
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand design for Rh(III)-catalyzed C–H activation: an unsymmetrical cyclopentadienyl group enables a regioselective synthesis of dihydroisoquinolones†
Todd K.
Hyster
,
Derek M.
Dalton
and
Tomislav
Rovis
*
Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA. E-mail: rovis@lamar.colostate.edu
First published on 1st October 2014
Abstract
We report the regioselective synthesis of dihydroisoquinolones from aliphatic alkenes and O-pivaloyl benzhydroxamic acids mediated by a Rh(III) precatalyst bearing sterically bulky substituents. While the prototypical Cp* ligand provides product with low selectivity, sterically bulky Cpt affords product with excellent regioselectivity for a range of benzhydroxamic acids and alkenes. Crystallographic evidence offers insight as to the source of the increased regioselectivity.
C–H activation mediated processes have provided a unique retrosynthetic approach to access a variety of substituted heterocycles.1 One tactic that has received increased attention is the coupling of π-components with heteroatom containing molecules.2 A variety of transition metals are capable of catalyzing this type of transformation, providing access to dozens of heterocyclic motifs.1–3 A challenge for these methods is controlling the regioselectivity of migratory insertion across alkenes and alkynes after the metallacycle forming C–H activation (eqn 1).
Steric and electronic effects are understood to control migratory insertion of unsymmetrical alkynes in Rh(III) catalyzed isoquinolone syntheses (eqn 1). When the substituents are electronically similar, the larger group resides β- to Rh in the metallacycle to avoid unfavorable steric interactions (selectivity is generally >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).4 When the substituents are electronically different, the more electron-donating group prefers being α- to rhodium in the metallacycle, presumably to stabilize the electron poor metal.5,6 The type of C–H bond being activated also plays an important role in the regioselectivity of migratory insertion; aromatic substrates typically provide synthetically useful regioselectivities when electronically different alkynes are used (>10:1) but alkenyl C–H activation leads to products with lower regioselectivities, presumably due to minimal steric interactions during migratory insertion.7,8 We found that sterically bulky di-tert-butylcyclopentadienyl ligand (Cpt) enhances the regioselectivity of the alkyne migratory insertion event in these cases, delivering regioselectivities (>10:1) modestly above those achievable by Cp* ligated Rh complexes (<6:1). However, when the alkyne migratory insertion was poorly selective with RhCp* (<3:1), RhCpt complex was ineffective at providing synthetically useful levels of selectivity. Furthermore, the Cpt ligand was only effective with aryl substituted alkynes, presumably because of strong steric interactions between the ligand and alkyne in the insertion event.
:1).4 When the substituents are electronically different, the more electron-donating group prefers being α- to rhodium in the metallacycle, presumably to stabilize the electron poor metal.5,6 The type of C–H bond being activated also plays an important role in the regioselectivity of migratory insertion; aromatic substrates typically provide synthetically useful regioselectivities when electronically different alkynes are used (>10:1) but alkenyl C–H activation leads to products with lower regioselectivities, presumably due to minimal steric interactions during migratory insertion.7,8 We found that sterically bulky di-tert-butylcyclopentadienyl ligand (Cpt) enhances the regioselectivity of the alkyne migratory insertion event in these cases, delivering regioselectivities (>10:1) modestly above those achievable by Cp* ligated Rh complexes (<6:1). However, when the alkyne migratory insertion was poorly selective with RhCp* (<3:1), RhCpt complex was ineffective at providing synthetically useful levels of selectivity. Furthermore, the Cpt ligand was only effective with aryl substituted alkynes, presumably because of strong steric interactions between the ligand and alkyne in the insertion event.

Migratory insertion of alkenes to access heterocycles using C–H activation chemistry is still relatively rare, with seminal studies by Glorius and Fagnou reporting the synthesis of dihydroisoquinolones.9–11 Similar to alkynes, alkenyl electron-donating groups favor the position adjacent to the metal in the metallacycle delivering high regioselectivity. In contrast to alkynes, aliphatic alkenes afford product with poor regioselectivity (2:1) (eqn 2).5h,12 We hypothesized competing steric and electronic effects cause the low regioselectivity, with steric effects favoring the formation of a 4-substituted product and electronics favoring the formation of a 3-substituted product.13 As a temporary solution to this problem, our group and others have employed tethering strategies to increase the regioselectivity of the migratory insertion event (eqn 3).14,15 Of course, regioselectivity controlled by the ligand on Rh would be the optimal solution to the selectivity problem (eqn 4).16 Consequently, we focused our attention toward developing an intermolecular variant of this reaction that would provide product with improved regioselectivity.
As a model system, we explored the impact ligands have on the coupling of O-pivaloyl-benzhydroxamic acid 1a with 1-decene 2a to provide dihydroisoquinolones 3a and 3a′. When Cp* is used as a ligand, the desired products are isolated in excellent yield but poor selectivity (2.4:1 3a:3a′) (Table 1, entry 1).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield (%) | Regioselectivity |
| a Reaction conditions: 1a (.2 mmol), 1-decene (.2 mmol), precatalyst (1 mol%), CsOAc (200 mol%), MeOH (0.1 M). b CpCF3 = 1-trifluoromethyl-2-3,4,5-tetramethylcyclopentadienyl. c Cp‡ = 1,2-di-phenyl-3,4,5-trimethylcyclopentadienyl. d Cpt = 1,3-di-t-butylcyclopentadienyl. | |||
| 1 | [RhCp*Cl2]2 | 90 | 2.4:1 |
| 2b | [RhCpCF3Cl2]2 | 85 | 2.4:1 |
| 3c | [RhCp‡Cl2]2 | 82 | 12:1 |
| 4d | [RhCptCl2]2 | 92 | 15:1 |

To determine the effect that ligand electronics have on product regioselectivity, we employed an electron deficient 1-trifluoromethyl-2,3,4,5-tetramethylcyclopentadienyl ligand originally developed by Gassman (CpCF3)17 and found that this catalyst provides 3a and 3a′ products in good yield but without an increase in selectivity (2.4:1) (Table 1, entry 2).18,19 Since ligand electronics did not appear to affect product regioselectivity, we tested an electron rich, sterically bulky di-phenyl-tri-methyl Cp ligand (Cp‡) and were pleased to find a remarkable increase in selectivity from 2.4:1 to 12:1 (3a:3a′). Pleased by this improvement, we tested the sterically bulky di-tert-butyl Cp ligand Cpt and were surprised to find that RhCpt provides the desired product in 91% yield with exquisite regioselectivity (15:1) (Table 1, entry 4).
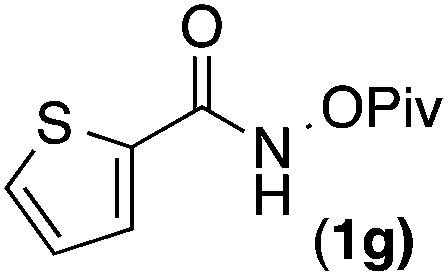
Having demonstrated that Cpt is able to substantially increase the regioselectivity of alkene migratory insertion, we explored the scope of O-pivaloyl benzyhydroxamic acids amenable to this reaction. We were pleased to find para-substituted benzhydroxamic acids are well tolerated (50–76% yields) with both Cp* and Cpt ligands. Notably, the low regioselectivity of <2.2:1 seen using [RhCp*Cl2]2 is dramatically improved with [RhCptCl2]2 giving excellent regioselectivities of >14:1 (Table 2, entries 1–4). Electron rich amides derived from gallic acid provide product in excellent yield and regioselectivity (Table 2, entry 5). Finally, heterocyclic amides are both well tolerated under the reaction conditions and responsive to the sterically bulky ligand, providing product with excellent regioselectivity (Table 2, entries 6–7).
|
|
||||
|---|---|---|---|---|
| Entry | Starting material | Yieldb (%) | Cp* | Cpt |
| a Reaction conditions: amide (.2 mmol), 1-decene (.2 mmol), precatalyst (1 mol%), CsOAc (200 mol%), MeOH (0.1 M). b Isolated yield of reaction using [RhCptCl2]2 as a precatalyst. | ||||

|
||||
| 1 | X = CF3 (1b) | 50 | 1.5:1 |
19:1 |
| 2 | X = Cl (1c) | 76 | 2.2:1 |
19:1 |
| 3 | X = OMe (1d) | 70 | 1.9:1 |
16:1 |
| 4 | X = Ph (1e) | 75 | 1.7:1 |
14:1 |
| 5 |

|
95 | 1.9:1 |
15:1 |
| 6 |
|
84 | 2.5:1 |
19:1 |
| 7 |

|
88 | 1.8:1 |
19:1 |

meta-Substituents also provide exquisite levels of regioselectivity for alkene migratory insertion when Cpt is used (>15:1) (Table 3). Interestingly, when Cp* is used the C–H activation occurs exclusively at the 6-position; we suggest that this selectivity is the result of steric interactions between the 2-substituent and metal complex during concerted metallation deprotonation. Surprisingly, when Cpt is used, the regioselectivity of the C–H activation actually decreases to afford a mixture of 2- and 6-substituted products. While on the surface the decrease of selectivity appears counterintuitive, it can be explained by the uneven distribution of steric bulk in the Cpt ligand, which we will comment on subsequently. Not only is this decrease in selectivity mechanistically intriguing, it also offers an exciting opportunity to potentially reverse the regioselectivity of C–H activation.
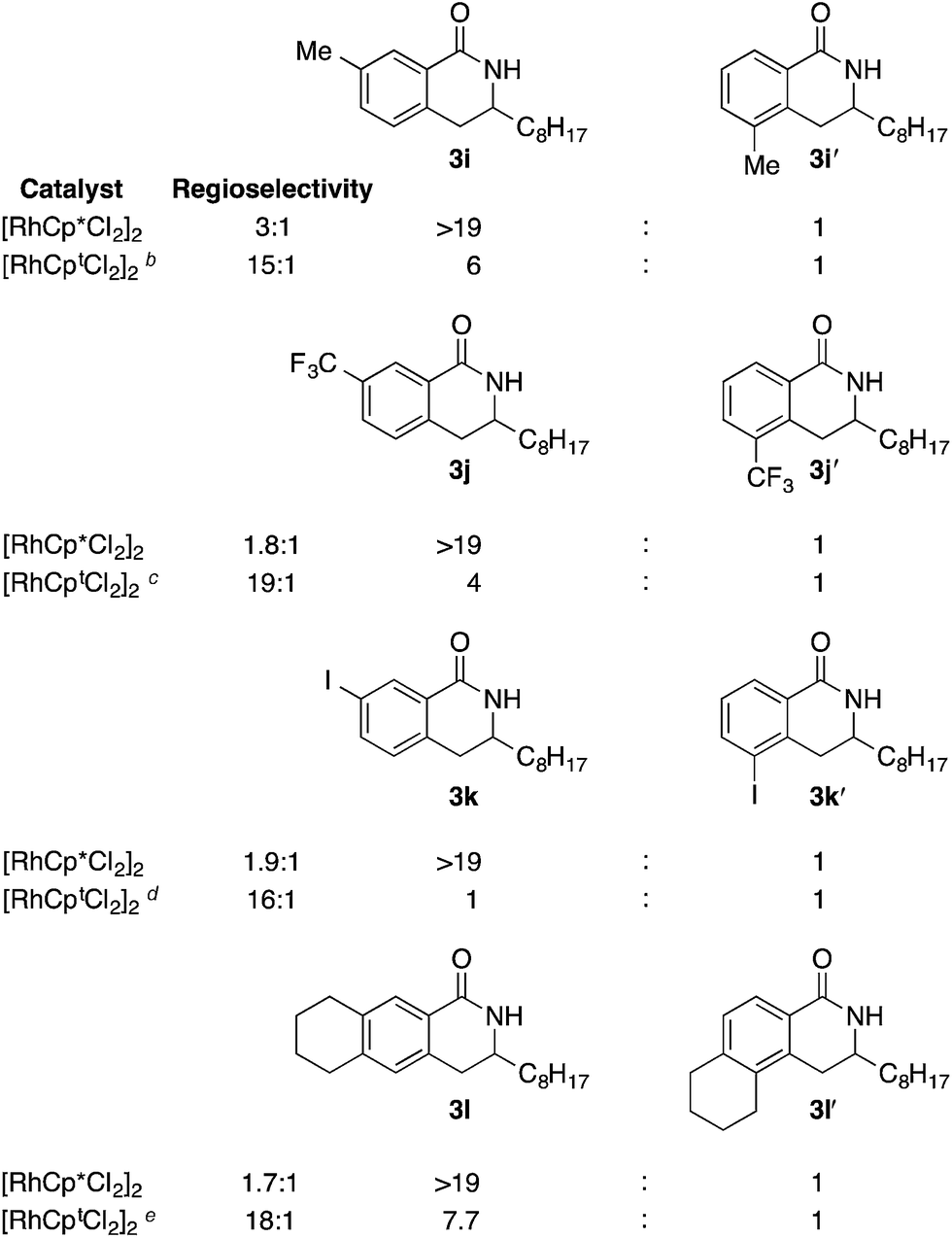
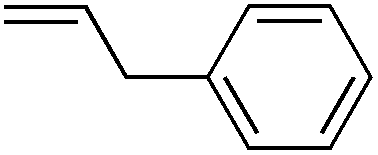
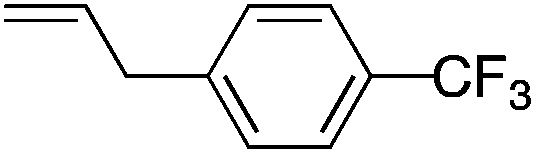
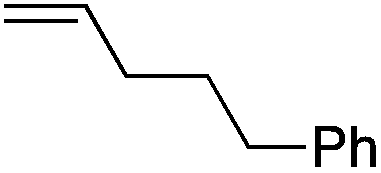
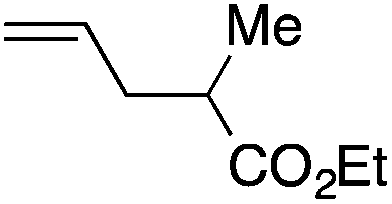
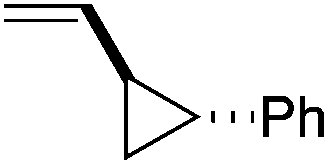
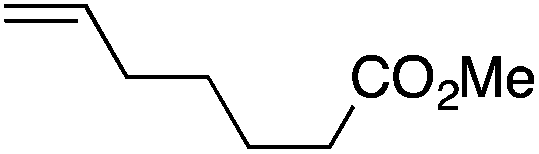
We next explored the alkene tolerance of the method. Allyl benzene 2b furnishes a 1.6:1 ratio of dihydroisoquinolone with RhCp* (Table 4, entry 1). When Cpt is employed, the regioselectivity increases to 4.5:1. Reducing the temperature to 0 °C further increases regioselectivity to 5.1:1. Placing an electron-donating group on the aromatic ring increases the selectivity of insertion (Table 4, entry 2). Aryl electron withdrawing substituents do not, however, provide a change in regioselectivity relative to phenyl (Table 4, entry 3). When the aromatic ring is moved further from the alkene the regioselectivity observed with Cpt increases and yields remain high (Table 4, entries 4 and 5). Ester and ketone functional groups are tolerated without a detrimental impact on regioselectivity (Table 4, entry 6, 9, and 10). Unfortunately, alkenes bearing stereocenters are ineffective at inducing diastereoselectivity with either ligand (Table 4, entries 6 and 8). Vinyl cyclopropanes participate with good levels of regioselectivity but poor diastereoselectivity (Table 4, entry 8). Unprotected alcohols provide product in excellent yield with no sign of oxidation (Table 4, entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | Alkene | Yieldb (%) | Cp* | Cpt |
|
a Reaction conditions: 1a (.2 mmol), alkene (.2 mmol), precatalyst (1 mol%), CsOAc (200 mol%), MeOH (0.1 M).
b Isolated yield of reaction using [RhCptCl2]2 as a precatalyst.
c Reaction conducted at 0 °C.
d Products isolated as a 1:1 ratio of diastereomers.
e Product isolated as a 2:1 ratio of diastereomers.
|
||||
| 1c |
|
85 | 1.6:1 |
5.1:1 |
| 2 |

|
68 | 1.6:1 |
9.4:1 |
| 3 |
|
70 | 1.3:1 |
5.5:1 |
| 4 |

|
95 | 2.3:1 |
14:1 |
| 5 |
|
85 | 1.6:1 |
8:1 |
| 6d |
|
92 | 1.2:1 |
7.2:1 |
| 7 |

|
80 | 1.4:1 |
12:1 |
| 8e |
|
93 | 1:1 |
11:1 |
| 9 |
|
89 | 2:1 |
14:1 |
| 10 |
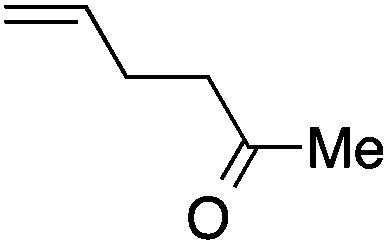
|
94 | 3:1 |
14:1 |
While it is desirable to achieve high regioselectivity for a single regioisomer, it is even more attractive to use a ligand to access alternate regioisomers. Currently, the only example of Rh(III)-catalyzed synthesis of 4-substituted dihydroisoquinolones is with potassium vinyltrifluoroborates where electronics are believed to control regioselectivity.20 We found that when vinylcyclohexane was submitted to a reaction with [RhCp*Cl2]2 as the precatalyst, the 3-substituted dihydroisoquinolone 4a was isolated in 90% yield with 11:1 regioselectivity (Fig. 1). However, when the same reaction was catalyzed by [RhCptCl2]2 the opposite isomer 4b was isolated in 75% yield and 10:1 (4b:4a) regioselectivity. Given this unexpected discovery, we were interested in gleaning insight into how Cpt influences regioselectivity of alkene migratory insertion. A competition experiment between vinyl cyclohexane 2m and 1-decene 2a run to 10% conversion favored the formation of dihydroisoquinolone 3a in >19:1 ratio as determined by 1H NMR. This experiment suggests that enhanced steric interactions between the substrate and ligand slow the rate of migratory insertion.
 | ||
| Fig. 1 Impact of ligand on reaction of vinyl cyclohexane. | ||
To investigate the steric differences between the RhCp* and RhCpt systems X-ray analysis was conducted on a 5-membered RhCpt metallacycle. While we were unable to obtain a 5-membered rhodacycle from our system, Jones and coworkers previously characterized 5-membered rhodacycle 5a from N-benzylidenemethanamine and [RhCp*Cl2]2.21 We found that a similar metallacycle 5b derived from [RhCptCl2]2 could be obtained in crystalline form under identical conditions and was evaluated by single crystal X-ray diffraction.
A comparison of the bond lengths and angles reveals several notable differences between our Cpt rhodacycle and the Cp* rhodacycle reported by Jones (Fig. 2). The Rh–Cp centroid distance in 5b is 0.011 Å longer than 5a which is either the result of increased steric interactions, or an artifact of Cpt being a less electron-donating ligand. While there are subtle differences in many bond lengths and angles, the most striking difference is the angle C3–Rh–Cl, which is 98.03° in 5b while only 90.09° in 5a. The angle increase is likely the result of steric interactions caused by the tert-butyl moiety being situated directly over the Rh–Cl bond. As alkene exchange presumably occurs with Cl, we suggest that steric interactions between the t-butyl of the ligand and the alkene substituent affect both the alkene coordination and 1,2-insertion events.
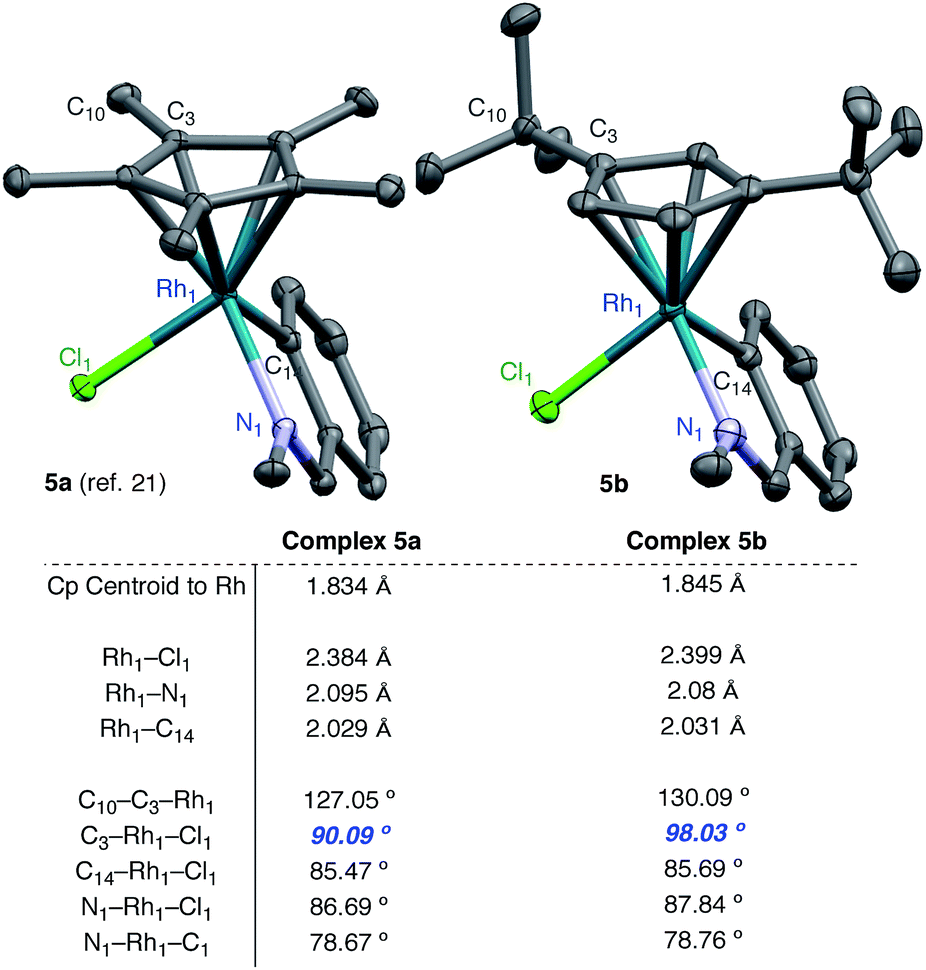 | ||
| Fig. 2 X-Ray analysis. | ||
Based on the X-ray crystal structure and regioselectivity data, we propose the following model for regioselectivity of the 1,2-migratory insertion of alkenes, where steric contributions from the t-butyl groups influence both alkene coordination and insertion events to give high selectivity. With small alkyl alkenes, we propose that steric interactions from one t-butyl of Cpt disfavor alkene coordination (I) and subsequent insertion to give the β-substituted product 3a′ (Fig. 3). Coordination of the alkene with the steric bulk oriented away from the t-butyl group finds minimized steric interactions during coordination (II). Subsequent migratory insertion from II places the alkyl substituent α to Rh in the transition state, which we propose is able to stabilize a buildup of partial positive charge, making the α-substituted product 3a both sterically and electronically favored with Cpt. In the case of the Cp* ligand with small alkyl alkenes, neither steric nor electronic interactions dominate so low selectivity is observed.
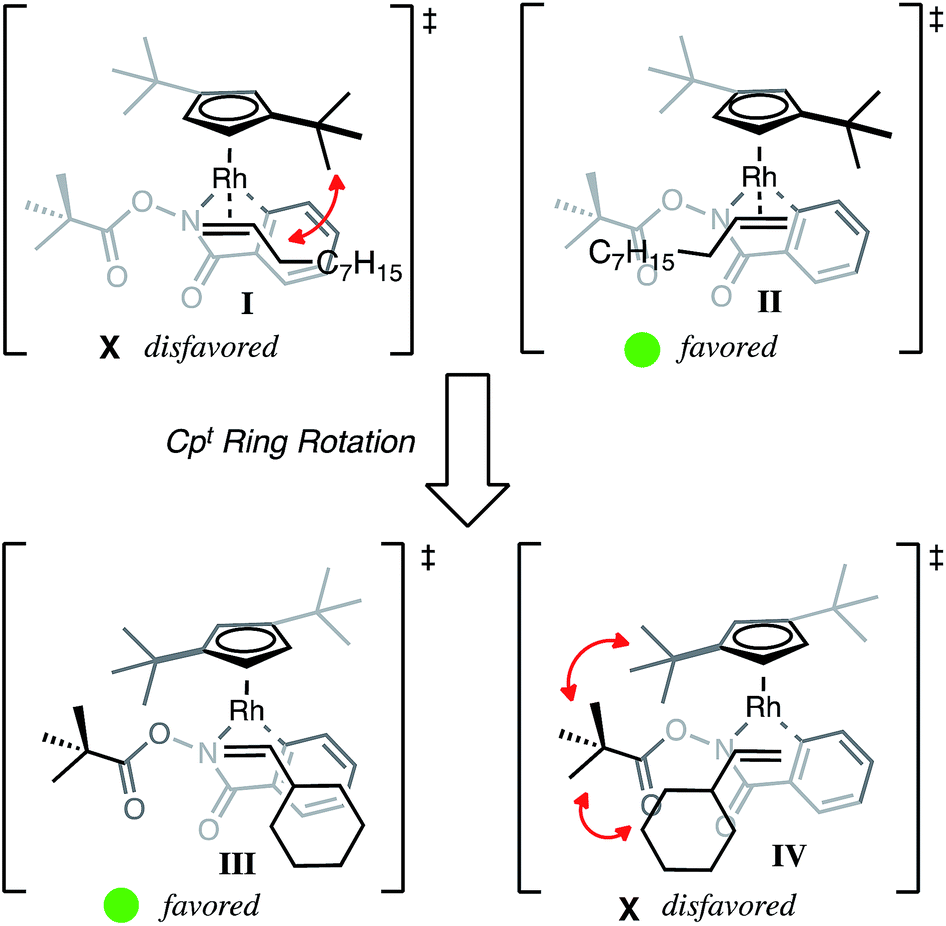 | ||
| Fig. 3 Rationale for selectivity. | ||
However if the size of the alkene substituent is significantly increased, as in the case of vinyl cyclohexane, then Cpt favors the opposite regioisomer. While certainly a puzzling result, we propose that the selectivity can be explained by Cpt rotation such that the t-butyl groups both occupy the space above the metallacycle. Cpt rotation gears the O-piv toward the alkene coordination site disfavoring alkene coordination to this side (IV) favoring the α-substituted product 3a. At the same time, alkene coordination (III) with the cyclohexyl opposite the O-piv minimizes steric interactions enabling insertion of the large alkene and preferential formation of β-substituted product 3a′. While not conclusive, the observation that cyclohexyl alkene reacts significantly slower than n-octyl alkene suggests that migratory insertion of the cyclohexyl alkene proceeds through a higher energy and potentially highly ordered transition state, such as Cpt rotation.
Conclusions
In conclusion, we found that sterically bulky di-tert-butyl cyclopentadienyl ligand (Cpt) is effective at increasing regioselectivity of alkene migratory insertion in the synthesis of dihydroisoquinolones by Rh(III) C–H activation catalysis. In contrast to previous cases where Cp* delivers modest levels of regioselectivity of migratory insertion (with alkynes), Cpt renders previously non-selective reactions (with alkenes) highly regioselective. Furthermore, ligand control (Cp* vs. Cpt) enables the highly selective synthesis of two regioisomeric products with vinylcyclohexanes. Finally, crystallographic evidence lends support for a possible explanation of the enhanced regioselectivity.Acknowledgements
We thank the NIGMS for generous support of this research (GM80442). T.R thanks Johnson Matthey for a generous loan of Rh salts. D.M.D. thanks the NIH Ruth Kirschstein predoctoral fellowship for funding.Notes and references
- (a) C. Zhu, R. Wang and J. R. Falck, Chem.–Asian J., 2012, 7, 1502 CrossRef CAS PubMed; (b) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31 CAS; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (d) D. M. D'Souza and T. J. J. Müller, Chem. Soc. Rev., 2007, 36, 1095 RSC; (e) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC.
- (a) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (b) L. Ackermann, Acc. Chem. Res., 2014, 47, 818 CrossRef PubMed.
- For early examples; see: (a) N. Guimond and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 12050 CrossRef CAS PubMed; (b) T. K. Hyster and T. Rovis, J. Am. Chem. Soc., 2010, 132, 10565 CrossRef CAS PubMed; (c) D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326 CrossRef CAS PubMed.
- For early examples; see: (a) K. Ueura, T. Satoh and M. Miura, Org. Lett., 2007, 9, 1407 CrossRef CAS PubMed; (b) K. Ueura, T. Satoh and M. Miura, J. Org. Chem., 2007, 72, 5362 CrossRef CAS PubMed; (c) D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474 CrossRef CAS PubMed; (d) S. Mochida, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 6295 CrossRef CAS PubMed; (e) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908 CrossRef CAS PubMed; (f) D. J. Schipper, M. Hutchinson and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6910 CrossRef CAS PubMed; (g) S. Rakshit, F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585 CrossRef CAS PubMed; (h) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449 CrossRef CAS PubMed.
- For rationale of alkyne electronics using Mayr's nucleophilicity scale; see: R. K. Friedman and T. Rovis, J. Am. Chem. Soc., 2009, 131, 10775 CrossRef PubMed.
- (a) T. K. Hyster and T. Rovis, Chem. Sci., 2011, 2, 1606 RSC; (b) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846 RSC.
- For additional examples of alkenyl Rh(III)-catalyzed C–H activation to synthesize heterocycles; see: (a) G. Song, D. Chen, C.-L. Pan, R. H. Crabtree and X. Li, J. Org. Chem., 2010, 75, 7487 CrossRef CAS PubMed; (b) Y. Su, K. Han, G. Song and X. Li, Org. Lett., 2010, 12, 5462 CrossRef CAS PubMed; (c) X. P. Zhang, D. Chen, M. A. Zhao, J. Zhao, A. Q. Jia and X. Li, Adv. Synth. Catal., 2011, 353, 719 CrossRef CAS; (d) P. C. Too, T. Noji, Y.-J. Lim, X. Li and S. Chiba, Synlett, 2011, 22, 2789 Search PubMed.
- (a) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350 CrossRef CAS PubMed; (b) Ref. 5h ; (c) H. Wang and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 7318 CrossRef CAS PubMed.
- For an enantioselective example using an artificial metalloenzyme; see: T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500 CrossRef CAS PubMed.
- For an enantioselective example using a chiral cyclopentadienyl ligand; see: (a) B. Ye and N. Cramer, Science, 2012, 338, 504 CrossRef CAS PubMed; (b) B. Ye and N. Cramer, J. Am. Chem. Soc., 2013, 135, 636 CrossRef CAS PubMed; (c) B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 7896 CrossRef CAS PubMed.
- In contrast, 1-octyne provides a single regioisomer, see ref. 5h.
- (a) H. A. Malik, G. J. Sormunen and J. Montgomery, J. Am. Chem. Soc., 2010, 132, 6304 CrossRef CAS PubMed; (b) T. Liu, J. Montgomery and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 6959 Search PubMed; (c) N. Wu, L. Deng, L. Liu, Q. Liu, C. Li and Z. Yang, Chem.–Asian J., 2012, 8, 65 CrossRef PubMed; (d) P. A. Wender and A. J. Dyckman, Org. Lett., 1999, 1, 2089 CrossRef CAS.
- (a) T. A. Davis, T. K. Hyster and T. Rovis, Angew. Chem., Int. Ed., 2013, 52, 14181 CrossRef CAS PubMed; (b) B. Ye, P. A. Donet and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 507 CrossRef CAS PubMed; (c) Z. Shi, M. Boultadakis-Arapinis, D. C. Koester and F. Glorius, Chem. Commun., 2014, 50, 2650 RSC; (d) T. K. Hyster and T. Rovis, Synlett, 2013, 24, 1842 CrossRef CAS PubMed.
- For other tethering stratagies; see: (a) X. Xu, Y. Liu and C.-M. Park, Angew. Chem., Int. Ed., 2012, 51, 9372 CrossRef CAS PubMed; (b) N. Quiñones, A. Seoane, R. Garcia-Fandiño, J. L. Mascareñas and M. Gulias, Chem. Sci., 2013, 4, 2874 RSC.
- (a) M. Itoh, K. Hirano, T. Satoh, Y. Shibata, K. Tanaka and M. Miura, J. Org. Chem., 2013, 78, 1378 Search PubMed; (b) K. Morimoto, M. Itoh, K. Hirano, T. Satoh, Y. Shibata, K. Tanaka and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 5359 CrossRef CAS PubMed; (c) Y. Shibata and K. Tanaka, Angew. Chem., Int. Ed., 2011, 50, 10922 CrossRef PubMed; (d) M. Shimizu, H. Tsurugi, T. Satoh and M. Miura, Chem.–Asian J., 2008, 3, 881 CrossRef CAS PubMed.
- P. G. Gassman, J. W. Mickelson and J. R. Sowa, J. Am. Chem. Soc., 1992, 114, 6942 CrossRef CAS.
- For catalytic uses of this ligand, see: (a) Ref. 7b ; (b) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735 CrossRef CAS PubMed.
- For Tanaka's electron deficient Cp ligand; see: (a) M. Fukui, Y. Hoshino, T. Satoh, M. Miura and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1638 CrossRef CAS; (b) Y. Hoshino, Y. Shibata and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1577 CrossRef CAS; (c) T. Piou and T. Rovis, J. Am. Chem. Soc., 2014, 136, 11292 CrossRef CAS PubMed.
- M. Presset, D. Oehlrich, F. Rombouts and G. A. Molander, Org. Lett., 2013, 15, 1528 CrossRef CAS PubMed.
- L. Li, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2008, 130, 12414 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterization. CCDC 1021286. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02590c |
| This journal is © The Royal Society of Chemistry 2015 |