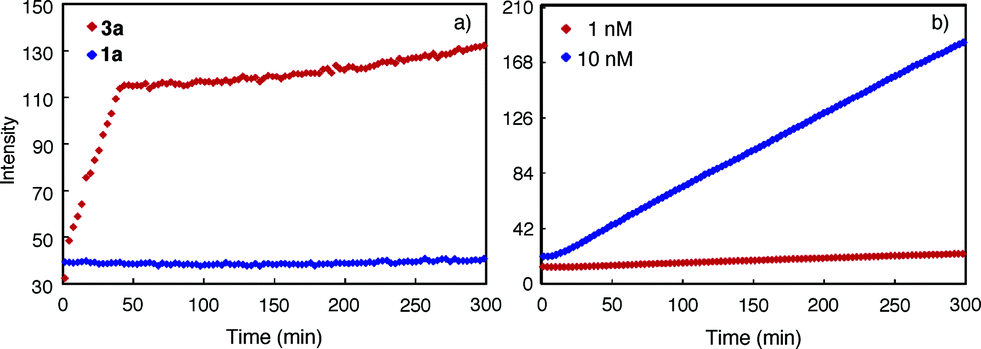 | ||
| Fig. 4 Change of fluorescence intensity at 450 nm as a function of time. (a) Upon mixing 3a or 1a dissolved in DMF with CHCl3. The final concentration of 1a or 3a is 1 μM and, (b) 3a (10 nM and 1 nM), in the mixed solvent of CHCl3 and DMF (1/1, v/v). The measurements were carried out using an excitation wavelength of 281 nm. | ||
The observed fluorescence emission and two-stage time course associated with the aggregation of 3a may be rationalized by a model that involves an initial (fast) assembling period during which the molecules of 3a undergo strong, backbone-mediated stacking, followed by a second (slow) phase in which the columnar stacks of 3a further pack via the surface interactions between columns (Fig. S14†). The initial phase is fast because, when 3a starts to aggregate, the concentration of monomer is high and that of the columns is negligible. The packing of columns is slower because the concentration of the columns is much lower than the monomers and it takes more time for the columns to diffuse and then to achieve optimum surface contact. The sharp transition from the first to the second phase thus indicates a threshold beyond which the packing of columns becomes dominant. At low concentrations, the second growth phase is no longer observable because the macrocyclic molecules are not able to stack into columns with the length and/or concentration needed for further packing. This also suggests that at low but aggregating concentrations, individually dissolved columnar stacks dominate.
Individually dissolved columns in solution
The likely presence of dispersed stacks of 3a in CHCl3 was probed with steady-state fluorescence anisotropy at 25 °C (see the ESI†). At 10 nM, a concentration at which 3a remains fully aggregated as shown by its emission spectrum (Fig. S15†), the aggregate of 3a, assumed to be a rotating “sphere”, has a diameter of 3.0 nm that is surprisingly close to that of 3a with side chains included. A plausible explanation is that the revealed diameter reflects the rotation of dispersed stacks of 3a around their long axes. In solution, only the self-spin of the cylindrical stacks is detected because such spin is much faster than the tumbling of the stacks around directions perpendicular to their long axes. Based on the data from fluorescence anisotropy and a refined model involving cylindrical stacks, the stacks of 3a, at 10 nM, have an average of six macrocyclic molecules (see the ESI†).Columnar assembly in the solid state
The columnar assembly of 3a was confirmed by XRD analysis on a solid sample prepared by drop-casting a solution in chloroform onto a glass plate. The obtained diffractogram contains a very intense peak at 25.8 Å that overshadows other peaks (Fig. 5). The 25.8 Å reflection and those at 14.7 Å, 13.0 Å, and 9.74 Å, with ratios of d-spacings being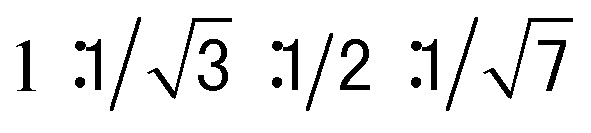 (Fig. 5a), are typical of columnar stacks of disc-like molecules that further packed on a hexagonal (colh) lattice (Fig. 5b).27 Based on the 25.8 Å peak, the hexagonal lattice parameter a, i.e., the diameter of the cylindrical stacks of 3a, was calculated to be 29.8 Å. The diameter determined by XRD agrees with that obtained from fluorescence anisotropy, which demonstrates that 3a stacks into cylindrical assemblies in both solution and the solid state. Moreover, a prominent peak at 3.66 Å, typical of π–π stacking, is observed. This peak can be attributed to the interplanar reflection between macrocyclic backbones within a column. Applying Scherrer's equation28 to this 3.66 Å reflection leads to a correlation length of 22 nm that corresponds to ∼60 continuously stacked macrocyclic units, which demonstrates the remarkable long-range ordering of the macrocycles within a column.
(Fig. 5a), are typical of columnar stacks of disc-like molecules that further packed on a hexagonal (colh) lattice (Fig. 5b).27 Based on the 25.8 Å peak, the hexagonal lattice parameter a, i.e., the diameter of the cylindrical stacks of 3a, was calculated to be 29.8 Å. The diameter determined by XRD agrees with that obtained from fluorescence anisotropy, which demonstrates that 3a stacks into cylindrical assemblies in both solution and the solid state. Moreover, a prominent peak at 3.66 Å, typical of π–π stacking, is observed. This peak can be attributed to the interplanar reflection between macrocyclic backbones within a column. Applying Scherrer's equation28 to this 3.66 Å reflection leads to a correlation length of 22 nm that corresponds to ∼60 continuously stacked macrocyclic units, which demonstrates the remarkable long-range ordering of the macrocycles within a column.
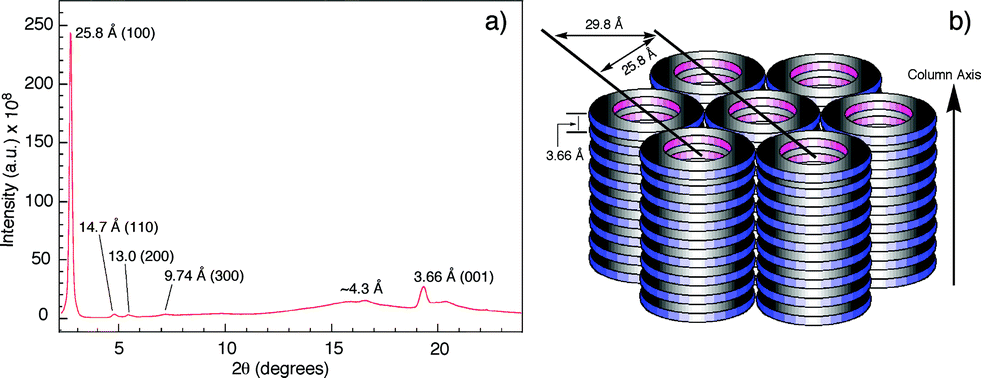 | ||
| Fig. 5 (a) Diffractogram of the solid sample of 3a. (b) Schematic drawing of the columnar packing of 3a and the hexagonal lattice. The hexagonal lattice parameter a is 29.8 Å. | ||
Transmembrane transport of proton (H+)
The tubular assembly of 3, with a non-deformable hydrophilic internal pore of ∼8 Å across, could serve as transmembrane channels when partitioning into lipid bilayers. Thus, a solution of large unilamellar vesicles (LUVs) enclosing the pH-sensitive dye HPTS was mixed with 3a and then subjected to an extravesicular acid (HCl) pulse. As shown in Fig. 6a, the decrease of fluorescence emission from the entrapped HPTS in the presence of 3a is similar to that caused by gramicidin, a well-known channel-forming peptide and is much faster than that of the control. Rupture of the LUVs upon adding Triton X-100, a nonionic surfactant, led to the nearly complete reduction of fluorescence emission. Under the same condition, macrocycle 3c also led to the same reduction of fluorescence emission, suggesting that the observed increase in proton influx was mediated by the inner pores, rather than the side chains, of the tubular assembly.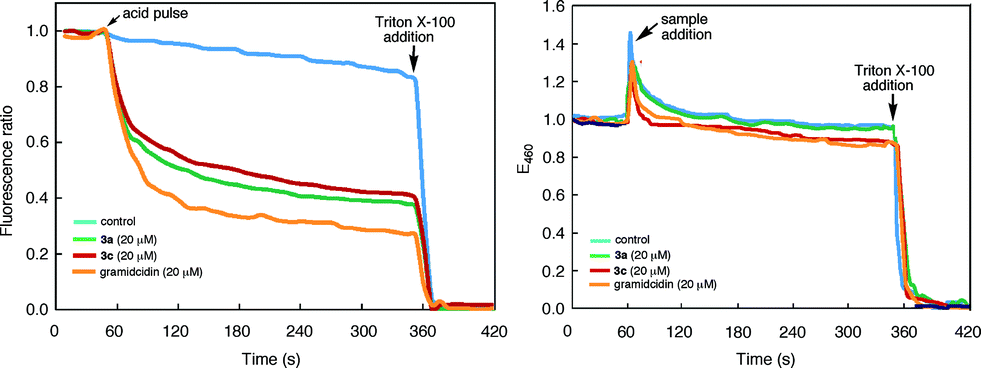 | ||
| Fig. 6 (a) Time-dependent changes in the (a) ratio of the emission intensities of 8-hydroxypyrene-1,3,6-trisulfonate (HPTS, 0.1 mM) encapsulated inside large unilamellar vesicles (LUVs) of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). Solutions of LUVs (KCl 145 mM, HEPES 100 mM, pH 7.0) were first mixed with 3a, 3c, gramicidin (added from 1 mM stock solutions in THF), or THF (control) and then incubated for 2 min, followed by a HCl (2 M) pulse. The ratio of emission intensities at 510 nm by exciting at 450 nm and 405 nm respectively was monitored over 5 min; (b) the fluorescence intensity of N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE) encapsulated inside LUVs of POPC. Solutions of Cl− free LUVs (10 mM MQAE, potassium gluconate 100 mM, HEPES 100 mM, pH 7.4) were first incubated with KCl (100 mM) in HEPES (100 mM, pH 7.4) buffer for 1 min. Stock solutions of 3a, 3c, gramicidin (1 mM in THF) and THF (control) were added to monitor the change of emission intensity at 460 nm (λex = 354 nm) for 5 min. The LUVs were ruptured by adding 200 μ(L of 1×X lysis buffer (1.55 mM Triton X-100 in pH 7.0 Tris-EDTA) (for (a)) or 200 μ(L of Triton X-100 (3.1 mM in H2O) (for (b)). | ||
The transport of anions, or the lack of which, through the nanopores of 3a or 3c was assessed by using LUVs enclosing MQAE, a chloride-sensitive fluorescence dye.29 It was found that, in the presence of a chloride gradient across the lipid bilayer, adding 3a, 3c or gramicidin failed to quench the fluorescence emission from the entrapped MQAE (Fig. 6b). As expected, rupturing the LUVs with Triton X-100 led to complete quenching of fluorescence emission from MQAE. These results demonstrate that the self-assembling pores of 3, with numerous inward pointing amide oxygens, and thus being electrostatically negative, facilitated the transport of cations while impeded the passage of anions.
Conclusions
Our study shows that the self-assembly of macrocycles 3 is remarkably strong, which affords a robust nanotubular motif that persists in both solution and the solid state. With their sub-nm inner pores, the tubular assemblies of 3 should be of wide use in constructing various nanostructures. For example, with their high stability and tunable solvent-compatibility (by adjusting side chains), the tubular stacks of 3 bode well for developing various mass-transporting channels when partitioned into biological membranes, as exemplified by the cation-transporting capabilities of 3a and 3c. The persistent nanotubular assemblies of 3 may also serve as a reliable supramolecular motif for fabricating nanoporous membranes, e.g., by blending with synthetic polymers. The high propensity of the tubular assemblies of 3 for parallel packing may lead to the next-generation membranes consisting of densely packed sub-nm pores. Furthermore, methods adopted in this study should be of general value for analysing hierarchical processes of other self-assembling systems, especially those involving extended or infinite stacks, which remains a major challenge.Acknowledgements
We thank the support of the National Science Foundation (CHE-1306326) and the Natural Science Foundation of China (91227109). Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CH11357.Notes and references
- (a) U. Koert, Phys. Chem. Chem. Phys., 2005, 7, 1502 RSC; (b) H. Bayley and P. S. Cremer, Nature, 2001, 413, 226 CrossRef CAS PubMed; (c) N. Sakai, J. Mareda and S. Matile, Acc. Chem. Res., 2008, 41, 1354 CrossRef CAS PubMed; (d) W. S. Childers, R. Ni, A. K. Mehta and D. G. Lynn, Curr. Opin. Chem. Biol., 2009, 13, 652 CrossRef CAS PubMed.
- (a) A. Harada, J. Li and M. Kamachi, Nature, 1993, 364, 516 CrossRef CAS; (b) J. M. Schnur, Science, 1993, 262, 1669 CAS; (c) N. Sakai, N. Majumdar and S. Matile, J. Am. Chem. Soc., 1999, 121, 4294 CrossRef CAS; (d) S. Tashiro, M. Tominaga, T. Kusukawa, M. Kawano, S. Sakamoto, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2003, 42, 3267 CrossRef CAS PubMed; (e) V. Percec, A. E. Dulcey, V. S. K. Balagurusamy, Y. Miura, J. Smidrkal, M. Peterca, S. Nummelin, U. Edlund, S. D. Hudson, P. A. Heiney, D. A. Hu, S. N. Magonov and S. A. Vinogradov, Nature, 2004, 430, 764 CrossRef CAS PubMed.
- A. Alexiadis and S. Kassinos, Chem. Rev., 2008, 108, 5014 CrossRef CAS PubMed.
- (a) G. Hummer, J. C. Rasaiah and J. P. Noworyta, Nature, 2001, 414, 188 CrossRef CAS PubMed; (b) K. Koga, G. T. Gao, H. Tanaka and X. C. Zeng, Nature, 2001, 412, 802 CrossRef CAS PubMed; (c) J. K. Holt, H. G. Park, Y. M. Wang, M. Stadermann, A. B. Artyukhin, C. P. Grigoropoulos, A. Noy and O. Bakajin, Science, 2006, 312, 1034 CrossRef CAS PubMed; (d) C. Y. Lee, W. J. Choi, J. H. Han and M. S. Strano, Science, 2010, 329, 1320 CrossRef CAS PubMed.
- (a) D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988 CrossRef CAS; (b) D. L. Gin, W. Q. Gu, B. A. Pindzola and W. J. Zhou, Acc. Chem. Res., 2001, 34, 973 CrossRef CAS PubMed; (c) N. Sakai and S. Matile, Chem. Commun., 2003, 2514 RSC; (d) H. M. Keizer and R. P. Sijbesma, Chem. Soc. Rev., 2005, 34, 226 RSC; (e) M. A. B. Block, C. Kaiser, A. Khan and S. Hecht, Top. Curr. Chem., 2005, 245, 89 CAS; (f) D. Pasini and M. Ricci, Curr. Org. Synth., 2007, 4, 59 CrossRef CAS; (g) B. Gong and Z. F. Shao, Acc. Chem. Res., 2013, 46, 2856 CrossRef CAS PubMed.
- (a) M. J. Mio, R. B. Prince and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 6134 CrossRef CAS; (b) H. Fenniri, B. L. Deng, A. E. Ribbe, K. Hallenga, J. Jacob and P. Thiyagarajan, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6487 CrossRef CAS PubMed; (c) Y. Kim, M. F. Mayer and S. C. Zimmerman, Angew. Chem., Int. Ed., 2003, 42, 1121 CrossRef CAS PubMed; (d) P. Jonkheijm, A. Miura, M. Zdanowska, F. J. M. Hoeben, S. De Feyter, A. P. H. J. Schenning, F. C. De Schryver and E. W. Meijer, Angew. Chem., Int. Ed., 2004, 43, 74 CrossRef PubMed; (e) Z. Q. Hua and C. F. Chen, Chem. Commun., 2005, 2445 RSC; (f) M. J. Zhou, P. R. Nemade, X. Y. Lu, X. H. Zeng, E. S. Hatakeyama, R. D. Noble and D. L. Gin, J. Am. Chem. Soc., 2007, 129, 9574 CrossRef CAS PubMed; (g) G. D. Pantos, P. Pengo and J. K. M. Sanders, Angew. Chem., Int. Ed., 2007, 46, 194 CrossRef CAS PubMed.
- (a) M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324 CrossRef CAS PubMed; (b) D. Seebach, J. L. Matthews, A. Meden, T. Wessels, C. Baerlocher and L. B. McCusker, Helv. Chim. Acta, 1997, 80, 173 CrossRef CAS; (c) G. Gattuso, S. Menzer, S. A. Nepogodiev, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed., 1997, 36, 1451 CrossRef CAS; (d) O. Y. Mindyuk, M. R. Stetzer, P. A. Heiney, J. C. Nelson and J. S. Moore, Adv. Mater., 1998, 10, 1363 CrossRef CAS; (e) H. Shimura, M. Yoshio and T. Kato, Org. Biomol. Chem., 2009, 7, 3205 RSC; (f) M. Fritzsche, A. Bohle, D. Dudenko, U. Baumeister, D. Sebastiani, G. Richardt, H. W. Spiess, M. R. Hansen and S. Höger, Angew. Chem., Int. Ed., 2011, 50, 3030 CrossRef CAS PubMed; (g) J. K. H. Hui, J. Jiang and M. J. MacLachlan, Can. J. Chem., 2012, 90, 1056 CrossRef CAS; (h) T. Hjelmgaard, O. Roy, L. Nauton, M. El-Ghozzi, D. Avignant, C. Didierjean, C. Taillefumier and S. Faure, Chem. Commun., 2014, 50, 3564 RSC.
- X. B. Zhou, G. D. Liu, K. Yamato, Y. Shen, R. X. Cheng, X. X. Wei, W. L. Bai, Y. Gao, H. Li, Y. Liu, F. T. Liu, D. M. Czajkowsky, J. F. Wang, M. J. Dabney, Z. H. Cai, J. Hu, F. V. Bright, L. He, X. C. Zeng, Z. F. Shao and B. Gong, Nat. Commun., 2012, 3, 949, DOI:10.1038/ncomms1949.
- (a) J. S. Moore, Acc. Chem. Res., 1997, 30, 402 CrossRef CAS; (b) U. H. F. Bunz, Y. Rubin and Y. Tobe, Chem. Soc. Rev., 1999, 28, 107 RSC; (c) C. Grave and A. D. Schlüter, Eur. J. Org. Chem., 2002, 67, 3075 CrossRef; (d) Y. Yamaguchi and Z.-I. Yoshida, Chem. - Eur. J., 2003, 9, 5430 CrossRef CAS PubMed; (e) S. Höger, Chem. - Eur. J., 2004, 10, 1320 CrossRef PubMed.
- (a) J. S. Moore and J. Zhang, Angew. Chem., Int. Ed., 1992, 31, 922 CrossRef; (b) J. Zhang, D. J. Pesak, J. L. Ludwick and J. S. Moore, J. Am. Chem. Soc., 1994, 116, 4227 CrossRef CAS; (c) S. Höger and V. Enkelmann, Angew. Chem., Int. Ed., 1995, 34, 2713 CrossRef; (d) V. Hensel, K. Lutzow, A.-D. Schlüter, J. Jacob, K. Gessler and W. Saenger, Angew. Chem., Int. Ed., 1997, 36, 2654 CrossRef CAS; (e) S. Höger, K. Bonrad, A. Mourran, U. Beginn and M. Moller, J. Am. Chem. Soc., 2001, 123, 5651 CrossRef PubMed; (f) K. Nakao, M. Nishimura, T. Tamachi, Y. Kuwatani, H. Miyasaka, T. Nishinaga and M. Iyoda, J. Am. Chem. Soc., 2006, 128, 16740 CrossRef CAS PubMed; (g) S. H. Seo, T. V. Jones, H. Seyler, J. O. Peters, T. H. Kim, J. Y. Chang and G. N. Tew, J. Am. Chem. Soc., 2006, 128, 9264 CrossRef CAS PubMed; (h) S. W. Sisco and J. S. Moore, J. Am. Chem. Soc., 2012, 134, 9114 CrossRef CAS PubMed; (i) F. Schlütter, F. Rossel, M. Kivala, V. Enkelmann, J.-P. Gisselbrecht, P. Ruffieux, R. Fasel and K. Müllen, J. Am. Chem. Soc., 2013, 135, 4550 CrossRef PubMed; (j) A. V. Aggarwal, A. Thiessen, A. Idelson, D. Kalle, D. Würsch, T. Stangl, F. Steiner, S.-S. Jester, J. Vogelsang, S. Höger and J. M. Lupton, Nat. Chem., 2013, 5, 964 CrossRef CAS PubMed.
- Examples of macrocyclic aromatic oligoureas: (a) F. Böhme, C. Kunert, H. Komber, D. Voigt, P. Friedel, M. Khodja and H. Wilde, Macromolecules, 2002, 35, 4233 CrossRef; (b) L. Xing, U. Ziener, T. C. Sutherland and L. A. Cuccia, Chem. Commun., 2005, 5751 RSC; (c) A. Zhang, Y. Han, K. Yamato, X. C. Zeng and B. Gong, Org. Lett., 2006, 8, 803 CrossRef CAS PubMed; (d) A. Gube, H. Komber, K. Sahre, P. Friedel, B. Voit and F. Böhme, J. Org. Chem., 2012, 77, 9620 CrossRef CAS PubMed; (e) Z. Wu, T. Hu, L. He and B. Gong, Org. Lett., 2012, 14, 2504 CrossRef CAS PubMed.
- Example of macrocyclic aromatic oligo(imines): P. D. Frischmann, G. A. Facey, P. Y. Ghi, A. J. Gallant, D. L. Bryce, F. Lelj and M. J. MacLachlan, J. Am. Chem. Soc., 2010, 132, 3893 CrossRef CAS PubMed.
- Examples of macrocyclic aromatic oligosulfonamides: (a) L. He, Y. An, L. Yuan, K. Yamato, W. Feng, O. Gerlitz, C. Zheng and B. Gong, Chem. Commun., 2005, 3788 RSC; (b) L. He, Y. An, L. Yuan, W. Feng, M. Li, D. Zhang, K. Yamato, C. Zheng, X. C. Zeng and B. Gong, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10850 CrossRef CAS PubMed.
- Examples of macrocyclic aromatic oligoamides: (a) L. H. Yuan, W. Feng, K. Yamato, A. R. Sanford, D. G. Xu, H. Guo and B. Gong, J. Am. Chem. Soc., 2004, 126, 11120 CrossRef CAS PubMed; (b) H. Jiang, J.-M. Léger, P. Guionneau and I. Huc, Org. Lett., 2004, 6, 2985 CrossRef CAS PubMed; (c) F. Campbell, J. Plante, C. Carruthers, M. J. Hardie, T. J. Prior and A. J. Wilson, Chem. Commun., 2007, 2240 RSC; (d) B. Qin, C. Ren, R. Ye, C. Sun, K. Chiad, X. Chen, Z. Li, F. Xue, H. Su, G. A. Chass and H. Zeng, J. Am. Chem. Soc., 2010, 132, 9564 CrossRef CAS PubMed.
- Examples of other rigid macrocycles: (a) J. L. Sessler, E. Tomat and V. M. Lynch, Chem. Commun., 2006, 4486 RSC; (b) K. Nakao, M. Nishimura, T. Tamachi, Y. Kuwatani, H. Miyasaka, T. Nishinaga and M. Iyoda, J. Am. Chem. Soc., 2006, 128, 16740 CrossRef CAS PubMed; (c) J. M. Holub, H. J. Jang and K. Kirshenbaum, Org. Lett., 2007, 9, 3275 CrossRef CAS PubMed; (d) Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649 CrossRef CAS PubMed; (e) S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704 CrossRef CAS PubMed.
- K. Yamato, M. Kline and B. Gong, Chem. Commun., 2012, 48, 12142 RSC.
- A. R. Sanford and B. Gong, Curr. Org. Chem., 2003, 7, 1649 CrossRef CAS.
- (a) L. H. Yuan, H. Q. Zeng, K. Yamato, A. R. Sanford, W. Feng, H. Atreya, D. K. Sukumaran, T. Szyperski and B. Gong, J. Am. Chem. Soc., 2004, 126, 16528 CrossRef CAS PubMed; (b) L. H. Yuan, A. R. Sanford, W. Feng, A. M. Zhang, J. S. Ferguson, K. Yamato, J. Zhu, H. Q. Zeng and B. Gong, J. Org. Chem., 2005, 70, 10660 CrossRef CAS PubMed.
- J. X. Cao, M. Kline, Z. Z. Chen, B. Luan, M. L. Lv, W. Zhang, C. X. Lian, Q. W. Wang, Q. F. Huang, X. X. Wei, J. G. Deng, J. Zhu and B. Gong, Chem. Commun., 2012, 48, 11112 RSC.
- (a) W. Feng, K. Yamato, L. Q. Yang, J. Ferguson, L. J. Zhong, S. L. Zou, L. H. Yuan, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2009, 131, 2629 CrossRef CAS PubMed; (b) J. S. Ferguson, K. Yamato, R. Liu, L. He, X. C. Zeng and B. Gong, Angew. Chem., Int. Ed., 2009, 48, 3150 CrossRef CAS PubMed.
- (a) R. D. Parra, H. Q. Zeng, J. Zhu, C. Zheng, X. C. Zeng and B. Gong, Chem. - Eur. J., 2001, 7, 4352 CrossRef CAS; (b) R. D. Parra, M. Furukawa, B. Gong and X. C. Zeng, J. Chem. Phys., 2001, 115, 6030 CrossRef CAS PubMed; (c) R. D. Parra, B. Gong and X. C. Zeng, J. Chem. Phys., 2001, 115, 6036 CrossRef CAS PubMed.
- Y. A. Yang, W. Feng, J. C. Hu, S. L. Zou, R. Z. Gao, K. Yamato, M. Kline, Z. H. Cai, Y. Gao, Y. B. Wang, Y. B. Li, Y. L. Yang, L. H. Yuan, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2011, 133, 18590 CrossRef CAS PubMed.
- X. X. Wu, G. X. Liang, G. Ji, H. K. Fun, L. He and B. Gong, Chem. Commun., 2012, 48, 2228 RSC.
- M. Kline, X. X. Wei and B. Gong, Org. Lett., 2013, 15, 4762 CrossRef CAS PubMed.
- J. B. Birk, Photophysics of Aromatic Molecules, Wiley-Interscience, New York, 1970 Search PubMed.
- (a) J. K. Rice, E. D. Niemeyer, R. A. Dunbar and F. V. Bright, J. Am. Chem. Soc., 1995, 117, 5832 CrossRef CAS; (b) R. B. Prince, J. G. Saven, P. G. Wolynes and J. S. Moore, J. Am. Chem. Soc., 1999, 121, 3114 CrossRef CAS.
- S. Kumar, Chem. Soc. Rev., 2006, 35, 83 RSC.
- B. D. Cullity and S. R. Stock, Elements of X-Ray Diffraction, Prentice-Hall, New Jersey, 3rd edn, 2001 Search PubMed.
- A. S. Verkman, Am. J. Physiol., 1990, 259, C375 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc02380c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |