
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerated Hantzsch electrospray synthesis with temporal control of reaction intermediates†
Ryan M.
Bain
a,
Christopher J.
Pulliam
a and
R. Graham
Cooks
*ab
aDepartment of Chemistry, Purdue University, West Lafayette, IN 47907, USA. E-mail: cooks@purdue.edu; Fax: +1-765-494-9421; Tel: +1-765-494-5263
bCenter for Analytical Instrumentation Development, Purdue University, West Lafayette, IN 47909, USA
First published on 9th September 2014
Abstract
Complex chemical reactions can occur in electrosprayed droplets on the millisecond time scale. The Hantzsch synthesis of 1,4-dihydropyridines was studied in this way using on-line mass spectral analysis to optimize conditions and characterize the product mixture. Changing the distance between the nanospray source and the MS inlet allowed exploration of reaction progress as a function of droplet time-of-flight. Desolvation of the charged microdroplets is associated with transformation from starting material to intermediates and eventually to product as the distance is increased. Results of the on-line experiments require a termination step that discontinuously completes the desolvation process and allows the generated gaseous ions to be used to characterize the state of the system at a particular time. The intermediates seen correspond to those known to occur in the bulk solution-phase reaction. Off-line collection of the sprayed reaction mixture allowed the recovery of 250 mg h−1 of desired reaction product from a single sprayer, permitting characterization by NMR and other standard methods. A thin film version of the accelerated reaction is described and it could be controlled through the temperature of the collection surface.
Introduction
This paper is concerned with accelerated chemical reactions occurring in charged microdroplets. Such reactions have previously been studied using electrospray and other spray-based ambient ionization techniques.1–5 Some of these studies used these rapid reactions to derivatize analytes for improved chemical analysis. In other cases, the rapid droplet reactions were used to study the course of chemical reactions and to identify reaction intermediates.6–8 In still other cases, the collection and characterization of the product(s) of microscale synthesis was of interest.9,10 For example, collection of product in milligram quantities from a Claisen–Schmidt base-catalyzed condensation was achieved by nanospraying a mixture of an indanone and an aldehyde under basic conditions.11 Accelerated reactions in confined volumes have been reported to occur in desorption electrospray ionization (DESI), in dropcasting, and in nanoelectrospray experiments. The emphasis on nanospray in this study, over DESI and other techniques, is because surface interactions are removed and purely homogeneous processes in droplets can be explored. Nanospray also allowed high-yield experiments to be performed by simply optimizing spray conditions (on-line) and subsequently collecting product on a surface (off-line).Accelerated reactions in droplets generated by electrospray are not to be confused with the use of electrospray and other droplet-based experiments to monitor the course of a reaction by mass spectrometry.12–26 An important fact, not sufficiently noted in the literature, is that under different conditions mass spectrometry can be used either to follow the course of a bulk phase chemical reaction or to accelerate a reaction and form products. The key factor in selecting between conditions which allow reaction monitoring vs. reaction acceleration is the nature of the desolvation process: continuous desolvation favors observation of accelerated reactions in droplets.
It will be evident from this study that accelerated reactions in electrospray droplets must be followed by a rapid discontinuous desolvation event (that does not promote reaction) in the ion source. This freezes out the state of the system. The Hantzsch reaction has been followed in bulk using electrospray ionization mass spectrometry (ESI-MS)27,28 but here it is studied as an accelerated reaction in individual droplets and (more briefly) in thin films. The choice of the Hantzsch reaction for study was made to allow comparison between reaction monitoring of bulk reactions, which has been reported by several authors, and accelerated reactions in droplets.
The Hantzsch synthesis of symmetric 1,4-dihydropyridine (DHP) derivatives has been of interest for over a century.29 Symmetric DHPs are synthesized from ethyl acetoacetate, ammonium acetate, and an aromatic aldehyde, as summarized in Scheme 1.30,31 Many DHP derivatives (9) serve as practical intermediates in the generation of the corresponding pyridines (10) through a simple oxidation of the DHP core.32–35 More information on the importance of the reaction can be found in the ESI.†
 | ||
| Scheme 1 Knoevenagel condensation product (4) reacts with the enamine ester (6) via a Michael addition to form 7, which undergoes proton transfers and intramolecular enamine formation to give 9. In a separate step, dehydrogenation yields the pyridine (10). All species are shown in their charged forms where appropriate. | ||
Results
Bulk-phase reaction and analysis
Traditional bulk-phase reactions were performed to determine the time required for synthesis with and without catalysts, as well as to measure yields. These reactions were monitored by absorption spectroscopy and run both at room temperature and under reflux in ethanol, with and without phenylboronic acid catalyst (10 mol%). Reactions conducted under reflux with phenylboronic acid were complete (∼85% yield) in just under 5 hours. Reactions unaided by catalyst or reflux were slower (Table S1†) and represent the conditions of the ESI sample before droplet creation and solvent evaporation. Samples under reflux with catalyst represent a traditional synthetic route for the reaction.Off-line reaction and analysis of electrosprayed material
Under optimized conditions, the accelerated nanoelectrospray experiment yielded 250 mg h−1 of the DHP product 9 from a single sprayer, corresponding to >90% yield. Off-line quantification of 9 was performed using UV-vis absorption spectroscopy and NMR proved valuable as an alternative form of structural characterization (Fig. S1†). The progress of the reaction was also monitored by TLC after spraying the reaction mixture onto a TLC plate.Effect of reagent choice
The effects of changing the amine or aldehyde reagent were investigated for both the bulk and accelerated electrospray Hantzsch reactions. The data are given in Tables S2 and S3.† Only ammonia or a primary amine can yield a dihydropyridine and accordingly, both the bulk and the accelerated experiments only yield product 9 when ammonia is the base; primary amines yield 5 and for secondary and tertiary amines only the reactants could be detected. Experiments with various aldehydes showed predictable results (Tables S4 and S5†).Distance, temperature and flow-rate effects in the on-line reaction
Increasing the distance the electrosprayed droplets must travel to reach the mass analyzer changes the product distribution dramatically. This shows that the reaction is dependent on experimental variables that control the extent of desolvation. As the distance between the ion transfer capillary and the spray source increases, a shift occurs from starting material 2 and its simple ammonia adduct 5 towards the Knoevenagel product 4, its precursor 3, and the Michael addition product 7, while eventually the DHP 9 and the pyridine 10 are seen. Species 6 is not seen in the mass spectra, which is consistent with the low basicity of enamines. Fig. S2 and Table S6† summarize these distance effects on the reaction progress. Distances were varied from 4 cm to 100 cm with approximate flight times (estimated assuming droplets are carried along by the nebulizing gas) of 200 microseconds to 5 milliseconds, respectively. An independent set of distance studies was performed using stainless steel tubes of varying lengths between the spray source and MS inlet. These tubes cause charge build up and confine the spray radially, increasing signals several fold. Product ion intensity is still maximized as distance is increased (Fig. 1 and Table S7†). It is clear that the extent of reaction is directly dependent on the distance that the droplets travel.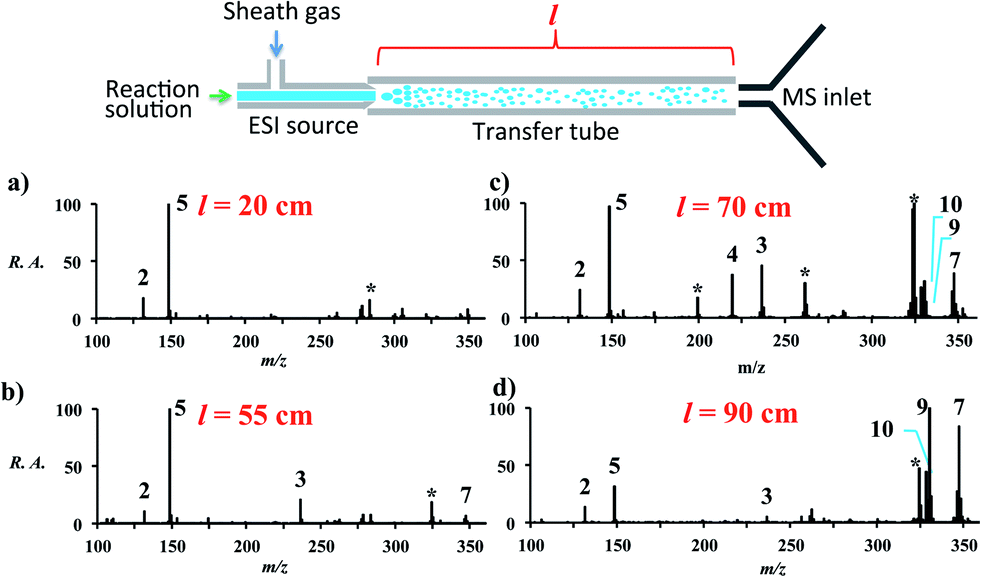 | ||
| Fig. 1 Extent of reaction as a function of distance (l) between the MS inlet and the spray source. The data are accounted for by desolvation to give smaller, more rapidly reacting droplets. * denotes known impurities thought to be from the guiding tube. | ||
Increasing the solution flow-rate (without adjusting other parameters) led to a decrease in the main product 9 and an increase in intermediates 4, 5, and 7. Working under conditions in which the extent of reaction was limited (l = 30 cm; room temperature) and then increasing the temperature of the tube from ambient to 80 °C resulted in a decrease in intensity of the early-stage intermediates (5 and 3) and an increase in the late-stage intermediate (7), Fig. 2. Product was not seen for this distance/temperature combination.
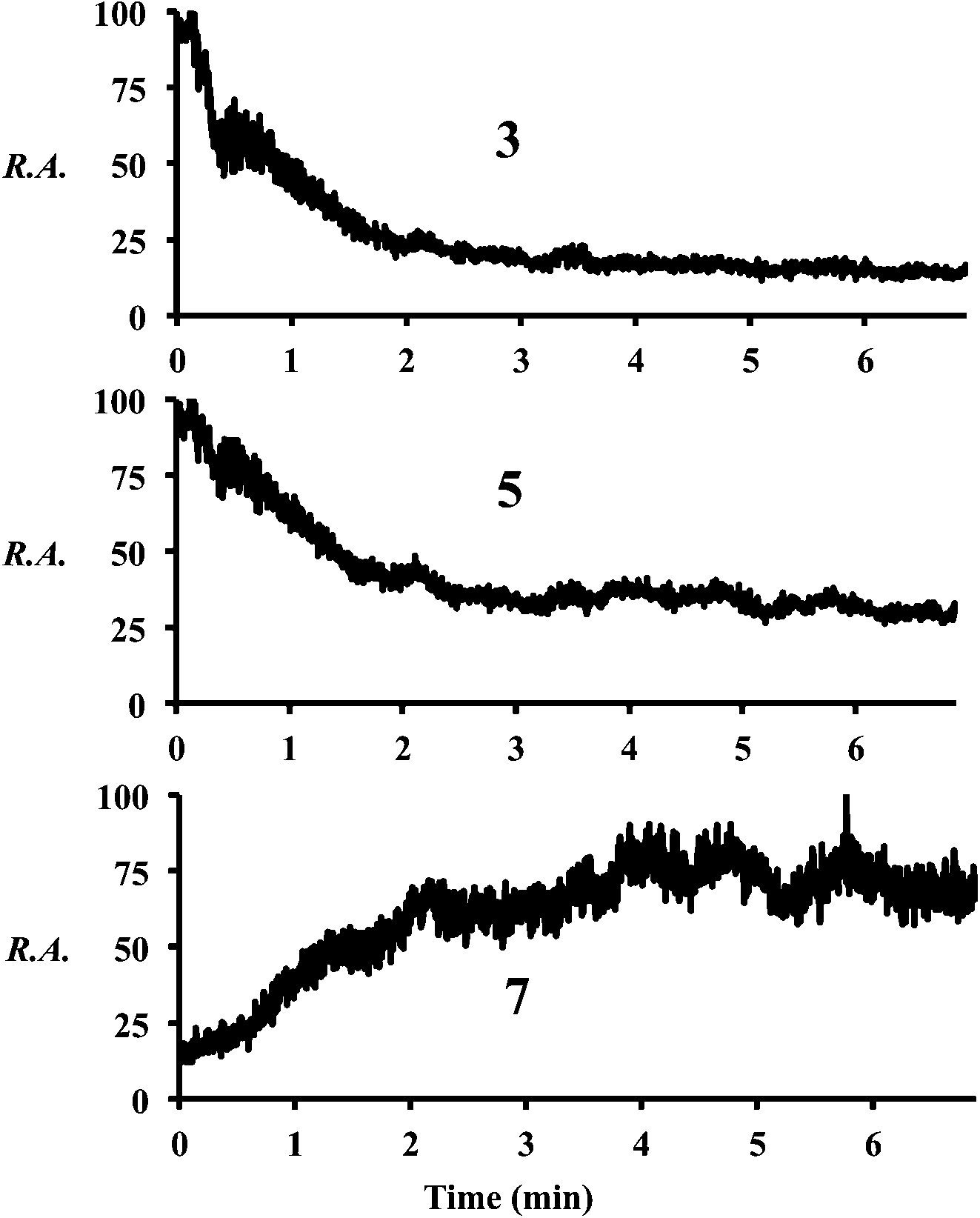 | ||
| Fig. 2 Chronograms showing relative abundances of compounds 3, 5, and 7 as the temperature is increased from room temperature to 80 °C (∼3 min) at l = 30 cm. | ||
Distance and temperature effects in the off-line reaction
Experiments were also performed with off-line collection of droplets and subsequent product analysis. In these experiments, the sprayer can be placed very close to the deposition surface where it generates a thin film of solution. At room temperature only product 9 was observed, while in an analogous experiment performed with the surface cooled to −77 °C or −17 °C no products were seen. Greater sprayer distances did yield product at these reduced temperatures, because some reaction occurs in the gas-phase droplets. A plot of the normalized relative intensity of each species present over a range of distances is shown in Fig. 3 (see Table S8 for mass spectra†). Simple dropcasting experiments showed no reaction, illustrating the contribution made by small electrospray droplets to reaction. | ||
| Fig. 3 Progress of the off-line cold surface reaction at various distances between the ESI source and the collection surface under conditions where reaction at the surface is minimized by operating at low temperature. | ||
Conversion of dihydropyridine to pyridine
The final Hantzsch reaction product is the dihydropyridine (DHP, 9) but conversion to the pyridine (10) can be achieved by increasing the temperature of the ion transfer capillary (part of the atmospheric pressure interface of the mass spectrometer). The temperature of the ion transfer capillary was used as an additional experimental variable: as the temperature of the capillary was raised from 150 °C to 350 °C, a shift in the spectrum occurred (Fig. 4 and Table S9†) with conversion from reaction product 9 to 10 by dehydrogenation. Both 9 and 10 were observed in three forms – as the [M + H]+, [M + Na]+ and [M + K]+ ions. The conversion from 9 to 10 was also attempted off-line by heating both the spray source and the collection surface, however, no product was observed.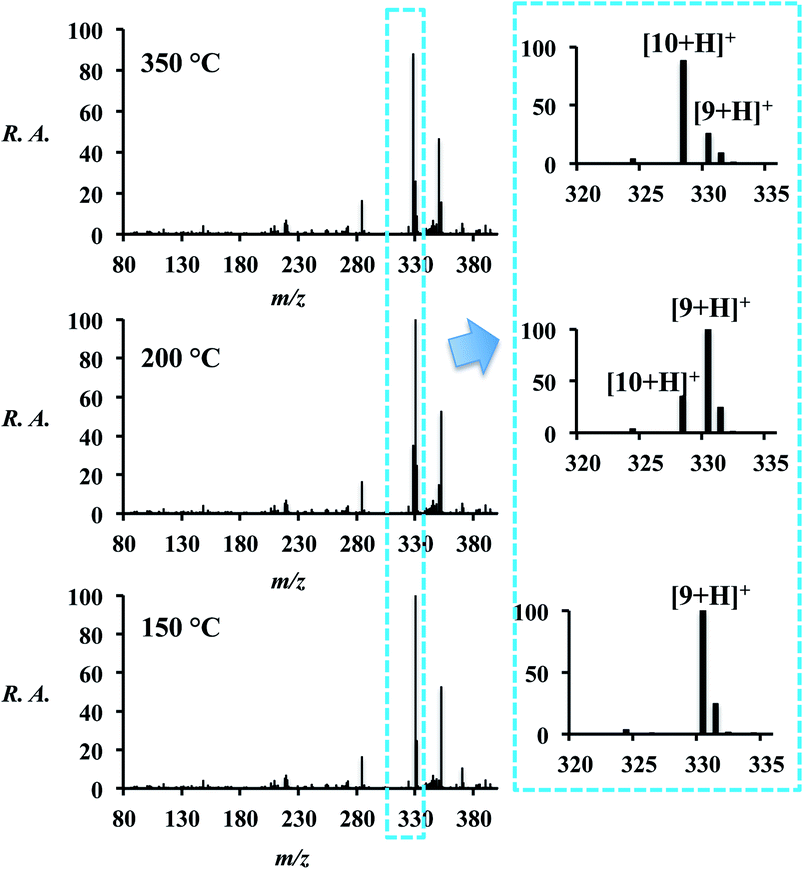 | ||
| Fig. 4 On-line reaction (l = 100 cm) at various ion transfer capillary temperatures. Spectra on the right are expansions showing 9 and 10. Note that 9 and 10 occur as protonated, sodiated, and potassiated forms. The signal at m/z 282 is due to background. | ||
Experimental
Spray source
All experiments used the same electrospray ionization source. The solution mixing and transfer system used three syringes (Hamilton) and two syringe pumps (Hamilton) with different flow rates to achieve molar ratios. High-pressure mixers were used to ensure complete mixing. Minimizing the amount of time after the reagents were mixed ensured that reaction occurred in the course of electrospray and not in the syringe. Many of the electrospray parameters had to be optimized including flow rates (gas and reagent), as summarized in Table S10.† Variables such as reagent and gas flow rate were crucial in controlling the rate of desolvation, as indicated above. All reagents were purchased from Sigma-Aldrich (Milwaukee, Wisconsin).On-line analysis of electrosprayed droplets
The charged droplets emerging from the spray source were directed pneumatically and electrically towards the inlet capillary of an ion trap MS (Thermo-Finnigan LTQ). Variables including the distance between the spray source and the ion transfer capillary, the capillary temperature, the glass/metal capillary length, the gas flow rate and the reagent flow rate were controlled as they lead to large changes in reaction yield. Table S10† gives the optimized values of these parameters.Off-line collection of electrosprayed droplets
The spray source was housed in an airtight poly(methyl methacrylate) box to minimize release of solvent vapor into the air and to provide the ability to explore moisture- and air-sensitive reactions (photographs in ESI†). Spray variables were those that optimized the on-line reaction, except that the effects of distance were systematically explored. Product collection was performed at room temperature on a grounded colander sitting atop a rotating beaker to increase surface area. Experiments were done under ambient conditions or using temperature-controlled surfaces (grounded copper foil cup floating atop a dry-ice/2-propanol bath, ∼−77 °C and a dry-ice/ethylene glycol bath, ∼−17 °C) to explore the role of confined volumes on the surface. Off-line mass spectral analysis showed that product formation decreased at −77 °C/−17 °C vs. room temperature. Quantitative and structural analysis of the collected spray product (as well as the products of the bulk reactions) was performed using a Cary 100 UV-vis spectrophotometer (λ = 378 nm, molar absorptivity 5.2 × 103 L mol−1 cm−1)36 and NMR (Bruker ARX 400 with 5 mm QNP probe), respectively [1H NMR (CDCl3): δ 1.26(6H), 2.32(6H), 2.33(3H), 4.08(4H), 4.99(1H), 5.67(1H), 7.20(5H)]. TLC analysis of the spray product was done by directly spraying onto a TLC plate with a ∼1 mm diameter hole cut in a piece of paper in between the sprayer and the plate (∼2 cm) to define the spot size.Bulk-phase reactions
Bulk-phase reactions were performed for comparison with reactions occurring in the electrosprayed droplets. These reactions were run at both 10 mM in ethanol and as specified by Debache et al.37 Both experiments yielded similar data.Discussion
Occurrence of the same intermediate species in the bulk-phase Hantzsch reaction and the accelerated reaction occurring in droplets establishes the underlying similarity in reaction mechanism. Exploration of both distance and temperature effects for the electrospray reaction emphasizes the role that droplet desolvation plays in reaction progress on the millisecond timescale. A particularly important finding is that the reported observations require a discontinuity in the droplet desolvation process. This discontinuity in the evaporation of solvent must occur in the MS inlet with sudden conversion of the solvated ions in the droplets into free gas-phase ions without contributing to reaction acceleration. Complete desolvation occurs independently of the extent of prior (droplet-phase) reaction. This explains how the formation of product can be directly dependent on distance: reactions occur only during the uninterrupted period of slow (millisecond) droplet desolvation. By contrast with these accelerated reactions in evaporating droplets, experiments on reaction monitoring27,28 are performed using conditions which do not allow this time for desolvation/reaction before discontinuous desolvation occurs, converting the sampled reaction mixture into gas-phase ions and allowing bulk reaction monitoring.The findings of the off-line distance studies show that the distances required to yield product under cold collection conditions are significantly greater than those when product is collected under ambient conditions. These data suggest that a second factor contributes to reaction rate enhancement in confined volume solutions. In addition to desolvation in microdroplets, desolvation from thin films on collection surfaces must also contribute. The rate enhancement effects occurring in droplets are shown by the distance effects of both the on-line and the cold collection off-line experiments. The lack of dependence on distance in the ambient off-line collection at room temperature suggests that the reaction is accelerated all the way to product 9 in the thin film confined volume on the surface, while the distances are such that there is little opportunity for reaction in the evaporating droplet.
Conclusion
An ESI-based droplet method was used to compare the accelerated Hantzsch synthesis in sprayed droplets with the parallel bulk-phase reactions. On-line chemical analysis in real-time allowed optimization of the sprayer conditions so that slow desolvation and its accompanying accelerated reaction could occur before the sudden and discontinuous desolvation event in the ion source. The on-line experiments allow the exploration of reaction progress as a function of distance between the sprayer and the ion transfer capillary. This distance controls the progress of the reaction and the degree of desolvation of the droplets. The discontinuity in desolvation before mass analysis in the on-line distance experiments requires a sudden completion of desolvation with release of free gas-phase ions and truncation of reaction. This event occurs in the atmospheric pressure interface or ion source. Accelerated reactions also occur in thin films and they can be suppressed by control of the surface temperature. The experiment as a whole illustrates the role both types of confined volumes (desolvating microdroplets and thin films) have in accelerating reaction progress. Off-line experiments using charged droplets yield product for this reaction at rates as high as 250 mg h−1. The study explains the apparently contradictory fact that the same process – droplet formation, evaporation and ion mass analysis – can be used to monitor the course of a bulk-phase reaction or to accelerate reactions of the same reagents in microdroplets.Acknowledgements
Authors acknowledge the support of Purdue University Department of Chemistry, particularly Ned Gangwer and Betty Dexter. Authors acknowledge discussions with Heather Osswald and Kinsey Bain and financial support provided by the National Science Foundation (CHE-1307264).Notes and references
- R. G. Cooks, Z. Ouyang, Z. Takats and J. M. Wiseman, Science, 2006, 311, 1566–1570 CrossRef CAS PubMed.
- M. E. Monge, G. A. Harris, P. Dwivedi and F. M. Fernández, Chem. Rev., 2013, 113, 2269–2308 CrossRef CAS PubMed.
- D. R. Ifa, C. Wu, Z. Ouyang and R. G. Cooks, Analyst, 2010, 135, 669–681 RSC.
- A. K. Badu-Tawiah, L. S. Eberlin, Z. Ouyang and R. G. Cooks, Annu. Rev. Phys. Chem., 2013, 64, 481–505 CrossRef CAS PubMed.
- R. D. Espy, M. Wleklinski, X. Yan and R. G. Cooks, TrAC, Trends Anal. Chem., 2014, 57, 135–146 CrossRef CAS PubMed.
- R. H. Perry, M. Splendore, A. Chien, N. K. Davis and R. N. Zare, Angew. Chem., Int. Ed. Engl., 2011, 50, 250–254 CrossRef CAS PubMed.
- R. H. Perry, K. R. Brownell, K. Chingin, T. J. Cahill III, R. M. Waymouth and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2246–2250 CrossRef CAS PubMed.
- G. Xu, B. Chen, B. Guo, D. He and S. Yao, Analyst, 2011, 136, 2385–2390 RSC.
- M. Girod, E. Moyano, D. I. Campbell and R. G. Cooks, Chem. Sci., 2011, 2, 501 RSC.
- R. M. Bain, C. J. Pulliam, Y. Xin, K. F. Moore, T. Müller and R. G. Cooks, J. Chem. Educ., 2014 DOI:10.1021/ed500288m.
- T. Müller, A. Badu-Tawiah and R. G. Cooks, Angew. Chem., Int. Ed. Engl., 2012, 51, 11832–11835 CrossRef PubMed.
- P. Dell'Orco, J. Brum, R. Matsuoka, M. Badlani and K. Muske, Anal. Chem., 1999, 71, 5165–5170 CrossRef.
- J. Brum, P. Dell'Orco, S. Lapka, K. Muske and J. Sisko, Rapid Commun. Mass Spectrom., 2001, 15, 1548–1553 CrossRef CAS.
- C. S. Allardyce, P. J. Dyson, J. Coffey and N. Johnson, Rapid Commun. Mass Spectrom., 2002, 16, 933–935 CrossRef CAS PubMed.
- M. Vairamani and M. L. Gross, J. Am. Chem. Soc., 2003, 125, 42–43 CrossRef CAS PubMed.
- S. Fürmeier and J. O. Metzger, J. Am. Chem. Soc., 2004, 126, 14485–14492 CrossRef PubMed.
- D. Fabris, Mass Spectrom. Rev., 2005, 24, 30–54 CrossRef CAS PubMed.
- L. S. Santos and J. O. Metzger, Angew. Chem., Int. Ed., 2006, 45, 977–981 CrossRef PubMed.
- H. Wang and J. O. Metzger, Organometallics, 2008, 27, 2761–2766 CrossRef CAS.
- P. Kebarle and U. Verkerk, Mass Spectrom. Rev., 2009, 28, 898–917 CrossRef CAS PubMed.
- E. L. Harry, A. W. T. Bristow, I. D. Wilson and C. S. Creaser, Analyst, 2011, 136, 1728–1732 RSC.
- K. Huvaere, B. Sinnaeve, J. Van Bocxlaer and L. H. Skibsted, J. Agric. Food Chem., 2012, 60, 9261–9272 CrossRef CAS PubMed.
- D. Schröder, Acc. Chem. Res., 2012, 45, 1521–1532 CrossRef PubMed.
- K. M. Roscioli, X. Zhang, S. X. Li, G. H. Goetz, G. Cheng, Z. Zhang, W. F. Siems and H. H. Hill, Int. J. Mass Spectrom., 2013, 336, 27–36 CrossRef CAS PubMed.
- R. L. Grimm, R. Hodyss and J. L. Beauchamp, Anal. Chem., 2006, 78, 3800–3806 CrossRef CAS PubMed.
- N. Gasilova, Q. Yu, L. Qiao and H. H. Girault, Angew. Chem., Int. Ed. Engl., 2014, 53, 4408–4412 CrossRef CAS PubMed.
- H. G. O. Alvim, G. A. Bataglion, L. M. Ramos, A. L. de Oliveira, H. C. B. de Oliveira, M. N. Eberlin, J. L. de Macedo, W. A. da Silva and B. A. D. Neto, Tetrahedron, 2014, 70, 3306–3313 CrossRef CAS PubMed.
- R. Kumar, N. H. Andhare, A. Shard and A. K. Sinha, RSC Adv., 2014, 4, 19111 RSC.
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1881, 14, 1637–1638 CrossRef.
- A. Katritzky, D. Ostercamp and T. Yousaf, Tetrahedron, 1987, 22, 5171–5186 CrossRef.
- A. Katritzky, D. Ostercamp and T. Yousaf, Tetrahedron, 1987, 42, 5729–5738 CrossRef.
- H. Adibi and A. R. Hajipour, Bioorg. Med. Chem. Lett., 2007, 17, 1008–1012 CrossRef CAS PubMed.
- J.-J. Xia and G.-W. Wang, Synthesis, 2005, 14, 2379–2383 Search PubMed.
- M. Nikoorazm, Sci. Iran., 2012, 20, 603–606 Search PubMed.
- O. De Paolis, J. Baffoe, S. Landge and B. Török, Synthesis, 2008, 21, 3423–3428 Search PubMed.
- J. Ramchander, G. Raju, N. Rameshwar, T. S. Reddy and A. R. Reddy, Spectrochim. Acta, Part A, 2012, 85, 210–216 CrossRef CAS PubMed.
- A. Debache, R. Boulcina, A. Belfaitah, S. Rhouati and B. Carboni, Synlett, 2008, 4, 509–512 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra for all on-line, off-line, and substituent studies as well as photographs of experimental setups are supplied. See DOI: 10.1039/c4sc02436b |
| This journal is © The Royal Society of Chemistry 2015 |