
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Divergence from the classical hydroboration reactivity; boron containing materials through a hydroboration cascade of small cyclic dienes†
Anna
Andreou
a,
Michal
Leskes
b,
Pablo G.
Jambrina
c,
Gary J.
Tustin
d,
Clare P.
Grey
b,
Edina
Rosta
c and
Oren A.
Scherman
*a
aMelville Laboratory for Polymer Synthesis, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: oas23@cam.ac.uk
bDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
cDepartment of Chemistry, Kings College London, Strand, London, WC2R 2LS, UK
dSchlumberger Cambridge Research, High Cross, Madingley Road, Cambridge, CB3 0EL, UK
First published on 6th August 2015
Abstract
The hydroboration 1,3- and 1,4-cyclic dienes has been systematically investigated. The behavior of such dienes towards mono and dihydroboration was monitored directly by 11B NMR to identify the actual boron species formed, as opposed to the most common analysis of the resultant oxidation products. Quantitative dihydroboration was achieved for the full range of cyclic dienes investigated including dienes, which were previously reported to be resistant to dihydroboration, leading to the formation of new boron-containing polymeric materials. The conditions favoring dihydroboration are reported as well as full characterisation of the materials. Furthermore, a hydroboration cascade mechanism is proposed for the formation of such boron-containing polymers, supported by both experimental and theoretical data.
1 Introduction
Hydroboration is undoubtedly a popular and widely used synthetic reaction.1,2 More specifically, hydroboration and subsequent oxidation of dienes to their corresponding diols has received much attention mainly on account of their wide usage in applications such as adhesives,3,4 polymeric materials,5,6 pharmaceuticals,7,8 boronic acid–diol sensors9,10 and as intermediates for further synthetic transformations such as in the synthesis of chiral ligands BINAP,11,12 DuPHOS13,14 amongst others. Despite the fact that dihydroboration of dienes followed by basic oxidation appears an attractive synthetic route to obtain diols, such compounds are typically prepared using epoxide hydrolysis,15,16 oxidation of alkenes,17,18 the Prévost reaction (anti diols)19–21 and the Woodward cis-hydroxylation (cis diols)22 giving rise to high stereo and regio control.It is possible that dihydroboration of cyclic dienes to generate diols has not been thoroughly explored as a result of the strong and contradicting conclusions drawn in a series of published studies by H. C. Brown et al.23–25 In these publications, the hydroboration of a series of cyclic dienes was explored. Brown et al. concluded that smaller cyclic rings, such as 1,3-cyclohexadiene 1, undergo dihydroboration only to a very small extent and were therefore concluded to be resistant based on the observation of selective formation of the monohydroboration products.23,25 In the case of 1, basic oxidation and gas chromatography (GC) analysis of the hydroboration mixture was reported to yield the formation of 2-cylohexene-1-ol 2 and 3-cyclohexene-1-ol 3 as shown in Fig. 1.25 Contradictingly, in a singular older report, H. C. Brown et al. mentioned the possibility of polymer formation via dihyrdroboration of cyclic dienes but have never investigated the outcome of such reactions.24 Research conducted by contemporaries in the field reported improved yields, however, generally following the same trend that smaller rings appeared to be resistant to dihydroboration.26 For example, when K. J. Saegebarth used a large excess (80%) of diborane(6) with 1,3-cyclopentadiene, an increased yield of cis-1,3-cyclopentanediol was obtained after oxidation, with monohydroboration, however, still being the main reaction. We found the exceptional resistance of smaller rings towards dihydroboration, even in the cases where excess hydroborating agent was used, rather puzzling. Although a diene is certainly a more electron rich system when compared to an alkene, the non-conjugated nature of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond present in the formed monohydroboration products could potentially compel them to be more reactive than the starting diene, on account of the non-conjugated double bond compared to the initial system. Additionally, both the cyclic diene and the borane reagents (both the starting borane and the formed products) are capable of undergoing further hydroboration reactions. Thus, one might expect the formation of polymeric and/or network materials, as was previously reported in the case of straight-chain dienes27–30 or the formation of oligomers through ring opening polymerization.31
C bond present in the formed monohydroboration products could potentially compel them to be more reactive than the starting diene, on account of the non-conjugated double bond compared to the initial system. Additionally, both the cyclic diene and the borane reagents (both the starting borane and the formed products) are capable of undergoing further hydroboration reactions. Thus, one might expect the formation of polymeric and/or network materials, as was previously reported in the case of straight-chain dienes27–30 or the formation of oligomers through ring opening polymerization.31
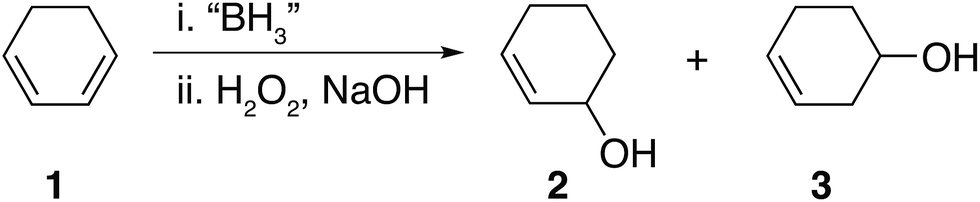 | ||
| Fig. 1 Hydroboration–oxidation of 1,3-cyclohexadiene 1 as reported by Brown et al.24,25 | ||
Unfortunately, almost all of these studies relied upon indirect observations of the subsequently isolated hydroxylated species formed after oxidation, rather than gaining direct evidence arising from the borane species. To date there have only been a few published hydroboration studies that analyze the reaction outcome directly, i.e. by identifying the boron species present.32,33 We describe herein a systematic investigation of the hydroboration of 1,3-cyclohexadiene 1 (for other cyclic dienes see ESI S10†) and its behavior towards mono and dihydroboration by direct observation of the boron species formed. The reaction conditions favoring dihydroboration lead to the formation of new boron-containing polymeric materials, and a mechanism for the formation of such materials is proposed and supported by both experimental and theoretical data.
2 Results and discussion
As a result of our interest in preparing substituted diboranes, we aimed to systematically investigate the hydroboration of cyclic dienes in order to identify the suitable conditions for dihydroboration of smaller rings. While the results and conclusions by Brown et al. were certainly a concern, we were encouraged by the singular literature report by Saegebarth, who reported up to 40% dihydroboration of 1,3-cyclopentadiene when a large excess of diborane(6) was used.26We first focused our attention on the number of equivalents of diene present in the reaction, as the large excess of 1 utilized by H. C. Brown et al.24,25 seemed unnecessary and was possibly the determining factor for the selective monohydroboration observed in their system. The number of equivalents of diene used was kept constant (1 equivalent) while the amount of borane was explored. Additionally, we investigated two modes of reagent addition: borane to diene and diene to borane. As can be clearly seen in Table 1, irrespective of borane molar equivalents and the mode of reagent addition, all entries yielded a white, insoluble precipitate. When THF was used as the solvent, a glassy material was formed as shown in Fig. 2, which gave rise to complications in characterisation of the products; therefore, in an attempt to eliminate the possibility of side reactions such as THF ring opening (known to be initiated by borane),34 diglyme was chosen as the reaction solvent (see ESI S6†). Moreover, the use of diglyme allowed for direct comparison of the reactions performed when two different borane sources were explored (BH3·SMe2vs. B2H6 prepared from NaBH4 and BF3·OEt2).
| Entry | Borane (molar equiv.) | Mode of reagent addition | Observation |
|---|---|---|---|
a The mode of reagent addition (borane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diene or diene:borane) yielded the same results.
b Observed during both modes of addition.
c Observed during BF3·OEt2 to NaBH4 addition.
d Observed during NaBH4 to BF3·OEt2. :diene or diene:borane) yielded the same results.
b Observed during both modes of addition.
c Observed during BF3·OEt2 to NaBH4 addition.
d Observed during NaBH4 to BF3·OEt2.
|
|||
| 1 | 0.33 | Borane:diene |
White precipitate formeda |
| Diene:borane |
Unreacted CC observeda |
||
| 2 | 0.50 | Borane:diene |
White precipitate formeda |
| Diene:borane |
No CC observeda |
||
| Unreacted boranea | |||
| 3 | 1.0 | Borane:diene |
White precipitate formeda |
| Diene:borane |
No CC observeda |
||
| Unreacted boranea | |||
| 4 | 2.0 | Borane:diene |
White precipitate formeda |
| Diene:borane |
No CC observeda |
||
| Unreacted boranea | |||
| 5 | 0.25 | BF3·OEt2 to NaBH4 | White precipitate formedb |
| NaBH4 to BF3OEt2 | Unreacted CC observedc |
||
| No CC observedd |
|||
| 6 | 0.5 | BF3·OEt2 to NaBH4 | White precipitate formedb |
| NaBH4 to BF3OEt2 | No CC observedc |
||
| Unreacted CC observedd |
|||
| 7 | 1.0 | BF3·OEt2 to NaBH4 | White precipitate formedb |
| NaBH4 to BF3·OEt2 | No CC observedb |
||
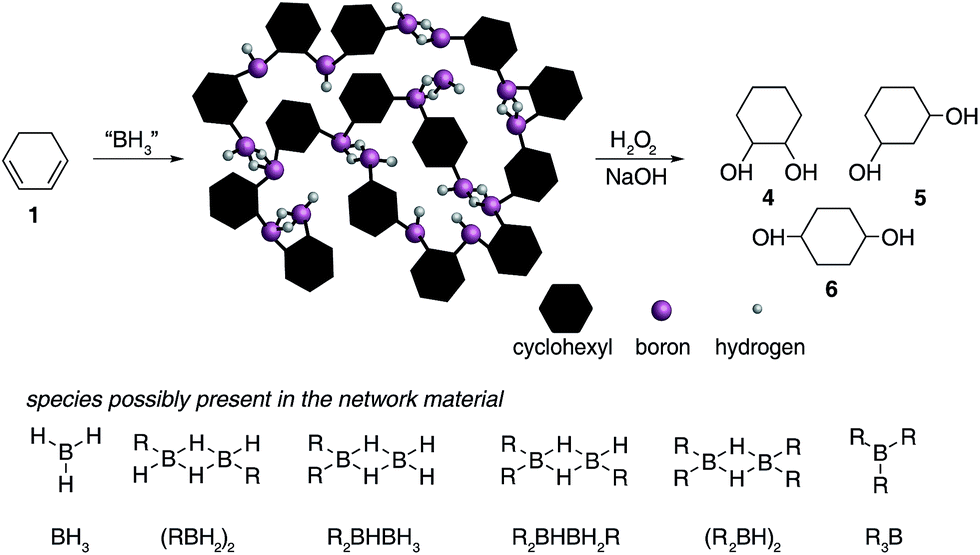 | ||
| Fig. 2 Hydroboration of 1,3-cyclohexadiene 1 leading to the formation of insoluble materials which contain a range of borane species. | ||
In order to fully characterise the reaction, the precipitates were collected by filtration (under nitrogen) and the filtrates were analysed by NMR. All B–H bonds appeared to undergo hydroboration, as unsaturated CC bonds were only observed in entry 1, while the borane starting reagent was fully consumed. Analysis of the filtrates obtained for entries 2–4 (molar borane equivalents of 0.5, 1.0 and 2.0, respectively), revealed large amounts of unreacted starting borane, seen as a quartet at −20 ppm in the 11B NMR. Furthermore, the absence of any CC bonds in the 1H NMR for these entries suggested that full dihydroboration has taken place utilising all CC bonds.
When the source of borane was changed to diborane(6), produced in situ, the same trend was observed (see Table 1). As diborane(6) was prepared in situ, two alternative modes of addition were employed, leading to either slow or fast diborane(6) gas production. This was achieved by either adding BF3·OEt2 to a solution containing both NaBH4 and 1, resulting in a slow release of the desired gas (as the reaction proceeds via the formation of the NaBH4BH3 complex first), or by addition of NaBH4 to a solution containing both BF3·OEt2 and diene 1, which resulted in the instantaneous release of diborane(6). As in the case of BH3·SMe2, all reactions yielded a white precipitate, with only NaBF4 being observed in the filtrate, appearing at approximately −0.4 ppm in the 11B NMR, indicating reaction completion.
As dienes can undergo cationic polymerisation initiated by BF3·Lewis base adducts,35 a control experiment was carried out to ensure that such a polymerisation was not the cause of the observed white precipitate. Thus, the reaction was performed in the absence of NaBH4 (see ESI S6.1†). The presence of BF3 alone did not lead to the formation of a white precipitate, confirming that the origin of these solid materials resulted indeed from the dihydroboration reactions.
2.1 Analysis and characterisation of the boron-containing precipitates
The solubility of the formed materials was investigated in a range of solvents. These materials, however, were found to be insoluble in acetic acid, acetone, acetonitrile, benzene, chloroform, cyclohexane, DCM, DMF, DMSO, 1,4-dioxane, 1,3-dioxane, ethyl acetate, diethyl ether, hexane and THF under a nitrogen atmosphere, even after being left for a week. Interestingly, these materials reacted with acetonitrile, methanol and triphenylphoshine (solution in diglyme) serving as indirect evidence that boron hydrides are present in these materials (see ESI S.7.1†).36As a result of the insoluble and air-sensitive nature of these materials, only a limited number of characterisation techniques including solid state NMR and FT-IR could be employed. Additionally, basic oxidation of the materials was also carried out. It is worth noting that the mass of these isolated materials was consistently above the maximum expected value (based on a yield of 100%, even after a week under vacuum) indicating the presence of trapped solvents, which was confirmed by solid state NMR. Moreover, this strongly suggested the formation of a polymeric network. Addition of Lewis bases such as PPh3 did not fully dissolve the solid material. Future work will further investigate additional reactions that might take place and the possible role of solvent in subsequent reactions.
C bonds (1800–1700 cm−1). All solids analysed resulted in similar spectra despite originating from a number of different reaction conditions. The FT-IR spectra of four selected materials are shown in Fig. 3, which correspond to the materials also analysed by solid state NMR (vide infra). The B–Ht bond stretches could indicate either presence of a R2BHBH3 species (see Fig. 2), which was found to form during the reaction between a (R2BH)2 species and unreacted borane or diborane(6) starting material, due to R2BHBH2R or RBH2BH2R, or unbridged R2BH species or even due to BH3 trapped inside the material network. The latter option, however, could be dismissed based on solid state NMR data (see Fig. 5c–f). Finally, the absence of any CC double bonds in both the filtrate and the solid confirmed that full dihydroboration had taken place.
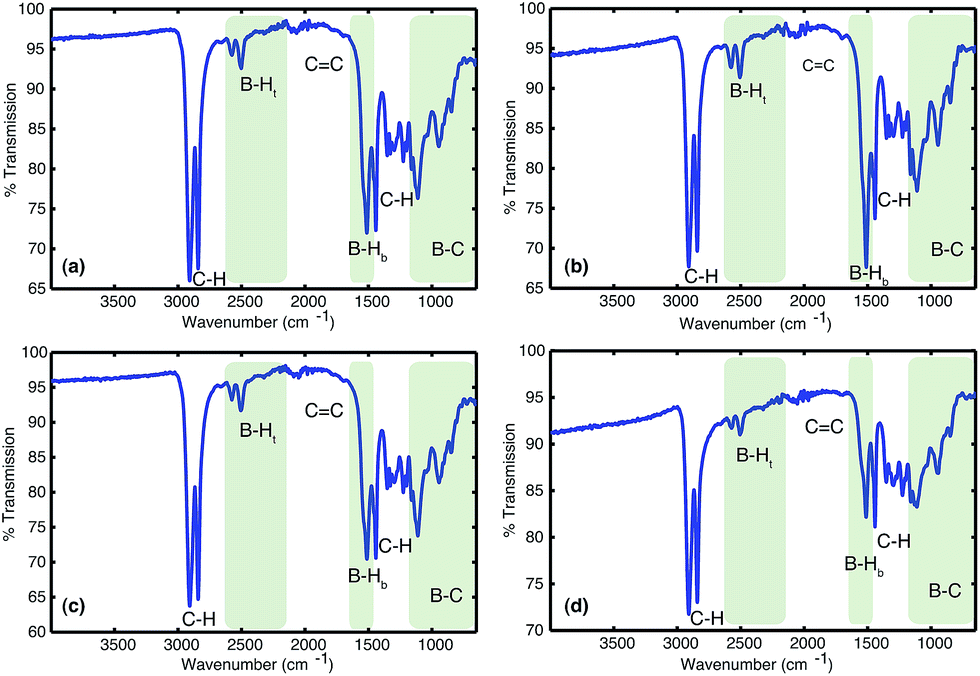 | ||
| Fig. 3 The FTIRs of the materials obtained from the reactions between (a) 1:2 molar equivalents of 1,3-cyclohexadiene 1 added to borane BH3·SMe2, (b) 1:1 molar equivalents of 1,3-cyclohexadiene 1 and diborane(6) B2H6 utilising slow gas release, (c) 2:1 molar equivalents of borane BH3·SMe2 added to 1,3-cyclohexadiene 1 and (d) 1:1 molar equivalents of 1,3-cyclohexadiene 1 and diborane(6) B2H6 utilising fast gas release. | ||
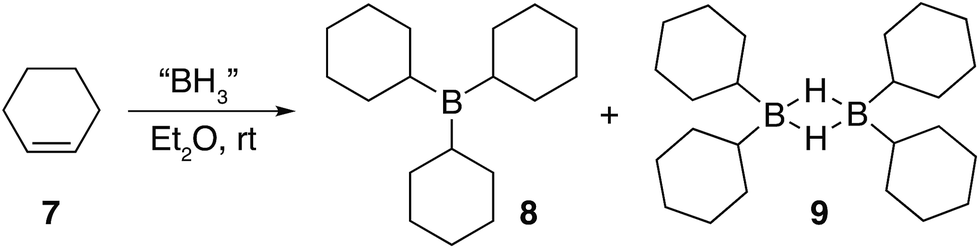 | ||
| Fig. 4 The main products formed during the hydroboration of 7. | ||
Three equivalents of cyclohexene 7 were reacted with a single equivalent of BH3·SMe2 leading to the formation tricyclohexylborane 8. However, when the borane was added quickly to 7, the product ratio significantly favoured the formation of the B–H–B bridged species 9 as confirmed by FT-IR (see ESI S.7.2.1†). Importantly, diethyl ether was chosen as the solvent to prepare model compounds 8 and 9 as opposed to diglyme as it enabled a straightforward method to isolate the two compounds. A white precipitate in ether was readily filtered and isolated to yield dicyclohexyl B–H–B bridged 9, and confirmed by solution NMR spectroscopy (+29 ppm in 11B NMR).37 Conversely, tricyclohexylborane 8 remained soluble in ether and could be isolated upon concentration followed by recrystallisation (+81 ppm in solution 11B NMR). The dominant formation of dicyclohexylborane 9 (R2BH)2, especially at the earlier stages of hydroboration, suggested that it was the kinetic product, which could be converted to the thermodynamic tricyclohexylborane 8 over time (see ESI S7.2.3, Fig. S18†), further supported by our theoretical calculations. DFT calculations using the B3LYP/6-311++G** level of theory have shown that in the presence of excess cyclohexene, 9 could hydroborate a third cyclohexene to form tricyclohexylborane 8 although with a high barrier, likely due to the steric effect of the substituent (20.4 kcal mol−1, see ESI S11, Table S1†). Interestingly, the powerful stabilisation offered by the B–H–B bridges provides 9 with enough stability in air over short periods of time (ca. 5 min) as opposed to the instantly pyrophoric and highly unstable tricyclohexylborane 8. The direct dependence on the rate of reagent addition towards the formation of kinetic product 9vs. thermodynamic product 8 led to materials with different physical properties and reactivities. For example, materials which are mainly consisted of the kinetic products bearing the boron hydride bridges (as opposed to the trialkylboranes, the thermodynamic products) are highly moisture sensitive but not pyrophoric. When these materials where exposed to moisture they transformed from solid to liquid clearly breaking the B–H–B bridges which hold the insoluble polymeric network together. We are currently investigating the applications of these materials especially as instant moisture scavengers.
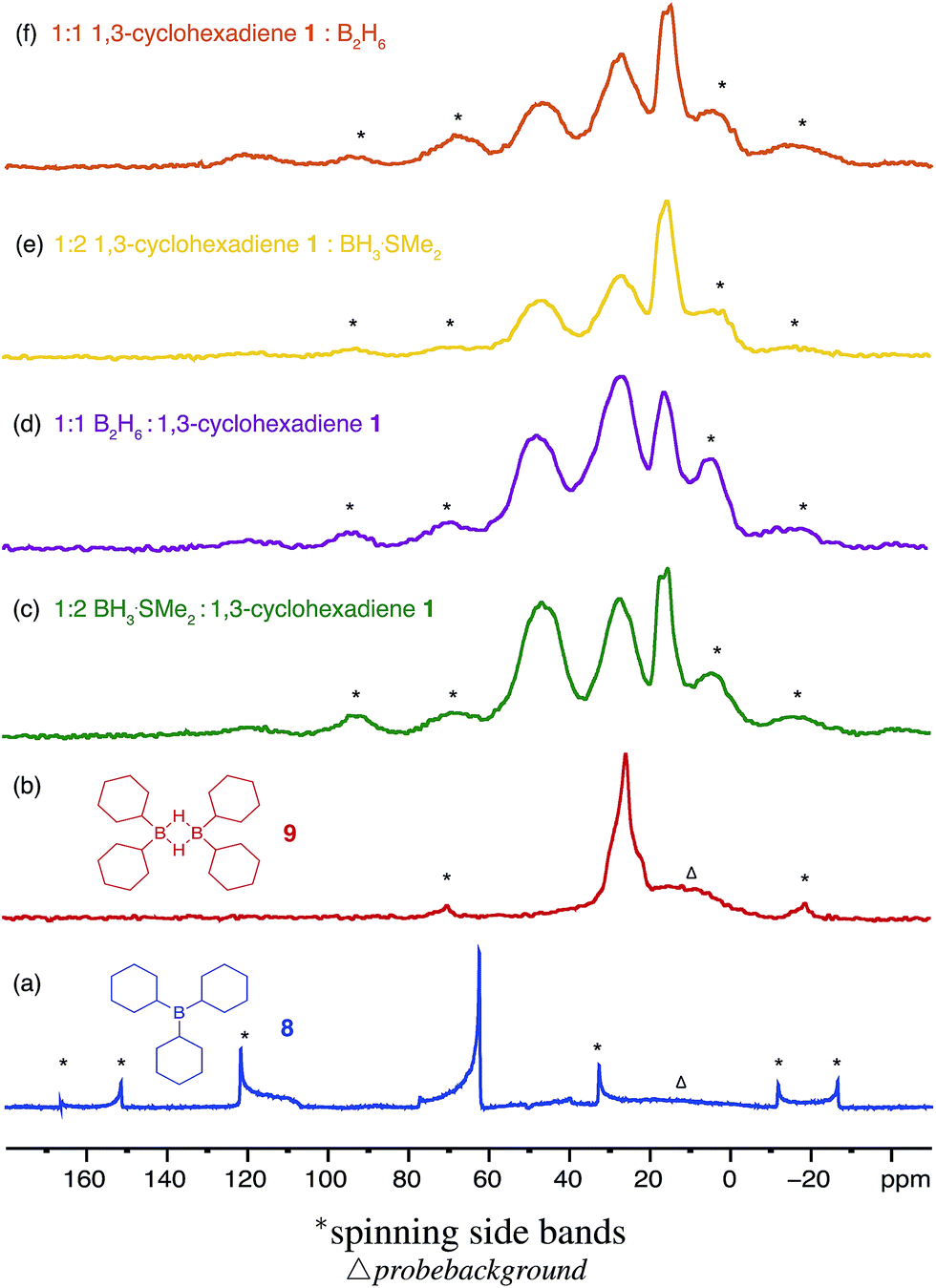 | ||
| Fig. 5 The solid state 11B NMR spectra of a range of different materials formed during the hydroboration of 1,3-cyclohexadiene 1 with 1 or 2 equivalents of borane BH3·SMe2 and diborane(6) B2H6 gas in diglyme against the model compounds (a) tricyclohexylborane 8 R3B and (b) dicyclohexylborane 9 (R2BH)2. | ||
The remaining signals appearing at approximately +50 and +18 ppm most likely correspond to other boron hydride species such as R2BHBH3 and R2BHBH2R (Fig. 2). This is further supported by the effect on the NMR signal intensities when the solid state 11B NMR is acquired with and without 1H decoupling (see ESI S.7.3, Fig. S20†), as well as by FT-IR and circumstantially by the reactivity of these materials with Lewis bases. Additionally, the presence of trapped diglyme solvent observed in the solid state 1H and 13C NMR spectra (see ESI S.7.3, Fig. S22 and S23†), lends strong support to the formation of polymeric networks. Most importantly, the absence of any unreacted CC bonds critically points to quantitative dihydroboration of 1 using a variety of borane sources and reaction conditions.
C bonds. The formation of this alcohol could be attributed to a diborane elimination side reaction similar to what is shown in Fig. 6. However, contrary to what was proposed by A. Hassner and B. H. Braun,38 we believe that such an elimination of 1,2-diboranes, such as 10, is highly unlikely in the absence of a driving force, such as a Lewis base. Perhaps, SMe2, THF (if present), diglyme or even a species formed during the reaction could mediate such an elimination. Protonolysis of the α-boron atom, in the 1,2-disubstituted diborane case, could also possibly lead to the formation of an oxidised alkylborane which resembles a carboxylic acid ([(RO)2B(OR)OH]−) thus eliminating one of the boron atoms and replacing it with a hydride. However, we believe that this is unlikely due to the large amounts of cyclohexanol 12 observed in some cases and the highly uncontrolled nature of this hydroboration system leading to a statistical mixture of 1,2-, 1,3- and 1,4-substitution patterns. Furthermore, during our studies we observed that β-hydride elimination of diboranes is possible, however, such an elimination followed by oxidation would not result in the formation of cyclohexanol but rather the unsaturated compound 2-cyclohexene-1-ol 2. Finally, elimination reactions caused by oxidation would also produce alcohols with CC double bonds rather than cyclohexanol and therefore must also be discounted. We are currently further investigating the mechanism of this elimination.
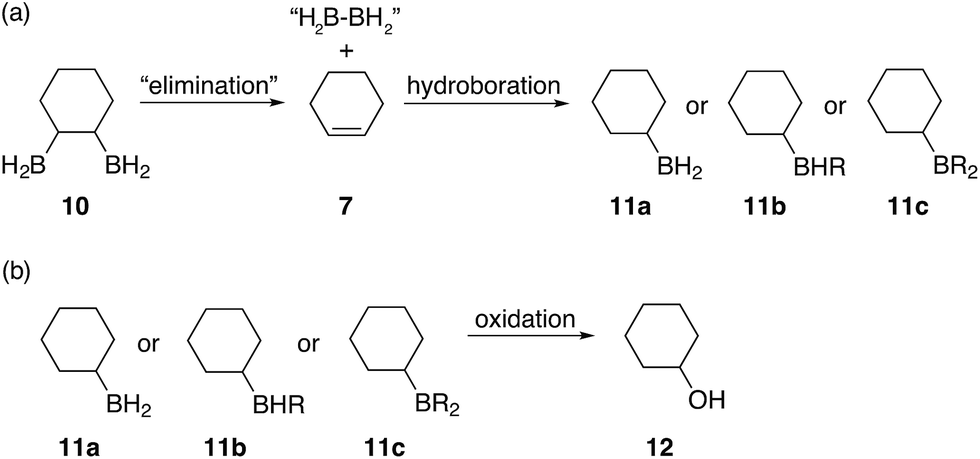 | ||
| Fig. 6 Proposed elimination mechanism for the formation of cyclohexanol when the boron containing polymers are oxidised. | ||
Several attempts were made to avoid the formation of the insoluble precipitates including lowering the reaction temperature (−78 °C), decreasing the concentration, or carrying out the reaction in non-ethereal solvents, known to considerably slow down the rate of hydroboration, such as DCM.39 Nevertheless, the insoluble materials formed in all cases, albeit at different rates, even when DCM was used as the solvent. The retarded rate of formation of these materials in DCM further suggested that a hydroboration cascade mechanism was indeed likely leading to chains and three dimensional networks as shown in Fig. 2.39 A number of additional observations further supported our hypothesis that the isolated materials were indeed borane-containing network polymers. The difunctional nature of 1 combined with the trifunctional nature of BH3 and the energetic stabilisation gained from the formation of B–H–B bridges is akin to an A2 + B3 step-growth polymerisation leading to insoluble, crosslinked networks. Moreover, the solids readily transform into liquids upon exposure to air, further suggesting that oxidation leads to facile disruption of the borane-hydrocarbon polymeric network. Finally, the dependence of the materials' physical appearance on the reaction solvent (glassy in THF and a white powder in diglyme) together with the high solvent content present in these materials are also indicative of the formation of a polymer network.
2.2 Proposed mechanism for the formation of the boron containing polymers
We believe that these materials are polymeric species formed via a cascade of hydroboration reactions. Detailed inspection of the boron species formed during hydroboration by NMR gave rise to the following proposed mechanism shown in Fig. 7. Initially, 1 undergoes monohydroboration to form either an allyl RBH2 species 13a or homoallyl RBH2 species 13b. The ratio of 13a to 13b is likely to be highly dependent on the particular diene structure and is beyond the scope of this study. However, it was expected that the allyl RBH2 species 13a is slightly more stable compared to the homoallyl 13b on account of conjugation with the CC bond. Indeed, during our calculations we found that the BH2 group is stabilised in an axial position, when SMe2 is absent, by interactions with the adjacent double bond that partly delocalises to the boron 2p orbital (see ESI S11, S53†). However, the TS2 barrier towards both 13a and 13b is almost identical, which cannot explain the selectivity. It is possible that the overall dynamics may determine the product selectivity as reported previously for the hydroboration of propene.40 Alternatively, the hydroboration product can rearrange and the BH2 group can isomerise, forming the more stable 13a axial product starting from the equatorial 13b isomer. Once 13a forms an adduct with SMe2 (or other electron donor molecules), the equatorial conformation is preferred. It is worth noting, that the formed RBH2 species most likely exist as dimers, which contain B–H–B bridges providing substantial stabilisation,41 however, for simplicity all borane species depicted in the proposed mechanism are drawn with terminal B–H bonds.
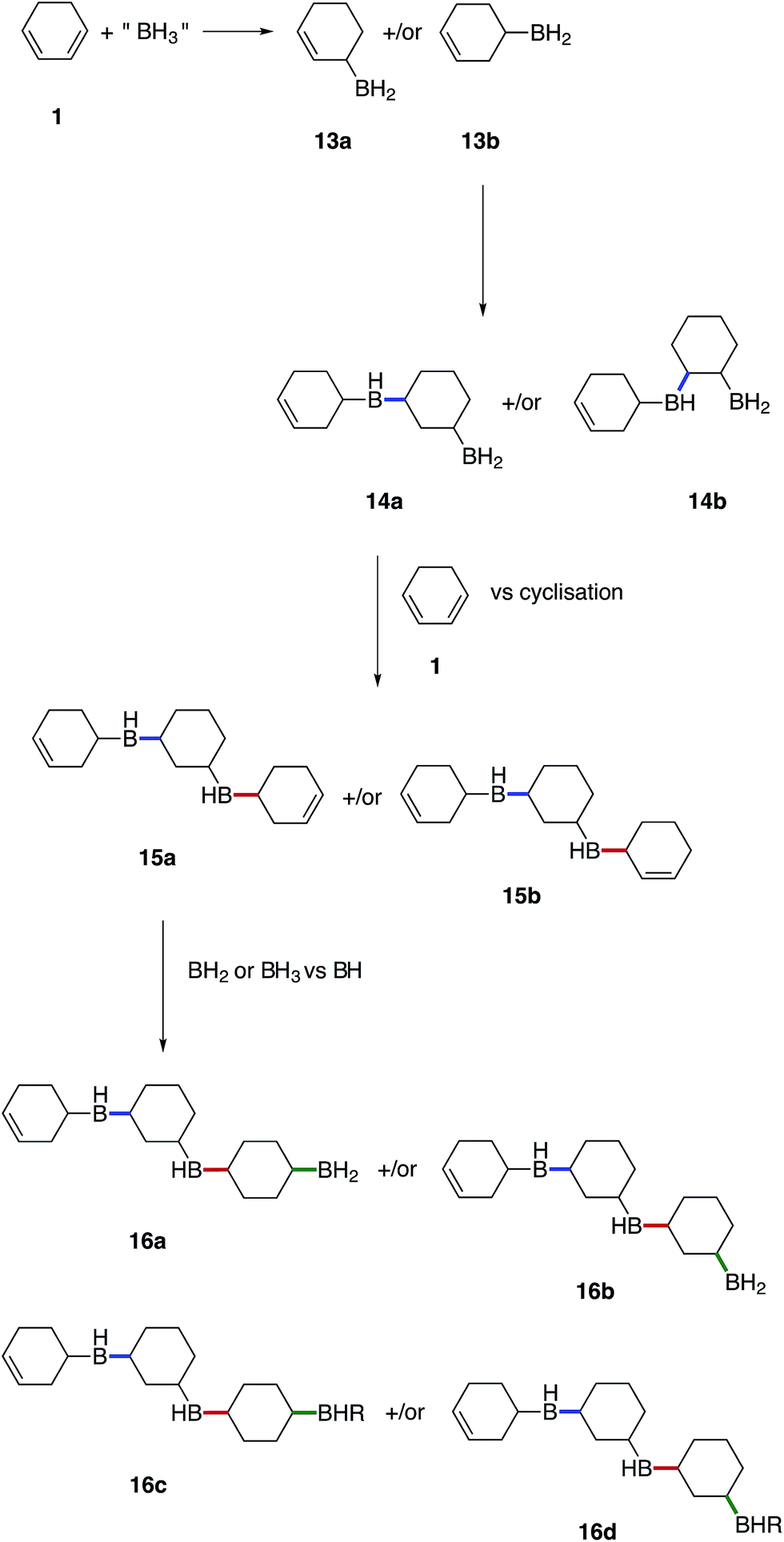 | ||
| Fig. 7 Proposed mechanism for the formation of insoluble boron containing hydrocarbons. | ||
Once hydroboration occurs, both the monohydroboration and dihydroboration species are much more reactive than the starting materials, conjugate diene 1 and borane. This was especially evident on account of the unreacted borane observed in solution after the consumption of all CC bonds, entries 2–4 in Table 1. Additionally, monohydroboration of 1 leads to the formation of an isolated CC bond, which was observed to be highly reactive (at least under our conditions) compared to the conjugated starting material 1, leading to a cascade of hydroboration reactions and concomitant formation of a cross-linked polymeric network as the monohydroboration species continue to react preferentially.
In general terms, hydroboration on double bonds is known to follow a two-step process.40 The first and usually rate-limiting step is the activation of BH3 (or substituted RBH2 species) by breaking the solvent–borane adduct or the borane dimer. The second step is the addition of the BH3 to the double bond and usually has a smaller barrier.40 Our calculations suggest that the formation of the solvent-free BH3 intermediate is stabilized by short-lived intermediate complexes formed with the CC bond containing reaction partner. We found that the barrier of the first step is reduced from +26.1 kcal mol−1 to +14.6 kcal mol−1 using BH2-cyclohexene, to +15.0 kcal mol−1 using 1,3-cyclohexadiene 1, and to +13.8 kcal mol−1 using cyclohexene (see ESI S11, Table S1†). It was evident that the energy difference between mono and dihydroboration is very small and therefore, the determining factor for the preferred reaction pathway is most likely influenced by the experimental conditions. The barrier of the first step to form solvent-free boranes is also significantly decreased when substituted boranes (RBH2) are used with electron-donating R groups, even when the free RBH2 is not further stabilised by adduct formation with a solvent molecule or a CC bond. With R = cyclohexyl (or cyclohexenyl), the barrier to generate a free BRH2 is +13.3 kcal mol−1 (or +13.1 kcal mol−1), which is +12.7 kcal mol−1 (or +13.0 kcal mol−1) smaller than for the dissociation of the BH3·SMe2 complex (see ESI S11 Table S2†). As a result, the formed monohydroboration products are expected to react further.
Subsequent hydroboration of 13a or 13b can take place via several different pathways, depending on the reaction conditions. The newly formed, solvent-free monoalkylboranes (13a or 13b) can further react rapidly and hydroborate another cyclohexadiene molecule (ΔE‡ = 1.0–2.2 kcal mol−1, see ESI S11 Table S3†), or other monocyclohexylboranes (ΔE‡ = 4.0–4.3 kcal mol−1, see ESI S11 Table S3†) leading to polymer formation. When excess borane is still available, BH3 can also hydroborate the remaining double bond on 13a or 13b, although with a somewhat higher energy barrier. For example, the barrier for the hydroboration of 13a is 17.7–18.3 kcal mol−1 (see ESI S11, Table S1†). Interestingly, this second hydroboration can also take place via an intramolecular mechanism through a RH2B–BH3 complex (ESI S11, Fig. S54†). The hydroboration reaction within this complex has some of the lowest barriers, 15.5 kcal mol−1 to form the kinetically favoured cis-1,2-diborane cyclohexane isomer (or 18.0 kcal mol−1 to form the thermodynamically favored cis-1,3-diborane cyclohexane, see ESI S11, Table S1†). The formed diboranes are further stabilised via B–H–B bridges. Potentially, this barrier is further reduced by forming a complex with another electron donating molecule, such as SMe2, that facilitates the release of an active BH3 (results are not shown). It is worth noting that the second hydroboration reaction can produce eight distinct diborane cyclohexane isomers in general if the chair-boat conformational change is not hindered, and many more in the conformationally restrained polymers. The formation of each isomer has different potential reaction mechanisms and corresponding rates. Interestingly, the intermolecular hydroboration, with the bridged cis-1,2-cyclohexane (see Fig. 6), is the kinetically favoured mechanism to form diborane over most of the other possibilities listed above (see ESI S11, Table S1†). The thermodynamically most stable product is the axial cis-1,3-cyclohexane, due to the favourable positions of the BH2 groups of this isomer to form B–H–B bridges. This intermolecular mechanism may provide an explanation of the relative abundance of cyclohexanol after basic oxidation of these materials, assuming a potential subsequent elimination reaction from the bridged diborane species (see Fig. 6).
Boron containing polymer formation from dienes has been reported previously by Brown et al. during the hydroboration of 1,3-butadiene.27–29,42 However, these polymers were found to form via cyclic hydroboration, which is not expected in the case of cyclic diene 1, as this would require the initial formation of highly unfavoured ring-in-ring structures (see ESI S8, Fig. S48†). As a result, cyclic hydroboration mechanisms and their products are not considered here.
2.3 Dihydroboration of other cyclic dienes
Intrigued by the results of our study on 1,3-cyclohexadiene, we expanded the substrate scope to include a range of different cyclic dienes including: α-terpinene, 1,3-cycloheptadiene, 1,3-cyclooctadiene, 1,3,5,5-tetramethyl-1,3-cyclohexadiene, γ-terpinene, 1,4-cyclohexadiene, 1,2,4,5-tetramethyl-1,4-cyclohexadiene, 1,5-cyclooctadiene and the acyclic 2,3-dimethyl-1,3-butadiene as a control and direct comparison to the reported polymer structures (see ESI S9, Fig. S49†). In a similar fashion to our observation with 1, all of the dienes listed above, including the acyclic diene, were susceptible to dihydroboration. Not all dienes above led to the formation of crosslinked polymeric networks, several resulted in a clear solution, which contained a mixture of boron hydride species and unreacted borane (see ESI S9, Fig. S50 and S51†). A clear pattern, however, was observed with simple cyclic dienes, such as 1, 1,3-cycloheptadiene, 1,3-cyclooctadiene, and 1,4-cyclohexadiene leading to the formation of the boron-containing crosslinked polymers whereas more sterically demanding dienes such as α-terpinene yielded a clear solution. Interestingly, γ-terpinene also led to the formation of a cross-linked material. Following this trend, hydroboration of 1,2,4,5-tetramethyl-1,4-cyclohexadiene yielded clear solutions while the reaction with 1,3,5,5-tetramethyl-1,3-cyclohexadiene first produced a clear solution, but observation of an insoluble material occurred after 2 h. Despite the absence of sterically demanding substituents, hydroboration of 1,5-cyclooctadiene resulted in a clear solution, yielding the commonly used reagent 9-BBN on account of the formation of a stable 6-member boron-containing cyclic structure. Finally, when acyclic diene, 2,3-dimethyl-1,3-butadiene was used in the reaction, a clear solution was obtained as previously reported.27–29 Therefore, it was clear that the position of the CC bond, in relation to the sterically demanding group, was crucial in the formation of these materials, which were not limited by ring size.
3 Conclusions
To conclude, boron containing polymers form when cyclic 1,3- or 1,4-dienes, free of sterically demanding groups are fully hydroborated, irrespective of the cyclic diene size, borane equivalents and mode of addition. When steric hindrance is present or cyclic borane species are favourably formed, as for example when straight chain of 1,5-dienes are utilised, clear solutions are observed. These materials were characterised by solid state FT-IR and NMR and were found to consist of boron hydride bridged species due to stabilisation reasons and the synthetic time scale utilised. Further supported by oxidation analysis, it was clear that hydroboration proceeds further than the monosubstituted borane adducts to form higher substituted, more stable, bridged borane species. Therefore, our results are in disagreement with the published conclusions on the study of dihydroboration of cyclic dienes by H. C. Brown et al.24,25 as we observed that dihydroboration of cyclic rings proceeds quantitatively irrespective of ring size or structure.Acknowledgements
The authors wish to thank Prof. D. S. Wright for helpful discussions. The authors would like to acknowledge the use of the EPSRC UK National Service for Computational Chemistry Software (NSCCS) at Imperial College London in carrying out this work. E. R. acknowledges computational support from the NIH Biowulf cluster, from CCPBioSim via the ARCHER UK National Supercomputing Service, and from King's ADA HPC cluster.References
- H. C. Brown, Organic synthesis via boranes, Wiley, 1st edn, 1975 Search PubMed.
- H. C. Brown, Tetrahedron, 1961, 12, 117–138 CrossRef CAS.
- X. Deng, F. Liu, Y. Luo, Y. Chen and D. Jia, Eur. Polym. J., 2007, 43, 704–714 CrossRef CAS PubMed.
- S. Gogolewski, Colloid Polym. Sci., 1989, 267, 757–785 CAS.
- D. Ratna and J. Karger-Kocsis, J. Mater. Sci., 2008, 43, 254–269 CrossRef CAS.
- F. Fenouillota, A. Rousseaua, G. Colomines, R. Saint-Loupe and J.-P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef PubMed.
- B. Saad, T. D. Hirt, M. Welti, G. K. Uhlschmid, P. Neuenschwander and U. W. Suter, J. Biomed. Mater. Res., 1998, 36, 65–74 CrossRef.
- R. B. Greewald, C. D. Conover and Y. H. Choe, Crit. Rev. Ther. Drug Carrier Syst., 2000, 17, 101–161 Search PubMed.
- W. Wang, X. Gao and B. Wang, Curr. Org. Chem., 2002, 6, 1285–1317 CrossRef CAS.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2012, 47, 1106–1123 RSC.
- A. Miyashita, A. Yasuda, H. Takaya, K. Toriumi, T. Ito, T. Souchi and R. Noyori, J. Am. Chem. Soc., 1980, 102, 7932–7934 CrossRef CAS.
- A. Miyashita, H. Takaya, T. Souchi and R. Noyori, Tetrahedron, 1984, 40, 1245–1253 CrossRef CAS.
- M. J. Burk, J. Am. Chem. Soc., 1991, 113, 8518–8519 CrossRef CAS.
- M. J. Burk, J. E. Feaster, W. A. Nugent and R. L. Harlow, J. Am. Chem. Soc., 1993, 115, 10125–10138 CrossRef CAS.
- M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 227, 936–938 CrossRef.
- C. A. G. M. Weijers, Tetrahedron: Asymmetry, 1997, 8, 639–647 CrossRef CAS.
- C. Döbler, G. Mehltretter and M. Beller, Angew. Chem., Int. Ed., 1999, 38, 3026–3028 CrossRef.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- C. Prévost, Comptes Rendus, 1933, 196, 1129–1131 Search PubMed.
- C. Prévost, Comptes Rendus, 1933, 196, 1661 Search PubMed.
- C. Prévost and B. Lutz, Comptes Rendus, 1933, 198, 2264 Search PubMed.
- R. B. Woodward, 1, 14-dimethyl-2-keto-6, 7-dihydroxy-2, 3, 4, 5, 6, 7, 8, 12, 13, 14-decahydrophenan-threne and process for preparing same, US2687435 A, United States Patent Office, 1954 Search PubMed.
- H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1959, 81, 5833 Search PubMed.
- G. Zweifel, K. Nagase and H. C. Brown, J. Am. Chem. Soc., 1962, 84, 183–189 CrossRef CAS.
- H. C. Brown and K. S. Bhat, J. Org. Chem., 1986, 51, 445–449 CrossRef CAS.
- K. L. Saegebarth, J. Org. Chem., 1960, 25, 2212–2213 CrossRef CAS.
- H. C. Brown, E. Negishi and S. K. Gupta, J. Am. Chem. Soc., 1970, 92, 2460–2467 CrossRef CAS.
- H. C. Brown, E. Negishi and P. L. Burke, J. Am. Chem. Soc., 1971, 93, 3400–3409 CrossRef CAS.
- H. C. Brown and E. Negishi, Pure Appl. Chem., 1972, 29, 527–546 CrossRef CAS.
- Y. Chujo, I. Tomita, Y. Hashiguchi, H. Tanigawa, E. Ihara and T. Saegusa, Macromolecules, 1991, 24, 345–348 CrossRef CAS.
- A. Hübner, Z.-W. Qu, U. Englert, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2011, 133, 4596–4609 CrossRef PubMed.
- R. Contreras and B. Wrackmeyer, Spectrochim. Acta, Part A, 1962, 38, 941–951 CrossRef.
- H. C. Brown and J. A. Sikorski, Organometallics, 1982, 1, 28–37 CrossRef CAS.
- M. Potyen, K. V. B. Josyula, M. Schuck, S. Lu, P. Gao and C. Hewitt, Org. Process Res. Dev., 2007, 11, 210–214 CrossRef CAS.
- E. V. Topchiev, S. V. Zavgorodnii and Y. M. Paushkin, Boron trifluoride and its compounds as catalysts in organic chemistry, Pergamon Press, London, 1st edn, 1959 Search PubMed.
- A. Andreou, PhD thesis, University of Cambridge, UK, 2013.
- H. C. Brown and G. J. Klender, Inorg. Chem., 1962, 1, 204–214 CrossRef CAS.
- A. Hassner and B. H. Braun, J. Org. Chem., 1963, 28, 261–262 CrossRef CAS.
- H. C. Brown and B. C. Subba Rao, J. Org. Chem., 1957, 22, 1136–1137 CrossRef CAS.
- Y. Oyola and A. D. Singleton, J. Am. Chem. Soc., 2009, 131, 3130–3131 CrossRef CAS PubMed.
- H. C. Brown and A. W. Moerikofer, J. Am. Chem. Soc., 1961, 83, 3417–3422 CrossRef CAS.
- H. C. Brown, E. Negishi and P. L. Burke, J. Am. Chem. Soc., 1972, 94, 3561–3567 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc02729a |
| This journal is © The Royal Society of Chemistry 2015 |