
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of σ-alkane complexes of manganese by NMR and IR spectroscopy in solution: (η5-C5H5)Mn(CO)2(ethane) and (η5-C5H5)Mn(CO)2(isopentane)
Olga
Torres
a,
James A.
Calladine
b,
Simon B.
Duckett
a,
Michael W.
George
*b and
Robin N.
Perutz
*a
aDepartment of Chemistry, University of York, York YO10 5DD, UK
bSchool of Chemistry, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: robin.perutz@york.ac.uk; Mike.George@nottingham.ac.uk
First published on 13th October 2014
Abstract
Irradiation of CpMn(CO)3 in liquid ethane at 135 K at 355 nm yields a photoproduct that exhibits ν(CO) bands in the IR spectrum shifted to low wavenumber with respect to CpMn(CO)3 that are indicative of a Mn(I) dicarbonyl. Parallel experiments employing in situ irradiation within an NMR probe (133 K, 355 nm photolysis) reveal the 1H NMR signals of this product and confirm its formulation as the σ-ethane complex CpMn(CO)2(η2-C1–H-ethane). The resonance of its coordinated C–H group is observed at δ −5.84 and decays with lifetime of ca. 360 s. Analogous photolysis experiments in isopentane solution with IR detection produce CpMn(CO)2(η2-C–H-isopentane) with similar IR bands to those of CpMn(CO)2(η2-C–H-ethane). 1H NMR spectra of the same species were obtained by irradiation of CpMn(CO)3 in a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 mixture of propane and isopentane; three isomers of CpMn(CO)2(η2-C–H-isopentane) were detected with coordination of manganese at the two inequivalent methyl positions and at the methylene group, respectively. The lifetimes of these isomers are ca. 380 ± 20 s at 135 K and do not vary significantly from each other. These σ-complexes of manganese are far more reactive than those of related CpRe(CO)2(alkane) complexes which are stable in solution at 170–180 K. The room temperature lifetimes of CpMn(CO)2(η2-C–H-ethane) and CpMn(CO)2(η2-C–H-isopentane), as determined by TRIR spectroscopy, are 2.0 ± 0.1 and 28 ± 1 μs, respectively.
:40 mixture of propane and isopentane; three isomers of CpMn(CO)2(η2-C–H-isopentane) were detected with coordination of manganese at the two inequivalent methyl positions and at the methylene group, respectively. The lifetimes of these isomers are ca. 380 ± 20 s at 135 K and do not vary significantly from each other. These σ-complexes of manganese are far more reactive than those of related CpRe(CO)2(alkane) complexes which are stable in solution at 170–180 K. The room temperature lifetimes of CpMn(CO)2(η2-C–H-ethane) and CpMn(CO)2(η2-C–H-isopentane), as determined by TRIR spectroscopy, are 2.0 ± 0.1 and 28 ± 1 μs, respectively.
Introduction
Alkane complexes1 have now been demonstrated both experimentally and theoretically to act as reaction intermediates in many fundamental transformations associated with transition metal centres: they include oxidative addition, and its reverse, reductive elimination,2,3 protonation,4 σ-complex assisted metathesis (σ-CAM) which can lead to alkane functionalisation,5 hydrogenation/dehydrogenation6 and substitution reactions.7 Such alkane complexes were first detected by UV/vis and IR spectroscopy in low temperature matrices and glasses in the 1970's.8,9 For many years, time-resolved spectroscopy, especially with IR detection, has provided strong evidence for the existence of σ-alkane complexes, via monitoring of the ν(CO) vibrations of metal carbonyls.10–13 This technique also revealed the variation in lifetime of the alkane complexes with the metal and spectator ligands. A breakthrough that enables the NMR detection of these complexes came through the use of UV irradiation inside the NMR probe to study complexes that had first been identified by IR spectroscopy as relatively long-lived.14–21 Low temperature protonation has added a thermal route to the formation of methane and, very recently, ethane complexes detected by NMR spectroscopy.4,22 These NMR studies have distinguished the different regioisomers of alkane complexes and revealed their diagnostic 1H and 13C chemical shifts and 2JCH coupling constants. The information from NMR spectroscopy has confirmed that these alkane complexes belong to the growing range of σ-bond complexes5,23,24 comprising dihydrogen,25 borane,26,27 silane28–31 and germane32 ligands as well as alkanes.The first examples of crystallographically characterised alkane complexes were discovered serendipitously in a coordination environment typical of a solvent of crystallisation.33,34 More recently, a rhodium(norbornane) complex containing two chelating Rh(η2-C,H) linkages has been made by hydrogenation of a norbornadiene complex in the solid state and characterised by single crystal X-ray crystallography (Rh–C1 2.494(10) Å; Rh–C2, 2.480(11) Å).6 The structures of some iron-based metal–organic frameworks that adsorb alkanes have been determined by powder neutron diffraction and yield far longer Fe–C distances of ca. 3 Å, suggesting weaker interactions.35
Some of the special properties of alkane complexes can be deduced from isotope exchange experiments on metal alkyl deuterides that result in metal (deuteroalkyl) hydride complexes.1,2,36 Of particular importance, is the ability of the metal to migrate from one CH2 or CH3 position to another within a σ-alkane complex, a phenomenon known as chain walking.37 These dynamic processes are usually investigated by product studies with isotope labelling. Recently, chain walking has been followed by dynamic 13C NMR spectroscopy for an ethane complex.22 On the other hand, geminal exchange of the coordination position of an η2-alkane complex is too fast to resolve by NMR spectroscopy.15,16
Alkane complexes represent an important route to alkane functionalisation as demonstrated for borylation,38,39 dehydrogenation40 and alkane metathesis.41,42 They are also likely to participate in the platinum chemistry of alkanes pioneered by Shilov.43,44 Simple alkanes are often very reactive towards metal ions in the gas phase, as shown by mass spectrometric methods.45 Their reactivity may be understood via alkane complexation and, in some cases, the σ-CAM mechanism.5,45 Alkane complexes are also postulated in Periana's most recent results on C–H functionalisation catalysed by heavy p-block metals.46 However, there are recent examples of methane functionalisation that involve very different chemical systems for which other mechanisms may apply.47–50
The study of alkane complexes and alkane functionalisation reactions by computational methods is an important adjunct to experiment providing geometric information and details of the potential energy surfaces.20,51–53 The highest level calculations (composite CCSD(T)/def2-QZVPP) on CpRe(CO)2(η2-CH4) give Re–C and Re–H distances of 2.60 and 1.92 Å, respectively, and a Re–CH4 complexation enthalpy of −62.0 kJ mol−1 at 298 K.54
We have demonstrated previously that we can identify suitable target alkane complexes for NMR spectroscopic investigation by first measuring their lifetimes at low temperature by TRIR or FTIR spectroscopy.18,19 We then investigate the same complexes by NMR spectroscopy using the same laser wavelength and the same temperature if the lifetime is estimated to be sufficient for NMR monitoring. Using this technique, we were able to measure the NMR spectra of the isomeric propane complexes, CpMn(CO)2(η2-C1–H-propane) and CpMn(CO)2(η2-C2–H-propane), the first example of the NMR characterisation of a first row transition metal alkane complex.21 The spectra of the corresponding butane complexes were also recorded.21 Recognising that the lifetime of the manganese propane complexes is only just long enough for NMR characterisation at 134 K, we selected three other alkanes suitable for investigation at such low temperatures, ethane, isopentane and methane and report here their reactivity towards photogenerated CpMn(CO)2 based on evidence from IR and NMR spectroscopy.
Experimental
Materials
For IR spectroscopy, CpMn(CO)3 (Aldrich), ethane and CO (BOC, CP grade) were used as received. Isopentane (Aldrich, 99%) was dried by refluxing over CaH2 for 24 h under an argon atmosphere. In all cases, the concentration of the CpMn(CO)3 was in the range 10−3 to 10−4 M, and the IR and UV absorbances at the probe and pump wavelengths were kept in the range 0.2–0.8. For NMR spectroscopy, chemicals were sourced as follows: ethane (Air Liquide, grade N2.5, 99.5%), isopentane (Aldrich, 99%), propane (BOC, instrument grade N2.5, 99.5%), propane-d2 (98% atom D, Isotec TM), methane (BOC research grade), CpMn(CO)3 (Strem). Propane and methane were used as received, but ethane was purified by passing through a U-tube containing Pt (5%)/charcoal catalyst (Johnson Matthey) on a high vacuum line at 195 K to trap ethylene, hydrogen and other reactive impurities.Room temperature TRIR spectroscopy
A flow system containing the isopentane solution was prepared on a Schlenk line and pressurized to 2 atm with CO. An infrared solution cell (Harrick Scientific Corp.) with CaF2 windows and 0.5 mm pathlength was used to record FTIR and TRIR spectral data. Fresh sample was flowed into the cell after each UV laser shot from a reservoir vessel into a receiving vessel, held at a slightly lower pressure. The activation energy measurements were performed in a variable temperature IR cell and a recirculating water bath was used to control the temperature. The solutions were allowed to come to thermal equilibrium before any measurements were made, and the internal IR cell temperature was monitored using a 1/16′′ K-type thermocouple.FTIR spectroscopy in low temperature solutions
A custom built, high-pressure, low-temperature copper cell (CaF2 windows, 2.5 mm pathlength), cooled by a CryoTiger cold-finger (Brooks Automation) was used for all low temperature FTIR and time-resolved measurements. Details of this apparatus are given elsewhere.55 For the experiments in liquid ethane, a small quantity of CpMn(CO)3 was placed in the cell and pressurized to 45 atm (no CO was added) using a hand-pump. For the isopentane experiment, a solution of CpMn(CO)3 pre-made using the method described above under 2 atm Ar was flowed into the cell (held at a slightly lower pressure). In both cases, the apparatus was cooled to 135 K and allowed to thermally equilibrate for 15 min before any measurements were taken.Time-resolved infrared spectroscopy
Microsecond TRIR spectra and kinetics were recorded on the Nottingham point-by-point quantum cascade laser (QCL) spectrometer.56 A Nd:YAG laser (Spectra Physics GCR-12; 355 nm) initiates the photochemical reaction and the laser-induced change in IR transmission through the sample, as a function of probe wavelength, is measured using an MCT detector cooled by liquid nitrogen and coupled to an oscilloscope and PC.
Rapid-scan FTIR spectroscopy was performed using an Agilent Cary 680 spectrometer. The cryostat was mounted on an external bench and the UV laser (SpectraPhysics GCR-12; 355 nm) aligned through the sample cell.55
Low-temperature NMR spectroscopy with in situ laser irradiation
NMR spectra were recorded on a Bruker Avance wide-bore 600 MHz spectrometer with solvent suppression achieved with the excitation sculpting pulse sequence with gradients zgesgp 1D sequence (Avance-version, Feb 2012), typically using about 24 scans.57 Some additional spectra were obtained with WATERGATE W5 using the pulse program zggpw5.581H NMR spectra were calibrated by adding a trace of benzene, taken as resonating at δ 7.15. Laser photolysis was carried out with a pulsed Nd:YAG laser (Continuum Surelite II) operating at 355 nm with 10 Hz repetition rate and a laser power of 85 mW. The laser radiation is directed through a hole in the probe housing onto the NMR tube. The sample is contained in a high-pressure sapphire NMR tube (IDEAS!, UVA, B.V.). The samples contained 1–2 mg of CpMn(CO)3 and approximately 0.4 mL of solvent that was transferred on a high pressure line (ethane and propane) or a high vacuum line (isopentane). A very dilute sample of [Ru(CO)2(Ph2PCH2CH2PPh2)(PPh3)] in C6D6 was used for real-time laser alignment in conjunction with para-hydrogen amplification.59 For kinetics, the integration of the relevant resonance was measured relative to that of benzene added in trace quantities. This apparatus is described in more detail elsewhere.19
Results and discussion
Infrared spectroscopic studies
The photochemistry of CpMn(CO)3 in several different alkane solvents including ethane has been investigated previously.13,21 A solution of CpMn(CO)3 in isopentane (2-methylbutane) at room temperature was investigated by TRIR spectroscopy with 355 nm photolysis. The transient difference spectrum measured 1.2 μs after the laser pulse (Fig. 1a, Table 1) reveals that bleaching of the parent ν(CO) bands occurs together with the formation of two new bands that are shifted 68 cm−1 (ν(CO)symm) and 53 cm−1 (ν(CO)antisymm) to lower wavenumber relative to the a1 and e modes of CpMn(CO)3, respectively. The shifts are consistent with a reduction in the number of CO groups with no change in oxidation state. They are assigned to the a′ and a′′ modes of the σ-alkane complex, CpMn(CO)2(isopentane) by comparison to the spectra of related alkane complexes formed by [CpMn(CO)2] and [CpRe(CO)2].13,19,21,60 In the presence of CO (2 atm) the Mn-isopentane complex reacts quickly and cleanly to regenerate the parent with a lifetime of 28 ± 1 μs (Fig. 1b). The rate of reaction of the σ-alkane complex with CO was measured as a function of temperature between 282 and 295 K and an activation energy (Ea) of 43 ± 2 kJ mol−1 obtained (Fig. 2). This activation energy can be used as a minimum value for the Mn–alkane bond strength, and is similar to those measured for analogous CpMn(CO)2(alkane) complexes (viz. Mn–heptane,60 39 kJ mol−1; Mn–methane,13 41 kJ mol−1). | ||
| Fig. 1 (a) Point-by-point TRIR difference spectrum of CpMn(CO)3 in isopentane in the presence of 2 atm CO, 1.2 μs after 355 nm photolysis. The solid line is a multi-Lorentzian fit of the spectrum. (b) Kinetic traces showing decay of the Mn–isopentane complex and reformation of the parent. | ||
| Complex | Solvent, temp/K | ν(CO)/cm−1 | Lifetime | E a (kJ mol−1) |
|---|---|---|---|---|
| a Ref. 13. | ||||
| CpMn(CO)3 | Liq. ethane, 298a | 2032, 1952 | ||
| Liq. ethane, 135 | 2029, 1946 | |||
| Isopentane, 298 | 2030, 1948 | |||
| Isopentane, 135 | 2028, 1944 | |||
| scCH4, 298a | 2035, 1955 | |||
| CpMn(CO)2(alkane) | Liq. ethane, 298a | 1967, 1902 | 2.0 ± 0.1 μs | 38 ± 2 |
| Liq. ethane, 135 | 1962, 1895 | 126 ± 6 s | ||
| Isopentane, 298 | 1962, 1896 | 28 ± 1 μs | 43 ± 2 | |
| Isopentane, 135 | 1958, 1890 | 250 ± 3 s | ||
| scCH4, 298a | 1972, 1908 | 400 ± 32 ns | 41 ± 2 | |
 | ||
| Fig. 2 Arrhenius plot for the reaction of CpMn(CO)2(isopentane) with CO measured from the decay of the σ–alkane complex ν(CO) band at 1896 cm−1. | ||
The reactivity of CpMn(CO)3 in isopentane was also investigated at low temperature by rapid-scan FTIR spectroscopy. A temperature of 135 K was selected in order to be consistent with the NMR experiments. Again, photolysis (355 nm) of an isopentane solution of CpMn(CO)3 leads to the formation of the σ–alkane complex, CpMn(CO)2(isopentane) with bands at 1958 and 1890 cm−1 (Fig. 3a). At 135 K, once irradiation is stopped, these bands are observed to decay with a lifetime of 250 ± 3 s (Fig. 3b) in the absence of added CO.
 | ||
| Fig. 3 Rapid-scan FTIR difference spectra obtained 2 s after 355 nm photolysis of CpMn(CO)3 at 135 K in (a) isopentane and (c) liquid ethane. Kinetic traces showing decay of (b) the Mn–isopentane complex at 1958 cm−1 and (d) the Mn–ethane complex at 1962 cm−1. | ||
The room temperature spectra and kinetics of CpMn(CO)2(ethane) have been reported previously.13 Analogous results to those for isopentane are obtained upon irradiation of a solution of CpMn(CO)3 in liquid ethane (650 psi) at 135 K. The rapid-scan FTIR difference spectrum measured 2 s after irradiation (Fig. 3c) illustrates ν(CO) bands due to the formation of CpMn(CO)2(ethane) at very similar wavenumbers of 1962 and 1895 cm−1. The Mn–ethane complex decays with a lifetime of 125 ± 6 s (Fig. 3d), about half the value of the isopentane analogue.
Studies with NMR spectroscopy
The lifetimes of the isopentane and ethane complexes measured by low temperature IR spectroscopy suggest that the NMR detection of these photoproducts is possible. A 1H NMR spectrum of a sample of CpMn(CO)3 dissolved in unpurified ethane at 133 K was first measured with solvent suppression at δ 0.65. In addition to the expected Cp resonance at δ 4.58, a considerable amount of ethylene was detected at δ 5.33 together with some hydrogen resonating at δ 4.55. The ethane was therefore purified by passing through a Pt/charcoal trap which removed the H2 impurity signal and greatly reduced the ethylene contamination. Integration indicated that the ethylene/CpMn(CO)3 ratio was ca. 1:0.7, although the ethane solvent is still in vast excess. Photolysis of CpMn(CO)3 (10–20 mmol dm−3) within the NMR probe for ca. 1–2 min using the pulsed laser operating at 355 nm in neat ethane at 133–135 K results in a decrease in intensity of the corresponding Cp resonance at δ 4.58 (s) and the appearance of a new resonance at δ 4.19 (s) together with another resonance in the high-field region at δ −5.84 (Fig. 4). The Cp resonance of the product is a clean singlet without any of the broadening or shoulders observed with propane and butane, reflecting the formation of a single isomer.21 These two new resonances can be attributed to the Cp and C1H3 resonances, respectively, of the product CpMn(CO)2(η2-C1–H-ethane) by comparison with previous studies of the photolysis of CpMn(CO)3 in neat propane.
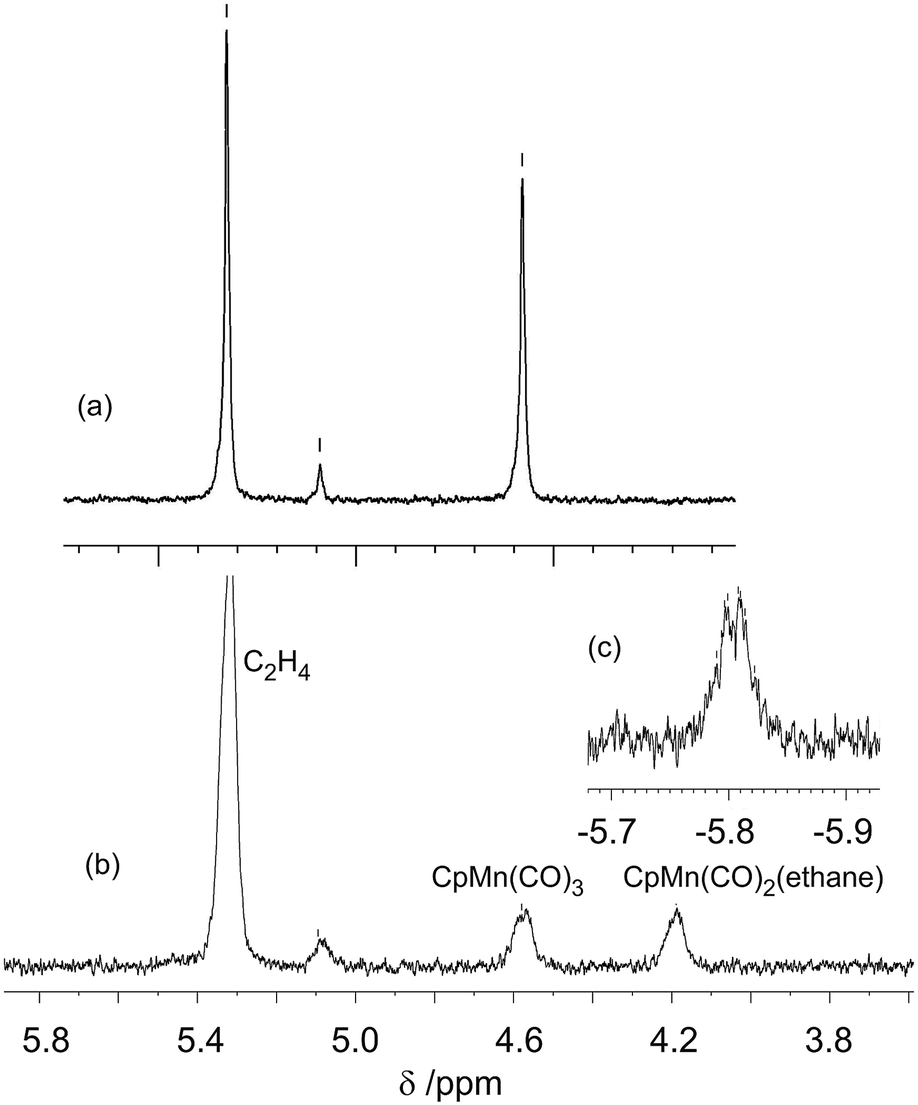 | ||
| Fig. 4 (a) 1H NMR spectrum of CpMn(CO)3 in ethane prior to irradiation in cyclopentadienyl region (b) spectrum after 355 nm laser irradiation. (c) Inset showing high field region after irradiation. | ||
The high field NMR resonance can be understood as arising from the average of the bound η2-C–H proton and the two geminal protons since there will be a very low barrier for exchange between them.16 This resonance is poorly resolved but may be interpreted as a binomial quartet with J = 7 Hz (Fig. 4c) arising from resolved coupling to the nuclei of the second CH3 group. The C2H3 resonance is not detected because of overlap with the solvent resonance. We estimated the lifetime of CpMn(CO)2(η2-C1–H-ethane) as ca. 360 s at 135 K which corresponds to a value of ΔG‡ of 39 kJ mol−1. The ethylene impurities in the ethane probably reduce the lifetime of the photoproduct. This estimate compares with 126 ± 6 s determined by TRIR spectroscopy above with unpurified ethane.
We then turned our attention to isopentane. The first attempt to characterise the products of the photolysis at 355 nm of CpMn(CO)3 in neat isopentane at 142 K was unsuccessful due to the poor solubility of the starting material in this solvent. In order to increase its solubility, we employed a propane/isopentane (60:40) mixture. We chose propane because of its advantageous spectroscopic properties and low freezing point, coupled with the fact that we have already reported the 1H NMR spectrum of the corresponding propane complexes. The 1H NMR spectrum measured before photolysis shows the expected Cp resonance at δ 4.58. After photolysis (200 s) of CpMn(CO)3 in a mixture of liquid propane and isopentane at 133 K a new Cp resonance is observed at δ 4.20 which exhibits a low field shoulder. Three new high field resonances associated with the Mn–CH moiety are observed in addition to the two resonances of CpMn(CO)2(η2-C1–H-propane) and CpMn(CO)2(η2-C2–H-propane). These three resonances appear at δ −5.77, −5.94 and −9.35 with integration ratios of 3:6:1 (Fig. 5b). They are consistent with the observation of three isomers of the photoproduct, namely CpMn(CO)2(η2-C4–H-isopentane), CpMn(CO)2(η2-C1–H-isopentane) and CpMn(CO)2(η2-C3–H-isopentane), respectively (Scheme 1). An experiment using 2,2-D2-propane in place of H8-propane eliminated the resonance at δ −8.2 due to CpMn(CO)2(η2-C2–H-propane). The kinetics of decay of the resonances at δ −5.77 and −5.94 were measured as 372 ± 22 s and 395 ± 23 s, respectively (Fig. 6), and compare to the lifetime of the FTIR signal of 250 ± 3 s. Note that these integrations should be considered as indicative because of the use of the solvent suppression sequence. For comparison, Ball has observed all three isomers of (η5-C5H4iPr)Re(CO)2(η2-C–H-pentane) and two isomers of CpRe(CO)2(η2-C–H-cyclohexane), whereas with (η6-C6Et6)W(CO)2(η2-C–H-pentane) and (η6-C6Et6)W(CO)2(η2-C–H-2,2-dimethylbutane) only the isomers associated with interactions with primary C–H bonds bound were evident.15,20
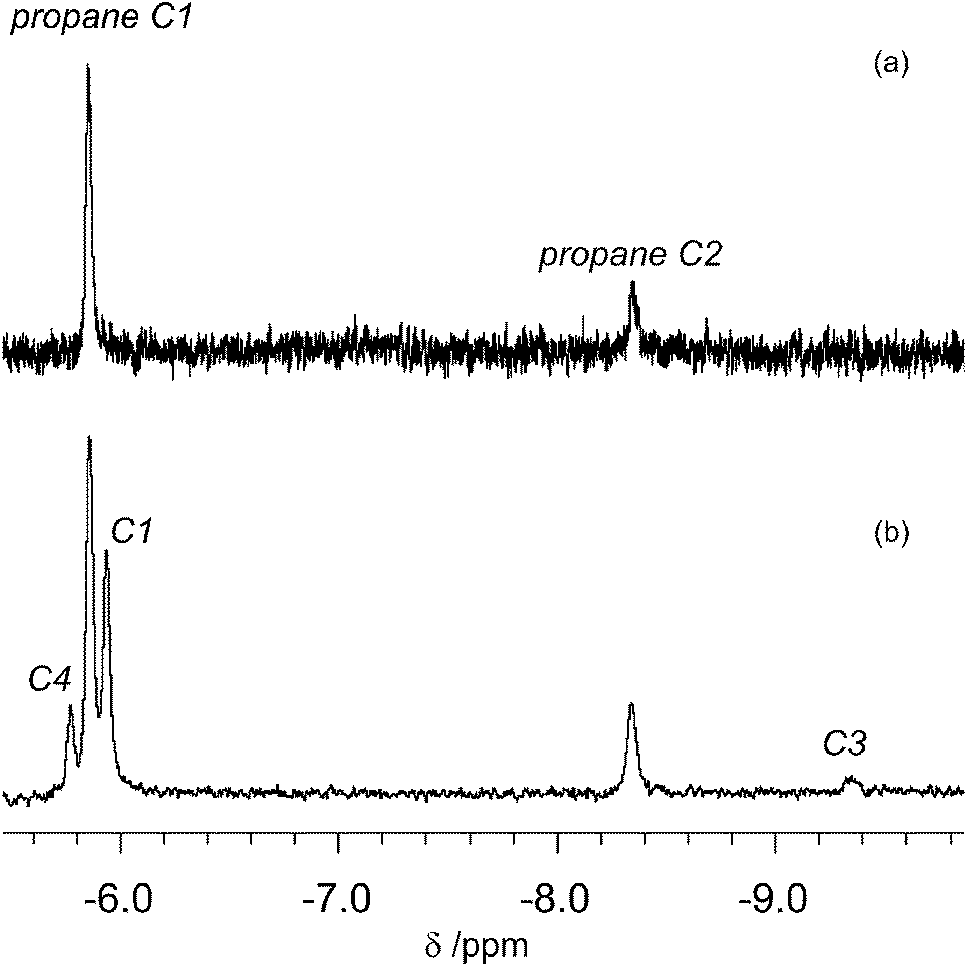 | ||
| Fig. 5
1H NMR spectrum in the high-field region obtained after 355 nm laser irradiation of (a) CpMn(CO)3 in liquid propane; (b) a 60:40 mixture of liquid propane and isopentane at 133 K. The labels indicate the positions of Mn coordination for propane (above) and isopentane (below). | ||
 | ||
| Scheme 1 Isomers of CpMn(CO)2(isopentane). The C2 isomer is not observed. | ||
 | ||
| Fig. 6 Time profile of the high field resonance integration of CpMn(CO)2(isopentane) relative to the internal benzene standard. ▲ δ −5.94 signal with the solid line showing the fit for exponential decay of lifetime 395 ± 23 s; ■ corresponding data for δ −5.77 signal and fit to a lifetime of 372 ± 22 s. | ||
We carried out similar photochemical experiments with CpMn(CO)3 dissolved in a 90:10 mixture of propane and methane at 133 K. Under these conditions, we were only able to detect the corresponding resonances of the propane complexes mentioned above. Given the signal-to-noise ratio obtained in these data we can deduce that the corresponding methane complex is less stable than the propane adduct.
Conclusions
A combination of IR spectroscopy and NMR spectroscopy at 135 K has been used to detect highly unstable ethane and isopentane complexes of the type CpMn(CO)2(η2-C–H-alkane). As expected, CpMn(CO)2(η2-C–H-ethane) exists as a single isomer whereas CpMn(CO)2(η2-C–H-isopentane) is detected as a mixture of three isomers by NMR spectroscopy. These species cannot be differentiated through their IR data, indicating that the electronic effects of the different coordination modes on the CpMn(CO)2 unit are very similar. The most sterically hindered isomer, with the tertiary C–H bond coordinated, viz. CpMn(CO)2(η2-C2–H-isopentane), is not observed. The only other ethane complex that has currently been observed by NMR spectroscopy is [(PONOP)Rh(η2-C–H-ethane)]+ where PONOP is a PNP pincer ligand.22 The chemical shifts of the coordinated η2-C–H-CH3 group of the manganese alkane complexes occur close to δ −6, whilst those of a coordinated η2-C–H-CH2 bond lie appreciably to higher field, in line with the reported observations on the analogous propane complex. These values represent averages of shifts for coordinated and uncoordinated C–H bonds because of rapid exchange of geminal protons. The CpMn(CO)2(η2-C–H-ethane) lifetime is ca. 360 s whilst that of two of the CpMn(CO)2(η2-C–H-isopentane) isomers are 395 ± 23 s and 372 ± 22 s. The similarity of the lifetimes could be interpreted in terms of interconversion of the isomers by a chain walking process that is slow on the NMR timescale and slow compared to that of the rhodium ethane complexes studied by Brookhart et al.22 Very satisfyingly, the lifetime of CpMn(CO)2(η2-C–H-ethane) equates to a value of ΔG‡ of 39 kJ mol−1 compared to 39 ± 2 kJ mol−1 for the activation energy (Ea) as measured by TRIR spectroscopy around room temperature.61 As demonstrated previously, the manganese complexes are vastly more reactive than their rhenium analogues which are stable in solution of the corresponding alkanes at 170–180 K.Acknowledgements
We thank the EPSRC (EP/D058031 and EP/D055768) and the Universities of Nottingham and York for funding. O.T. would like to thank the Ministerio de Ciencia e Innovación and the Fundación Española para la Ciencia y la Tecnología for funding. M.W.G. gratefully acknowledges receipt of a Royal Society Wolfson Merit Award.Notes and references
- C. Hall and R. N. Perutz, Chem. Rev., 1996, 96, 3125 CrossRef CAS PubMed.
- M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471 CrossRef CAS PubMed.
- W. D. Jones, Acc. Chem. Res., 2003, 36, 140 CrossRef CAS PubMed.
- W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Science, 2009, 326, 553 CrossRef CAS PubMed.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578 CrossRef CAS PubMed.
- S. D. Pike, A. L. Thompson, A. G. Algarra, D. C. Apperley, S. A. Macgregor and A. S. Weller, Science, 2012, 337, 1648 CrossRef CAS PubMed.
- L. Biber, L. D. Reuvenov, T. Revzin, T. Sinai, A. Zahavi and R. H. Schultz, Dalton Trans., 2007, 41 RSC; R. Krishnan and R. H. Schultz, Organometallics, 2001, 20, 3314 CrossRef CAS; R. H. Schultz, Organometallics, 2004, 23, 4349 CrossRef.
- R. N. Perutz and J. J. Turner, J. Am. Chem. Soc., 1975, 97, 4791 CrossRef CAS.
- M. Poliakoff and J. J. Turner, Dalton Trans., 1974, 2276 RSC.
- J. A. Calladine, K. Q. Vuong, X. Z. Sun and M. W. George, Pure Appl. Chem., 2009, 81, 1667 CrossRef CAS.
- M. K. Kuimova, W. Z. Alsindi, J. Dyer, D. C. Grills, O. S. Jina, P. Matousek, A. W. Parker, P. Portius, X. Z. Sun, M. Towrie, C. Wilson, J. X. Yang and M. W. George, Dalton Trans., 2003, 3996 RSC.
- M. Besora, J.-L. Carreon-Macedo, A. J. Cowan, M. W. George, J. N. Harvey, P. Portius, K. L. Ronayne, X.-Z. Sun and M. Towrie, J. Am. Chem. Soc., 2009, 131, 3583 CrossRef CAS PubMed.
- A. J. Cowan, P. Portius, H. K. Kawanami, O. S. Jina, D. C. Grills, X. Z. Sun, J. McMaster and M. W. George, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6933 CrossRef CAS PubMed.
- S. Geftakis and G. E. Ball, J. Am. Chem. Soc., 1998, 120, 9953 CrossRef CAS.
- D. J. Lawes, S. Geftakis and G. E. Ball, J. Am. Chem. Soc., 2005, 127, 4134 CrossRef CAS PubMed.
- G. E. Ball, C. M. Brookes, A. J. Cowan, T. A. Darwish, M. W. George, H. K. Kawanami, P. Portius and J. P. Rourke, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 6927 CrossRef CAS PubMed.
- D. J. Lawes, T. A. Darwish, T. Clark, J. B. Harper and G. E. Ball, Angew. Chem., Int. Ed, 2006, 45, 4486 CrossRef CAS PubMed.
- S. B. Duckett, M. W. George, O. S. Jina, S. L. Matthews, R. N. Perutz, X. Z. Sun and K. Q. Vuong, Chem. Commun., 2009, 1401 RSC.
- J. A. Calladine, O. Torres, M. Anstey, G. E. Ball, R. G. Bergman, J. Curley, S. B. Duckett, M. W. George, A. I. Gilson, D. J. Lawes, R. N. Perutz, X.-Z. Sun and P. C. Vollhardt, Chem. Sci., 2010, 1, 622 RSC.
- R. D. Young, A. F. Hill, W. Hillier and G. E. Ball, J. Am. Chem. Soc., 2011, 133, 13806 CrossRef CAS PubMed; R. D. Young, D. J. Lawes, A. F. Hill and G. E. Ball, J. Am. Chem. Soc., 2012, 134, 8294 CrossRef PubMed.
- J. A. Calladine, S. B. Duckett, M. W. George, S. L. Matthews, R. N. Perutz, O. Torres and K. Q. Vuong, J. Am. Chem. Soc., 2011, 133, 2303 CrossRef CAS PubMed.
- M. D. Walter, P. S. White, C. K. Schauer and M. Brookhart, J. Am. Chem. Soc., 2013, 135, 15933 CrossRef CAS PubMed.
- G. J. Kubas, Metal Dihydrogen and σ-Bond Complexes, Kluwer Academic Publishers, New York, 2001 Search PubMed.
- R. H. Crabtree, Angew. Chem., Int. Ed., 1993, 32, 789 CrossRef.
- G. J. Kubas, J. Organomet. Chem., 2014, 751, 33 CrossRef CAS.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed.
- G. Alcaraz, M. Grellier and S. Sabo-Etienne, Acc. Chem. Res., 2009, 42, 1640 CrossRef CAS PubMed.
- J. Y. Corey and J. Braddock-Wilking, Chem. Rev., 1999, 99, 175 CrossRef CAS PubMed.
- J. Y. Corey, Chem. Rev., 2011, 111, 863 CrossRef CAS PubMed.
- Z. Y. Lin, Struct. Bonding, 2008, 130, 123 CrossRef CAS.
- G. I. Nikonov, Adv. Organomet. Chem., 2005, 53, 217 CrossRef CAS.
- J. L. Vincent, S. Luo, B. L. Scott, R. Butcher, C. J. Unkefer, C. J. Burns, G. J. Kubas, A. Lledós, F. Maseras and J. Tomàs, Organometallics, 2003, 22, 5307 CrossRef CAS.
- D. R. Evans, T. Drovetskava, R. Bau, C. A. Reed and P. D. W. Boyd, J. Am. Chem. Soc., 1997, 119, 3633 CrossRef CAS.
- I. Castro-Rodriguez, H. Nakai, P. Gantzel, L. N. Zakharov, A. L. Rheingold and K. Meyer, J. Am. Chem. Soc., 2003, 125, 15734 CrossRef CAS PubMed.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606 CrossRef CAS PubMed; S. J. Geier, J. A. Mason, E. D. Bloch, W. L. Queen, M. R. Hudson, C. M. Brown and J. R. Long, Chem. Sci., 2013, 4, 2054 RSC.
- D. D. Wick, K. A. Reynolds and W. D. Jones, J. Am. Chem. Soc., 1999, 121, 3974 CrossRef CAS.
- W. D. Jones, Inorg. Chem., 2005, 44, 4475 CrossRef CAS PubMed; T. O. Northcutt, D. D. Wick, A. J. Vetter and W. D. Jones, J. Am. Chem. Soc., 2001, 123, 7257 CrossRef PubMed.
- J. F. Hartwig, K. S. Cook, M. Hapke, C. D. Incarvito, Y. Fun, C. E. Webster and M. B. Hall, J. Am. Chem. Soc., 2005, 127, 2538 CrossRef CAS PubMed.
- C. S. Wei, C. A. Jimenez-Hoyos, M. F. Videa, J. F. Hartwig and M. B. Hall, J. Am. Chem. Soc., 2010, 132, 3078 CrossRef CAS PubMed.
- J. Choi, A. H. Roy MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761 CrossRef CAS PubMed.
- A. S. Goldman, A. H. Roy, Z. Huang, R. Ahuja, W. Schinski and M. Brookhart, Science, 2006, 312, 257 CrossRef CAS PubMed.
- D. C. Leitch, Y. Choi Lam, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2013, 135, 10302 CrossRef CAS PubMed.
- J. E. Bercaw, G. S. Chen, J. A. Labinger and B. L. Lin, J. Am. Chem. Soc., 2008, 130, 17654 CrossRef CAS PubMed; J. E. Bercaw, G. S. Chen, J. A. Labinger and B. L. Lin, Organometallics, 2010, 29, 4354 CrossRef.
- G. S. Chen, J. A. Labinger and J. E. Bercaw, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6915 CrossRef CAS PubMed.
- M. Schlangen and H. Schwarz, Dalton Trans., 2009, 10155 RSC.
- B. G. Hashiguchi, M. M. Konnick, S. M. Bischof, S. J. Gustafson, D. Devarajan, N. Gunsalus, D. H. Ess and R. A. Periana, Science, 2014, 343, 1232 CrossRef CAS PubMed.
- R. A. Periana, R. A. Mironov, O. D. J. Taube, G. Bhalla and C. J. Jones, Science, 2003, 301, 814 CrossRef CAS PubMed.
- E. V. Kudrik, P. Afanasiev, L. X. Alvarez, P. Dubourdeaux, M. Clémancey, J.-M. Latour, G. Blondin, D. Bouchu, F. Albrieux, S. E. Nefedov and A. B. Sorokin, Nat. Chem., 2012, 4, 1024 CrossRef CAS PubMed.
- A. Caballero, E. Despagnet-Ayoub, M. M. Díaz-Requejo, A. Díaz-Rodríguez, M. E. González-Núñez, R. Mello, B. K. Muñoz, W.-S. Ojo, G. Asensio, M. Etienne and P. J. Pérez, Science, 2011, 332, 835 CrossRef CAS PubMed.
- M. V. Kirillova, M. L. Kuznetsov, P. M. Reis, J. A. L. da Silva, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, J. Am. Chem. Soc., 2007, 129, 10531 CrossRef CAS PubMed.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 112, 749 CrossRef PubMed.
- B. A. Vastine and M. B. Hall, Coord. Chem. Rev., 2009, 253, 1202 CrossRef CAS.
- M. D. Walter, P. S. White, C. K. Schauer and M. Brookhart, New J. Chem., 2011, 35, 2884 RSC.
- B. Chan and G. E. Ball, J. Chem. Theory Comput., 2013, 9, 2199 CrossRef CAS.
- J. A. Calladine, A. Love, P. A. Fields, R. G. M. Wilson and M. W. George, Appl. Spectrosc., 2014, 68, 324 CrossRef CAS PubMed.
- J. A. Calladine and M. W. George, Spectrosc. Eur., 2009, 21, 6 CAS.
- T.-L. Hwang and A. J. Shaka, J. Magn. Reson., 1995, 112, 275 CrossRef CAS.
- M. L. Lui, X. A. Mao, C. H. Ye, H. Huang, J. K. Nicholson and J. C. Lindon, J. Magn. Reson., 1998, 132, 125 CrossRef.
- R. A. Green, R. W. Adams, S. B. Duckett, R. E. Mewis, D. C. Williamson and G. G. R. Green, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 67, 1 CrossRef CAS PubMed.
- G. I. Childs, C. S. Colley, J. Dyer, D. C. Grills, X. Z. Sun, J. X. Yang and M. W. George, J. Chem. Soc., Dalton Trans., 2000, 1901 RSC.
- A. J. Cowan, PhD Thesis, University of Nottingham, 2007 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |