DOI:
10.1039/C4SC02793K
(Edge Article)
Chem. Sci., 2015,
6, 436-441
Polycyclic aromatic azomethine ylides: a unique entry to extended polycyclic heteroaromatics†
Received
10th September 2014
, Accepted 21st October 2014
First published on 22nd October 2014
Abstract
Based on polycyclic aromatic azomethine ylides (PAMYs), a metal-free “cycloaddition-planarization-sequence” is proposed, providing a unique entry to extended nitrogen-containing polycyclic aromatic hydrocarbons (N-PAHs). This method is highly versatile, as the structure of unprecedented N-PAHs can be tailored by the dipolarophile in the crucial 1,3-cycloaddition-reaction. Linear, as well as five- and six-membered cyclic dipolarophiles are successfully used. The geometric and optoelectronic nature of N-PAHs are investigated by UV-vis absorption and single crystal structure analysis. Remarkably, the newly synthesized N-PAHs demonstrate varying absorption profiles, covering the whole visible light range with rich photophysical properties, for example, fluorescent quantum yields up to 54%.
Introduction
Azomethine ylides and polycyclic aromatic hydrocarbons
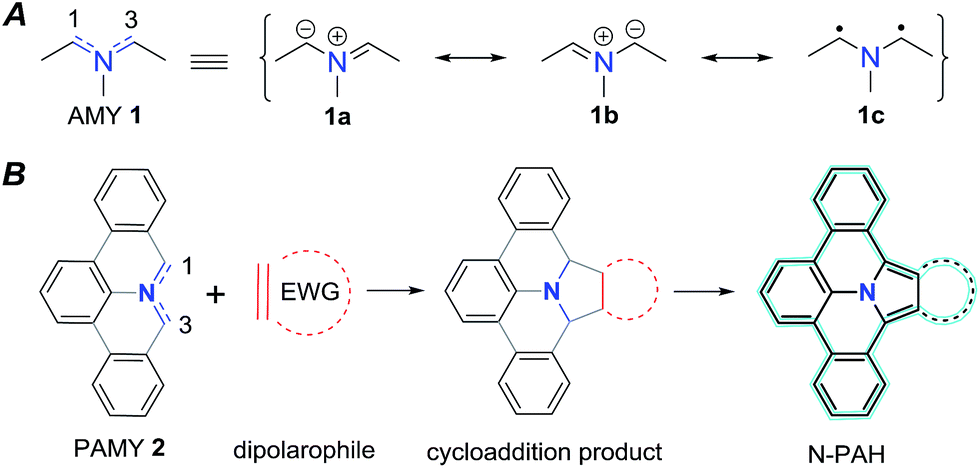
Azomethine ylides (AMY 1, Fig. 1) are prime examples of 1,3-dipolar compounds.1,2 Their structure is isoelectronic to that of the allyl anion with the negative charge equally distributed over two carbon atoms adjacent to the central nitrogen atom (see Fig. 1). Besides the two ionic Lewis structures (1a and 1b), the diradical (1c) structure contributes to the overall ground state as well.3–6 Calculations have been used to quantify the diradical character of AMYs and to correlate it to their chemical reactivity in 1,3-dipolar cycloaddition reactions.7,8 In general, AMYs are highly reactive and only a few examples of stabilized AMYs have hitherto been isolated.9–16 The high reactivity makes AMYs essential for facile construction of five-membered heterocycles, and a variety of methods, such as deprotonation or desilylation of precursor derivatives have been developed.17 However, the synthesis of AMY conjugated with multiple aromatic rings (PAMY 2) has not been described, although they are an attractive building block for the synthesis of nitrogen-containing polycyclic aromatic hydrocarbons (N-PAHs) as proposed in Fig. 1.
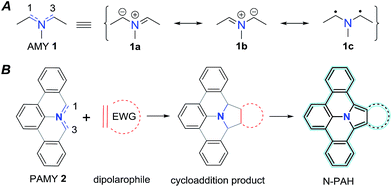 |
| | Fig. 1 An azomethine ylide (AMY, 1) together with its two ionic (1a–b) and its diradical (1c) Lewis structures. Schematic representation of a polycyclic aromatic azomethine ylide (PAMY, 2) used in a 1,3-dipolar cycloaddition reaction (click-reaction) with an electron-poor dipolarophile. After oxidation of the cycloaddition product, extended π-conjugated nitrogen containing N-PAHs are obtained. | |
Results and discussion
1,3-Dipolar cycloaddition reaction and planarization
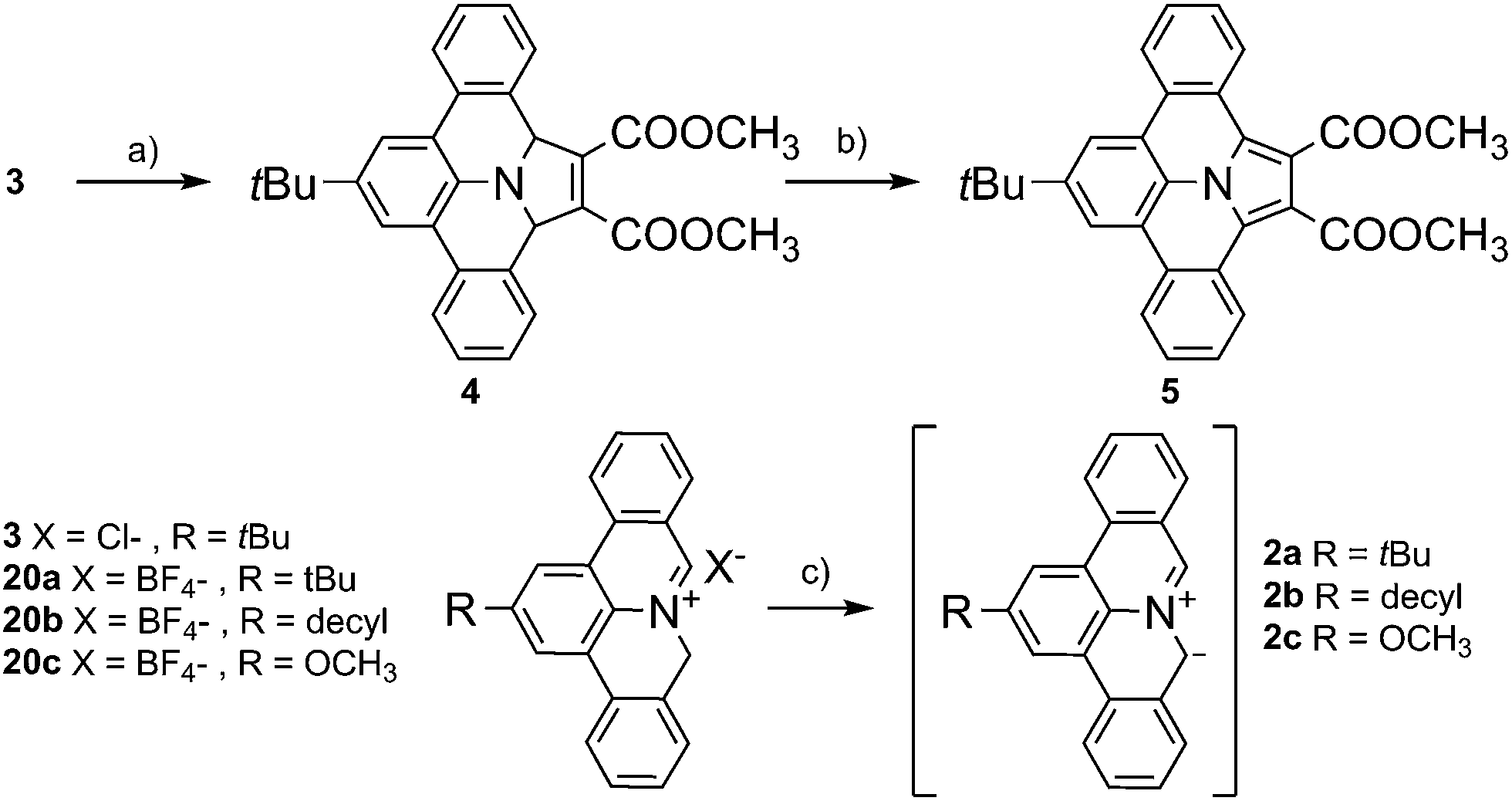
With our initial motif to explore biradicals based on 9a-azaphenalene,18 we recently found that treatment of 2-(tert-butyl)-8H-isoquinolino[4,3,2-de]phenanthridin-9-ium chloride (3) with triethyl amine in the presence of dimethoxy acetylene dicarboxylate (6, DMAD) resulted in direct formation of dimethyl-8-(tert-butyl)-2a,13b-dihydrobenzo[7,8]indolizino[6,5,4,3-def]phenanthridine-1,2-dicarboxylate (4) via a selective 1,3-dipolar cycloaddition reaction between DMAD and in situ generated PAMY 2a (see Scheme 1). The X-ray crystallographic analysis of 4 validated the selective 1,3-addition and revealed a tilted geometry with minor π-conjugation along the meta-terphenylene plane. The polycyclic feature of compound 4 strongly suggests that it can be further converted to an extended N-PAH. Therefore, in this work, we subjected compound 4 to oxidative dehydrogenation by treating with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ). A fully planar, π-extended N-PAH, 8-(tert-butyl)-benzo[7,8]indolizino[6,5,4,3-def]phenanthridine-1,2-dicarboxylate (5) was obtained in 82% yield, which was unambiguously characterized by NMR, MS, and single crystal analysis. Notably, this sequence can be conducted without purification of the intermediate cycloaddition product.
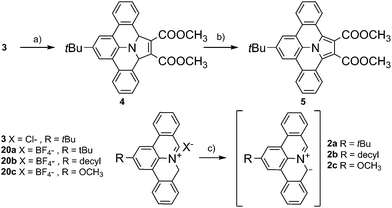 |
| | Scheme 1 Synthetic route to N-PAH 5 based on 3. Mild oxidation of 4 by DDQ results in planar N-PAH 5 with extended π-conjugation over the whole molecule. (a) DMAD, TEA, dichloromethane, 25 °C; (b) DDQ, toluene, 25 °C; (c) TEA, dichloromethane, 25 °C. | |
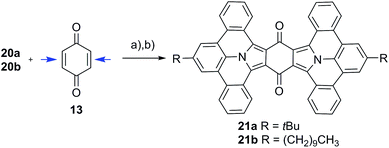 |
| | Scheme 2 Twofold addition of PAMYs to both double bonds in 13 (indicated by blue arrows). The reaction was induced by treatment of precursor 20a or 20b with triethyl amine, followed by planarization with DDQ to obtain large N-PAHs 21a–b; (a) triethyl amine, dichloromethane, 25 °C; (b) DDQ, toluene. | |
Scope of reaction
Encouraged by the successful synthesis of N-PAH 5, we conceive that our synthetic protocol of using PAMYs it is not only applicable to linear ethyne DMAD (6), but also to a series of tilted (10, 11), pentacyclic (12) and hexacyclic ethylenes (13–14) as dipolarophiles. In this way, unprecedented nitrogen containing N-PAHs 15–19b can be accessed (see Fig. 2). Moreover, the scope of this concise, metal-free reaction sequence can be further broadened by the use of other PAMYs with long alkyl chains (2b), or electron-donating substituents (2c). Like AMYs, PAMYs are highly reactive and thus need to be generated in situ from a suitable precursor. To obtain PAMYs 2a–c with tert-butyl-, decyl- and methoxy-substituents, the corresponding precursor derivatives 20a–c were synthesized from 2,6-dibromo-anilines (see Fig. S2, ESI†). Finally, the key step for the preparation of N-PAHs 5 and 15–19b, the 1,3-dipolar cycloaddition reaction of in situ formed PAMYs with dipolarophiles 6–14, was induced by dropwise addition of triethyl amine to solutions of the corresponding precursors 20a–c. Afterwards, the crude cycloaddition product was directly oxidized with DDQ in toluene, to afford the planarized N-PAHs 5 and 15a–19b in good to excellent yields of 48–95%. As shown in Table 1, the yields of N-PAHs 5, 15–19b are summarized for each precursor (20a–c) and dipolarophile (6–14). The linear ethyne 6, as well as the tilted ethynylenes 10, and 11 participated in the 1,3-dipolar cycloaddition reaction with the in situ generated PAMY 2a and afforded the same N-PAH 5 after planarization. This is in agreement with our expectation that linear unsaturated carbon–carbon bonds such as ethynes and ethynylenes with electron withdrawing groups undergo 1,3-dipolar cycloaddition reaction with the electron rich PAMYs, independent of their molecular geometry. On the contrary, the electron rich ethynes such as diphenyl acetylene (8) and monophenyl acetylene (9), which are frequently used in Diels–Alder reactions of inverse electron-demand, failed to undergo the 1,3-dipolar cycloaddition reaction. This result indicates that the electronic effect has a large influence on the reactivity of cycloaddition reaction. Importantly, 1,2-bis(perfluorophenyl)ethyne (7) was found to be suitable as the dipolarophile, showing that other non-carbonyl electron-withdrawing groups can be used as well. In this case, the planarized N-PAH 16 is obtained as colourless crystals in 48% yield.
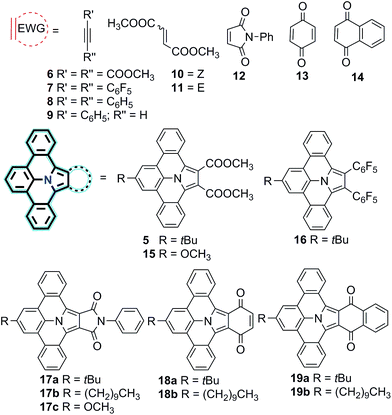 |
| | Fig. 2 Scope of N-PAHs 15a–19b synthesized dependent on the dipolarophiles 6–14 used in the 1,3-dipolar cycloaddition reaction. | |
Table 1 Precursors and dipolarophiles used in the cycloaddition reactions and isolated yields of N-PAHs 15a–19b, obtained after oxidation with DDQ
| Precursor |
Dipolarophile |
N-PAH |
Yielda (%) |
|
Isolated yield of N-PAHs after planarization.
|
|
20a
|
6
|
5
|
82 |
|
7
|
16
|
48 |
|
8
|
— |
— |
|
9
|
— |
— |
|
10
|
5
|
95 |
|
11
|
5
|
83 |
|
12
|
17a
|
64 |
|
13
|
18a
|
78 |
|
14
|
19a
|
90 |
|
20b
|
12
|
17b
|
91 |
|
13
|
18b
|
69 |
|
14
|
19b
|
89 |
|
20c
|
3
|
15
|
68 |
|
9
|
17c
|
74 |
Inspired by the successful synthesis of 5 and 16, the cyclic five-membered dipolarophile N-phenylmaleimide 12 was thus examined in combination with precursor 20a yielding N-PAH 17a as yellow solid in 64% yield after two steps. In contrast to N-PAHs 5, 15 and 16, the conjugation of N-PAH 17a is extended over an additional five-membered pyrrole cycle. Probably due to the larger conjugated structure, the solubility of N-PAH 17a in conventional organic solvents is considerably lower than that of N-PAHs 5 and 16. However, when 20b was used as precursor, a decyl chain could be introduced via PAMY 2b in N-PAH 17b, which enhanced solubility compared to the tert-butyl substituent in N-PAH 17a.
We further extend our synthetic protocol to larger six-membered dipolarophiles, such as 1,4-benzoquinone (13) and 1,4-napthoquinone (14). Both 13 and 14 indeed afforded N-PAHs 18a and 18b as red and N-PAHs 19a and 19b as orange powders, respectively, in 78–90% yield. Encouraged to further streamline the two-step procedure, we tested to conduct cycloaddition and oxidation in one-pot. Exemplified on 20a as precursor and 14 as dipolarophile, N-PAH 19a was achieved in one-pot using toluene as inert solvent (see Fig. S5, ESI†).
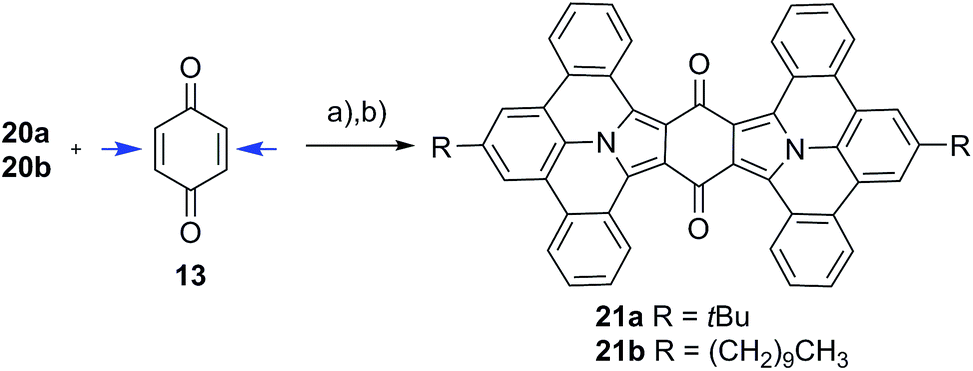
Twofold addition
Compound 13 can be considered as “double-dipolarophile” and allows for a twofold 1,3-dipolar cycloaddition reaction by simply adjusting the ratio of PAMY precursor to dipolarophile to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Scheme 2). After planarization with DDQ, highly extended N-PAHs 21a–b are obtained as red powders in excellent yields of 82%. In N-PAHs 21a–b, 48 π-electrons are delocalized throughout the molecule. For comparison, the famous PAH hexa-peri-hexabenzocoronene (HBC) only consists of 42 delocalized π-electrons.19–21 More importantly, 21a–b contain two (pyrrolic) nitrogen atoms at the interior of the planar PAH-skeleton, which is usually synthetically much more challenging than introducing nitrogen at the more accessible peripheries.22 The few examples that have been reported to date, strongly suggested that N-PAHs with central nitrogen atoms may pave the way to developing new building blocks in nanoelectronics and supramolecular assemblies.23–28
:1 (Scheme 2). After planarization with DDQ, highly extended N-PAHs 21a–b are obtained as red powders in excellent yields of 82%. In N-PAHs 21a–b, 48 π-electrons are delocalized throughout the molecule. For comparison, the famous PAH hexa-peri-hexabenzocoronene (HBC) only consists of 42 delocalized π-electrons.19–21 More importantly, 21a–b contain two (pyrrolic) nitrogen atoms at the interior of the planar PAH-skeleton, which is usually synthetically much more challenging than introducing nitrogen at the more accessible peripheries.22 The few examples that have been reported to date, strongly suggested that N-PAHs with central nitrogen atoms may pave the way to developing new building blocks in nanoelectronics and supramolecular assemblies.23–28
Structure
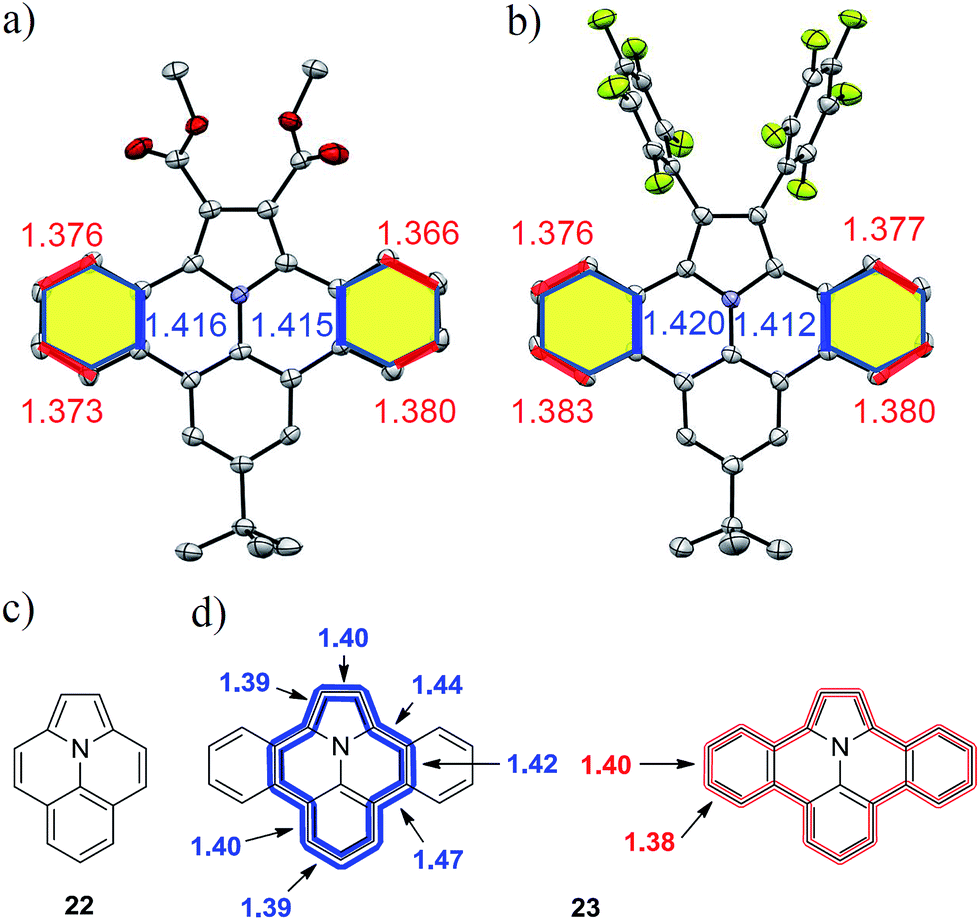
Although the solubility of the larger derivatives 21a–b is limited to high-boiling organic solvents such as tetrachloroethane, all intermediates and precursor compounds could be analyzed by 1H-, 13C-NMR-spectroscopy and HR-ESI mass spectrometry. Single crystals of the planararized N-PAHs 5 and 16 suitable for X-ray crystallographic analysis were grown by slow evaporation of solutions in dichloromethane, which clearly revealed their fully planar structures (Fig. 3a and b).
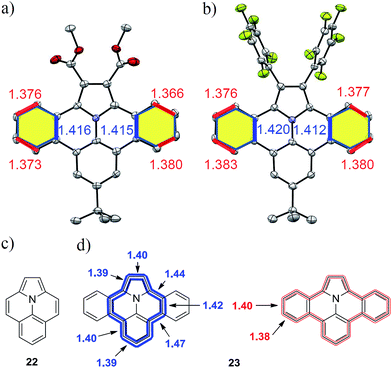 |
| | Fig. 3 Crystal structures of N-PAHs (a) 5 and (b) 16 with selected bond lengths. Chemical structures of ullazine (22) and dibenzoullazine (23) with calculated bond length along the inner (blue) and outer (red) perimeter. | |
Both compounds showed different carbon–carbon bond lengths in the annulated benzene rings (highlighted yellow). Those along the inner perimeter (marked blue) had a length of 1.41 Å, whereas a bond length of 1.38 Å was found at the outer perimeter (marked red) (Fig. 3a and b). This indicates a more pronounced double bond character in comparison to the typical bond length of 1.39 Å in benzene for the outer perimeter. On the other hand, the average bond length of 1.40 Å in the inner perimeter is in agreement with that in nitrogen containing heterocycles like pyridine (1.40 Å). Similar, the C2–C2a and C13b–C14 distances, as well part of the inner perimeter, are elongated to 1.39 Å from 1.38 Å in pristine pyrrole.29 Indeed, the inner perimeter of 5 and 16 can be identified as indolizino[6,5,4,3-aij]quinoline (22), which is an electronic isomer of pyrene with the more common name “Ullazine” based on the nomenclature of Bali and Zeller (see Fig. 3).30 Recently, derivatives of ullazine 22 attracted attention owing to their both electron donating and accepting properties and therefore their potential use as efficient sensitizers in dye-sensitized solar cells.31–33 In the dibenzoannulated derivative 23, the ullazine core is stabilized by two additional benzenes, while the annulene character of 22 along the inner perimeter (blue) is still maintained. Therefore, we propose the name dibenzo[d,k]ullazine for N-PAH 23, whose structural motif is found in all N-PAHs 5, 15–19b and twofold in 21a–b.
Photophysical properties
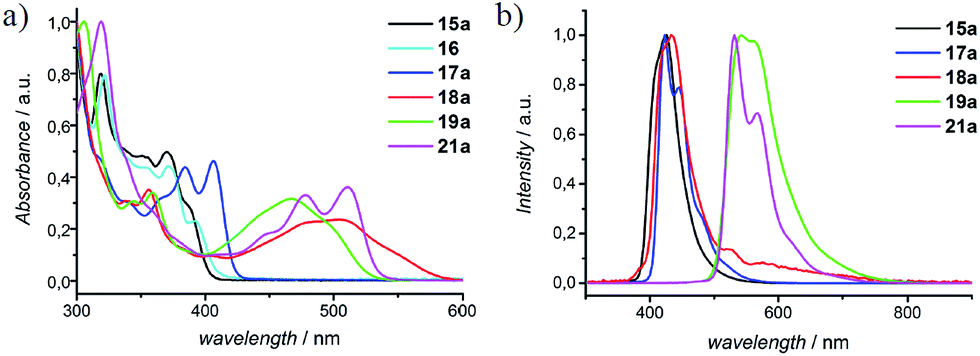
In Fig. 4, UV-vis absorption spectra of representative N-PAHs 5, 16, 17a, 18a, 19a, and 21a are displayed. N-PAHs 5 and 16 show the absorption maxima at λmax = 388 nm and 394 nm, respectively, and both exhibit high molar extinction coefficients (ε) of 687 m2 mol−1. The pyrrole-annulated compound 17a shows three pronounced transitions with a maximum absorption bathochromically shifted to λmax = 406 nm. Annulation with quinones leads to a much stronger bathochromic shift with a broad absorption up to 600 nm for 18a and a less shifted but more pronounced absorption maximum at λmax = 498 nm for 19a. In contrast to 19a, which shows a strong fluorescence emission at 541 nm, the emission maximum of 18a is found at 435 nm, close to that of N-PAHs 5 and 17a. Therefore, we assume the S1 state of 18a to be closer to the smaller N-PAHs 5 and 16–17a and attribute the broad absorbance to an intramolecular charge-transfer band. Furthermore, for the large N-PAH 21a, the absorption pattern shows three pronounced transitions with an absorption maximum at λmax = 510 nm and a high value of ε = 1665 m2 mol−1. This absorption spectra resemble that of N,N′-diphenyl-3,4,9,10-perylentetracarboxylic-3,4:9,10-diimide (PDI), a benchmark system in dye chemistry and industrial pigments. Moreover, all N-PAHs 5, 15–17a and 21a have good fluorescence quantum yields in a range from 14% to 54% obtained from the comparative method of Williams et al., using 9,10-diphenylanthracene or Rhodamin 6G as ref. 34. To evaluate HOMO/LUMO energies of N-PAHs 15a–21a, cyclic voltammetry was conducted using ferrocene as reference. The combined optoelectronic data are summarized in Table 2.
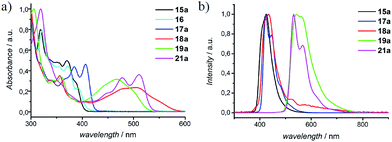 |
| | Fig. 4 Normalized (a) UV-vis absorption and (b) fluorescence spectra of selected N-PAHs 5, 16, 17a, 18a, 19a, and 21a, representing the effect of annulation and/or substitution. | |
Table 2 Optical and electrochemical data
|
|
λ
max
[nm] |
λ
em
[nm] |
Φ
F
|
E
(0–0)
[eV] |
U
[V] |
E
HOMO
[ev] |
E
LUMO
[ev] |
|
λ
max: absorption maximum at longest wavelength.
λ
em: emission wavelength.
Obtained from the comparative method of Williams et al., using 9,10-diphenylanthracene as reference at excitation wavelength λex = 380 nm.
Rhodamin 6G at λex = 470 nm. 9,10-diphenylanthracene as standard at excitation wavelength λex = 325 nm.
Measured at the intersection of the normalized absorbance and emission spectra.
Redox potentials from CV are reported vs. Fc/Fc+ (0.1 M nBu4NPF6 in CH3CN), scan rate 50 mV s−1.
HOMO values were derived from the first measured oxidation potential.
LUMO values were evaluated by ELUMO = EHOMO(CV) + E(0–0).
HOMO value was evaluated by EHOMO = ELUMO(CV) − E(0–0).
LUMO values were derived from the first measured reduction potential.
|
|
5
|
388 |
424 |
0.45c |
3.16 |
0.75 |
−5.55 |
−2.39 |
|
15
|
392 |
427 |
— |
3.12 |
0.74 |
−5.54 |
−2.42 |
|
16
|
393 |
428 |
0.51c |
3.14 |
0.77 |
−5.57 |
−2.50 |
|
17a
|
407 |
423 |
0.54c |
3.02 |
0.84 |
−5.64 |
−2.50 |
|
17c
|
408 |
423 |
0.38c |
3.00 |
— |
— |
— |
|
18a
|
555 |
434 |
— |
3.15 |
−1.38; 0.89 |
−5.69 |
−3.42j |
|
19a
|
502 |
541 |
0.34d |
2.45 |
−1.62; 0.85 |
−5.65 |
−3.95j |
|
21a
|
512 |
532 |
0.14d |
2.41 |
−1.86 |
−5.30i |
−2.94j |
Density functional calculations
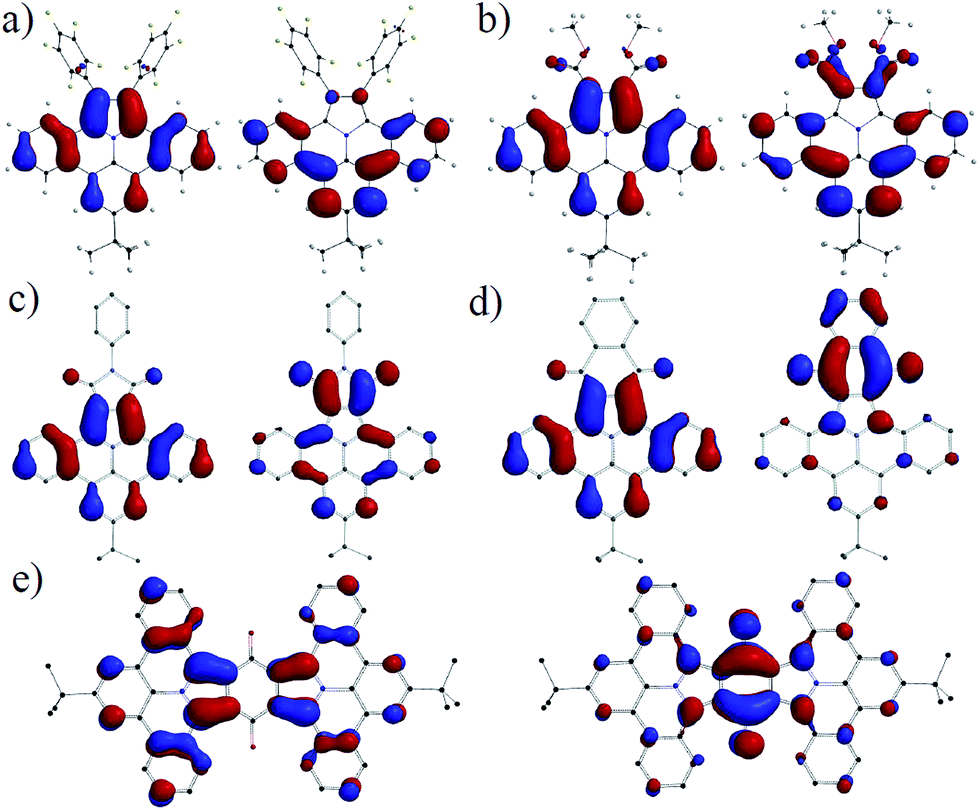
To relate the optical and geometrical properties of N-PAHs 5, 15–19b and 21 to their electronic ground state, we performed density functional theory (DFT) with Gaussian09 using b3lyp functional on 6-31g(d,p) level.35,36 The geometries of 17a–19a, 21a and 23 were derived from the crystal structures of 16, optimized on AM1 and computed with DFT b3lyp at the 6-31g(d,p) level. The graphical representations of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) in Fig. 5 both show a bisecting nodal plane, through the 8-position and the nitrogen atom for all N-PAHs. For comparison, ullazine 22 possesses such a nodal plane only in the HOMO.33,37 As a result, substituents introduced in the 8-position such as tert-butyl, decyl, and methoxy do not exert an influence on the optoelectronic properties, which is in agreement with the experimental results.
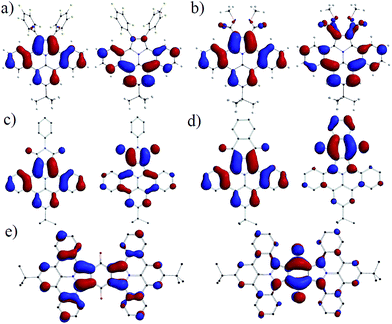 |
| | Fig. 5 Graphical representation of HOMOs (left) and LUMOs (right) of N-PAHs (a) 16, (b) 5, (c) 17a, (d) 19a and (e) 21a. The calculations were performed with DFT B3LYP 6-31g(d,p) based on geometries derived from crystal structure data obtained for 15a and 16. | |
In stark contrast, substitution and/or annulation in the 1,2-position for 5, 16, 17a, 19a and 21a lead to a redistribution in the orbital landscape. For N-PAHs 5 and 16, HOMO and LUMO are located on the main part of the molecule similar to those of the parent molecule 23 and only little coefficients are located on the pentafluorophenyl substituents. The highly planar nitrogen containing heterocycle of the ullazine family, consisting of 24 delocalised π-electrons, can be considered as a donor molecule, which will facilitate a strong intramolecular charge-transfer if an acceptor is present. By introducing carbonyl groups such as carboxylic esters in 5, the LUMO is shifted from the main core of dibenzoullazine 23 to the part of the molecule containing the electron-withdrawing groups. However, the free rotation around the C–C bond causes only minor conjugation over theses substituents in the HOMO and the LUMO. Therefore, no charge transfer can be detected and the λmax for 5, 15 and 16 is closely around 390 nm. This situation changes if the 1,2-substituents are forced into conjugation by formation of five-(17a–c)- or six-(18a–19b)-membered rings. Here an efficient charge-transfer can be observed. The HOMO is localized on the donor part while the LUMO relies exclusively on the acceptor (five-, six-membered ring) unit. This resembles a classic donor–acceptor (D–A) pattern and a dipole is induced along the nodal plane of the molecule with increasing acceptor strength. The increasing dipole moment and the charge transfer are origin of the bathochromic shift of λmax ranging from ∼20 nm for 17a to ∼170 nm (18a) compared to 5. In 21a the HOMO is located on two donor units (D–A–D), while the LUMO is centered on the middle quinone core. A charge transfer occurs from both sides. The extension of the overall conjugated system explains an additional bathochromic shift of 10 nm for 21a compared to 19a. The high symmetry of the HOMO and LUMO in combination with a certain rigidity of the planar core in 17a might explain the high fluorescent quantum yields, because of large transfer integral and low relaxation of the excited state via motion.
Conclusions
In summary, we established the use of planar polycyclic aromatic azomethine ylides (PAMYs) and demonstrated their versatile potential in the construction of large nitrogen containing polycyclic aromatic hydrocarbons for the first time. We provide a general approach to PAMYs, exemplified for three differently substituted pentacyclic derivatives based on readily available 2,6-dibromo-anilines. By cycloaddition reaction with a series of dipolarophiles and subsequent oxidation of the intermediate, it was possible to synthesize six classes of novel N-PAHs in a one-pot reaction, which all show good optoelectronic properties, such as high extinction coefficients and fluorescence quantum yields up to 54%. These unprecedented N-PAHs can further be functionalized in various ways giving access to promising materials, for example as organic sensitizers for solar-cell application.33
Acknowledgements
We acknowledge support by the EC Graphene Flagship (contract no. CNECT-ICT-604391), MoQuaS and ERC Nanograph. We thank Dr Schollmeyer (University of Mainz) for X-ray crystal-structure analysis. R.B. thanks the Fond der Chemischen Industrie for a Chemiefonds fellowship.
Notes and references
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
-
L. M. Harwood and R. J. Vickers, in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, John Wiley & Sons, Inc., 2003, pp. 169–252 Search PubMed.
- L. R. Domingo, E. Chamorro and P. Perez, Lett. Org. Chem., 2010, 7, 432–439 CrossRef CAS.
- S. D. Kahn, W. J. Hehre and J. A. Pople, J. Am. Chem. Soc., 1987, 109, 1871–1873 CrossRef CAS.
- R. Beugelmans, L. Benadjila-Iguertsira and G. Roussi, J. Chem. Soc., Chem. Commun., 1982, 544–545 RSC.
- A. Padwa, W. Dent, H. Nimmesgern, M. K. Venkatramanan and G. S. K. Wong, Chem. Ber., 1986, 119, 813–828 CrossRef CAS.
- B. Braida, C. Walter, B. Engels and P. C. Hiberty, J. Am. Chem. Soc., 2010, 132, 7631–7637 CrossRef CAS PubMed.
- D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10187–10198 CrossRef CAS PubMed.
- E. Lopez-Calle, M. Keller and W. Eberbach, Eur. J. Org. Chem., 2003, 2003, 1438–1453 CrossRef.
- Y. Terao, M. Aono and K. Achiwa, Heterocycles, 1988, 27, 981–1008 CrossRef CAS PubMed.
- J.-P. Fleury, J.-P. Schoeni, D. Clerin and H. Fritz, Helv. Chim. Acta, 1975, 58, 2018–2026 CrossRef CAS.
- L. Toupet and Y. Délugeard, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 1935–1936 CrossRef.
- R. Grigg, J. F. Malone, T. Mongkolaussavaratana and S. Thianpatanagul, J. Chem. Soc., Chem. Commun., 1986, 421–422 RSC.
- J. J. D'Amico, B. R. Stults, P. G. Ruminski and K. V. Wood, J. Heterocycl. Chem., 1983, 20, 1283–1286 CrossRef.
- C. Romming and P. Kolsaker, Acta Chem. Scand., Ser. B, 1978, 32, 679–682 CrossRef PubMed.
- S. Takahashi and H. Kanō, J. Org. Chem., 1965, 30, 1118–1122 CrossRef CAS.
- C. Najera and J. M. Sansano, Curr. Org. Chem., 2003, 7, 1105–1150 CrossRef CAS.
- R. Berger, A. Giannakopoulos, P. Ravat, M. Wagner, D. Beljonne, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 10520–10524 CrossRef CAS PubMed.
- A. Stabel, P. Herwig, K. Müllen and J. P. Rabe, Angew. Chem., Int. Ed. Engl., 1995, 34, 1609–1611 CrossRef CAS.
- K. Müllen, ACS Nano, 2014, 8, 6531–6541 CrossRef PubMed.
- R. Rieger and K. Müllen, J. Phys. Org. Chem., 2010, 23, 315–325 CAS.
- P. O. Dral, M. Kivala and T. Clark, J. Org. Chem., 2012, 78, 1894–1902 CrossRef PubMed.
- M. Takase, T. Narita, W. Fujita, M. S. Asano, T. Nishinaga, H. Benten, K. Yoza and K. Müllen, J. Am. Chem. Soc., 2013, 135, 8031–8040 CrossRef CAS PubMed.
- F. Schlütter, F. Rossel, M. Kivala, V. Enkelmann, J.-P. Gisselbrecht, P. Ruffieux, R. Fasel and K. Müllen, J. Am. Chem. Soc., 2013 Search PubMed.
- M. Kivala, W. Pisula, S. H. Wang, A. Mavrinskiy, J. P. Gisselbrecht, X. L. Feng and K. Mullen, Chem.–Eur. J., 2013, 19, 8117–8128 CrossRef CAS PubMed.
- D. Wu, R. Liu, W. Pisula, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 2791–2794 CrossRef CAS PubMed.
- M. Takase, V. Enkelmann, D. Sebastiani, M. Baumgarten and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 5524–5527 CrossRef CAS PubMed.
- F. Tran, B. Alameddine, T. A. Jenny and T. A. Wesolowski, J. Phys. Chem. A, 2004, 108, 9155–9160 CrossRef CAS.
- U. Nygaard, J. T. Nielsen, J. Kirchheiner, G. Maltesen, J. Rastrup-Andersen and G. O. Sørensen, J. Mol. Struct., 1969, 3, 491–506 CrossRef.
- H. Balli and M. Zeller, Helv. Chim. Acta, 1983, 66, 2135–2139 CrossRef CAS.
- J. Feng, Y. Jiao, W. Ma, M. K. Nazeeruddin, M. Grätzel and S. Meng, J. Phys. Chem. C, 2013, 117, 3772–3778 CAS.
- A. Dualeh, R. Humphry-Baker, J. H. Delcamp, M. K. Nazeeruddin and M. Graetzel, Adv. Energy Mater., 2013, 3, 496–504 CrossRef CAS.
- J. H. Delcamp, A. Yella, T. W. Holcombe, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2013, 52, 376–380 CrossRef CAS PubMed.
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067–1071 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, USA, 2009.
- E. R. Davidson and D. Feller, Chem. Rev., 1986, 86, 681–696 CrossRef CAS.
- F. Gerson and A. Metzger, Helv. Chim. Acta, 1983, 66, 2031–2043 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1022817 and 1022816. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02793k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a