
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transition-metal-free controlled polymerization for poly(p-aryleneethynylene)s†
Takanobu
Sanji
*,
Asahi
Motoshige
,
Hideaki
Komiyama
,
Junko
Kakinuma
,
Rie
Ushikubo
,
Satoru
Watanabe
and
Tomokazu
Iyoda
Iyoda Supra-Integrated Material Project (iSIM), Exploratory Research for Advanced Technology (ERATO), Japan Science and Technology Agency (JST), Tokyo Institute of Technology, 4259-S2-3 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan. E-mail: sanji.t.aa@m.titech.ac.jp
First published on 29th October 2014
Abstract
A transition-metal-free controlled polymerization for the attainment of poly(p-aryleneethynylene)s is developed. The polymerization of 1-pentafluorophenyl-4-[(trimethylsilyl)ethynyl]benzene with a catalytic amount of fluoride anions proceeds in a chain-growth-like manner to afford polymers with controlled molecular weights and low polydispersity indexes. The mechanism involves a pentacoordinated fluorosilicate as a key intermediate. The anionic “living” nature of this process is applied to block copolymerization and also surface-terminated polymerization.
Introduction
Among the most extensively studied families of molecular optoelectronic materials, such as organic field-effect transistors (FETs) and organic electroluminescent (EL) devices, poly(p-aryleneethynylene)s (PAEs) are one of the most important types of materials.1 The polymers are commonly prepared by polycondensations using Sonogashira- or Stille-type cross couplings2 or by an alkyne metathesis.3 In all cases, however, the molecular weight (MW) and polydispersity index (PDI) of the polymers are not controlled because the polycondensations proceed by a step-growth mechanism. In contrast to these conventional step-growth polycondensations, chain-growth polymerizations have been demonstrated recently, but they require transition-metal catalysts.4 There remains a need to achieve additional synthetic methods for well-defined polymers with control of the MW, the polydispersity, modification of the end group, and also block copolymerization, which could offer new architectures and materials.5Here, we describe a transition-metal-free controlled polymerization for the attainment of PAEs. The polymerization proceeds in a chain-growth-like manner to afford the polymers with controlled MWs and low PDIs. This could be an alternative synthetic method for well-controlled PAEs.
Results and discussion
We designed 1-pentafluorophenyl-4-[(trimethylsilyl)ethynyl]benzene 1 as a monomer and examined its polymerization with a catalytic amount of fluoride anions (Scheme 1). This is because fluoride anions were found to catalyze silylacetylene activation for subsequent reaction with a number of electrophiles,6,7 and also because regioselective SNAr reactions of perfluoroaryl groups with nucleophiles8,9 are well studied. Very recently, we demonstrated the transition-metal-free polymerization of 2-perfluoroaryl-5-trimethylsilylthiophenes promoted by fluoride anions to afford polymers with controlled MWs and low PDIs.10 In contrast, the reported polycondensation of hexafluorobenzene and 1,4-bis[(trimethylsilyl)ethynyl]benzene was not controlled.11 Recently, Bielawski et al. reported a controlled Pd catalyzed transfer polycondensation for poly(p-phenyleneethynylene).12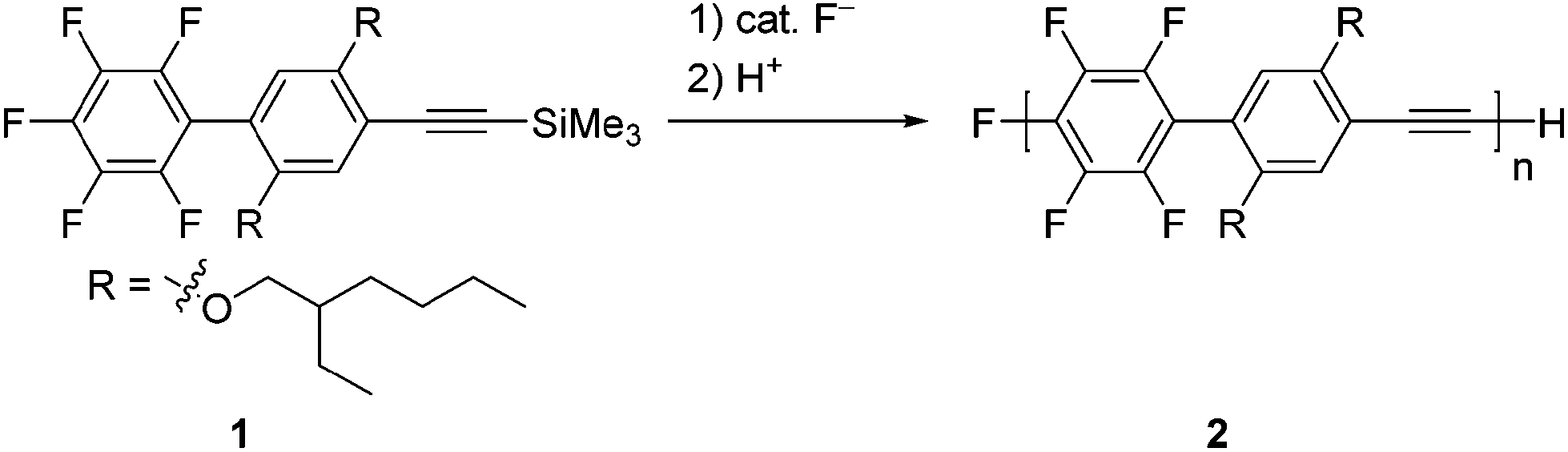 | ||
| Scheme 1 Polymerization of 1. | ||
A catalytic amount of fluoride anions smoothly promoted the polymerization of 1. Table 1 summarizes the polymerization results. For example, the reaction of 1 with 5 mol% tetrabutylammonium fluoride (TBAF)13 as a fluoride anion source in tetrahydrofuran (THF) at room temperature for 2 h led to poly(p-tetrafluorophenylene-phenylene-ethynylene) 2 with a number-averaged MW (Mn) of 4400 and a PDI of 1.31 in an 81% isolated yield (entry 2). The MWs measured by size-exclusion chromatography (SEC)-multi-angle light scattering (MALS) were almost the same as those measured by SEC. When the polymerization of 1 was examined with respect to varying the mol% of TBAF, the MW of the polymer linearly increased with an increasing monomer to TBAF ratio (entries 1–4), while the PDIs were relatively low (≤2). Addition of 1 to a solution of TBAF also afforded the polymer (entry 5). However, tetramethylammonium fluoride was not a good initiator because some remained undissolved in THF (entry 6). Potassium fluoride or cesium fluoride in the presence of 18-crown-6 or cryptand[2.2.2] did not give a good result (entries 7–9). Tetrabutylammonium difluorotriphenylsilicate was not effective for the polymerization of 1 (entry 10).
| Entry | F− (mol%) | Reaction conditions | Yield (%) | M n , | PDIb,c |
|---|---|---|---|---|---|
| a All reactions were run with [1] = 0.3 mmol in 5 mL solvent. b The number-average MW (Mn) and PDI were determined by SEC using polystyrene standards. c The numbers in parentheses are the weight-averaged MW (Mw) and the PDI determined by SEC-MALS. d A THF solution of 1 was added to a TBAF solution. | |||||
| 1 | Bu4NF (TBAF) (10) | THF, rt, 2 h | 34 | 3950 (3600) | 1.26 (1.24) |
| 2 | Bu4NF (TBAF) (5) | THF, rt, 2 h | 81 | 4400 (4500) | 1.31 (1.31) |
| 3 | Bu4NF (TBAF) (2) | THF, rt, 2 h | 52 | 7400 (8900) | 1.68 (1.68) |
| 4 | Bu4NF (TBAF) (1) | THF, rt, 2 h | 82 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 (14500) 500 (14500) |
2.11 (1.57) |
| 5 | Bu4NF (TBAF) (5) | THF, rt, 2 hd | 48 | 7600 | 1.72 |
| 6 | Me4NF (5) | THF, rt, 2 h | 71 | 4500 | 4.15 |
| 7 | KF (5)/18-C-6 (10) | THF, 80 °C, 20 h | 1 | 1400 | 1.08 |
| 8 | CsF (5)/18-C-6 (10) | Toluene, 80 °C, 20 h | 75 | 13700 |
3.88 |
| 9 | KF (5)/cryptand[2.2.2] (10) | THF, rt, 2 h | 0 | — | — |
| 10 | Bu4N+[Ph3SiF2]− (5) | THF, rt, 2 h | 27 | 2500 | 1.48 |
The obtained polymer has a structure with high regioregularity, as demonstrated by 1H, 13C, and 19F NMR analyses (Fig. S1 in the ESI†). The 1H NMR spectrum of 2 shows major signals at around 0.8, 1.4, and 4.0 ppm owing to the side chain on the phenylene units, along with a small signal at 3.7 ppm arising from the ethynyl group at the polymer end. In the 19F NMR spectrum of the polymer, two strong signals and three weak signals were found, which are assigned to the 1,4-tetrafluorophenylene units in the main chain and the pentafluorophenyl group at the polymer end, respectively. Thus, the NMR spectra are consistent with a high regioregularity for the polymer main chain, indicating that the polymerization process itself must be highly regioselective. In addition, because the polymer ends are designated as the pentafluorophenyl and the ethynyl groups, the integral ratio of the peaks from the side chain on the main chain and the end group in the 1H and 19F NMR spectra provide a Mn of ca. 5000, which is in reasonable agreement with the MW estimated by SEC (Table 1, entry 2). Furthermore, matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectra also indicate that the polymer has pentafluorophenyl and ethynyl groups at its ends (Fig. S2 in the ESI†).
The controlled MWs and relatively low PDIs found in Table 1 indicate that the polymerization proceeds in a chain-growth-like manner under the specified conditions. To provide further evidence for this, we monitored the polymerization of 1 with 2.5 mol% TBAF as a function of monomer conversion. Fig. 1 shows the Mn and PDI as functions of monomer conversion. The polymerization of 1 was fast, with the conversion of 1 being up to 50% after a few minutes (Table S1 and Fig. S3 in the ESI†). As shown in Fig. 1, the linear relationship between Mn and monomer conversion, and the relatively low PDI, confirm the chain-growth-like process.
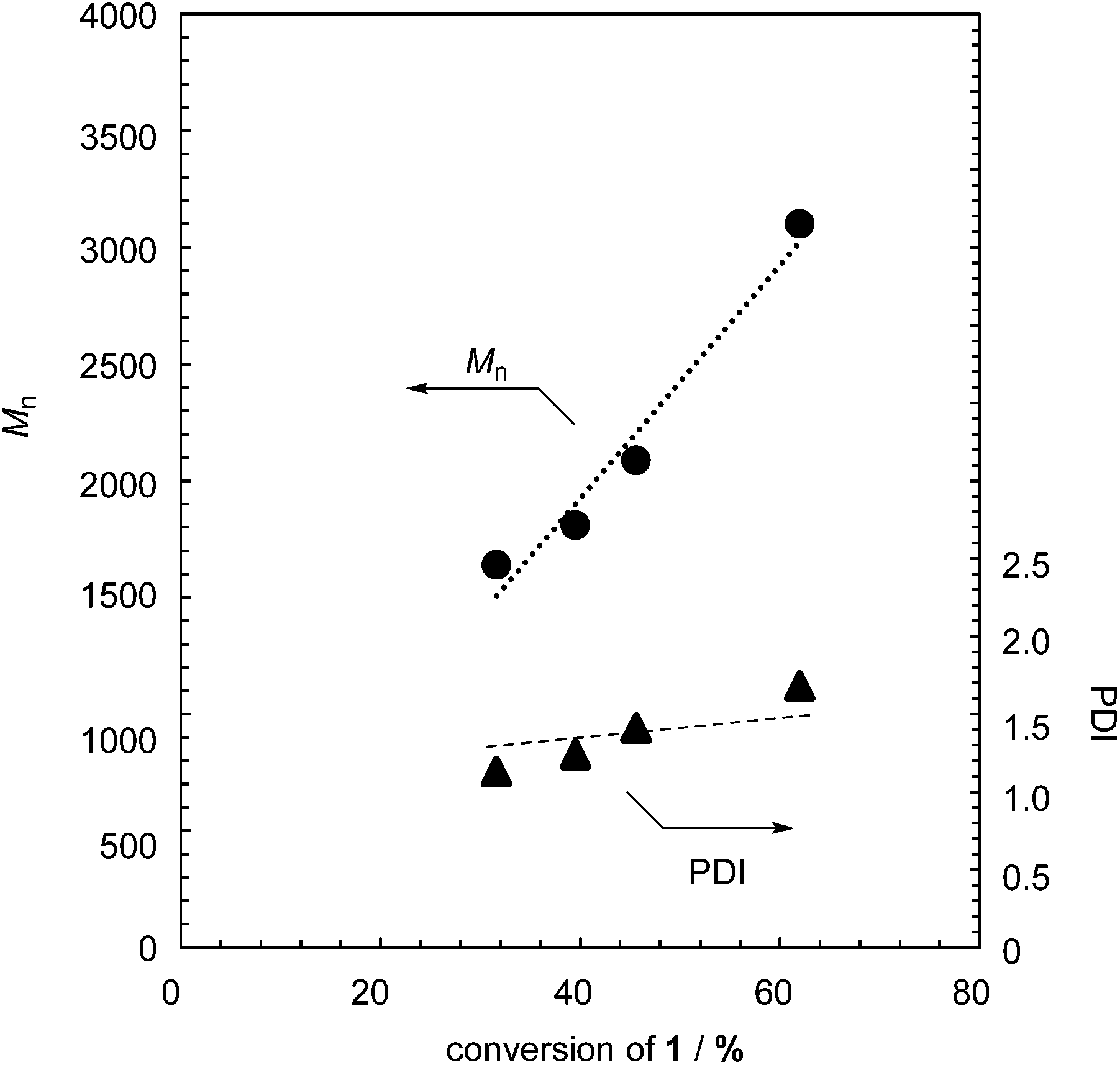 | ||
| Fig. 1 M n and PDI of formed polymer 2 as functions of monomer conversion in the polymerization of 1 with 2.5 mol% TBAF. | ||
Scheme 2 shows a possible polymerization mechanism. In the initial reaction step, a fluoride anion attacks the trimethylsilyl group of 1 to form a pentacoordinate silicate. The silicate is quite reactive and could regioselectively attack the 4-position of the pentafluorophenyl group of 1 to reproduce a fluoride anion. The fluoride anion would then transfer intramolecularly to the trimethylsilylethynyl group at the polymer end, where there may be an anion–π interaction that produces an associated pair.14 Then, the fluoride anion catalyzes the polymerization to give polymer 2. In the polymerization, the reactivity of the trimethylsilyl group and/or the silicate at the polymer end is changed by replacing the 4-position of the pentafluorophenyl group of 1. Watson et al. discussed the change in reactivity in the polymerization of 1,4-bis[(trimethylsilyl)ethynyl]benzene and hexafluorobenzene by substitution with fluoride anions.11 Further description of the polymerization mechanism requires further studies.
 | ||
| Scheme 2 Possible polymerization mechanism of 1. | ||
According to the polymerization mechanism, all of the propagating polymer chains contain reactive pentacoordinate silicate at a chain terminus. Indeed, a pentacoordinated alkynylsilicate, which is prepared in situ by the reaction of 1-(4-trimethylsilyl)phenyl-2-(triethoxysilyl)acetylene 3 and potassium t-butoxide,15 efficiently initiated the polymerization of 1 in the presence of cryptand[2.2.2] to give polymer 4 with a Mn of 12300 and a PDI of 2.08 in 89% yield (Scheme 3).16 The 1H NMR spectrum of 4 shows peaks at around 0.3 and 3.4 ppm arising from the trimethylsilyl and ethynyl groups at the polymer ends, respectively, where the observed integral ratio is in good agreement with the calculated value (calcd for –SiMe3/–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH 9.0, found 8.8) within the experimental errors (Fig. S4 in the ESI†).17
CH 9.0, found 8.8) within the experimental errors (Fig. S4 in the ESI†).17
 | ||
| Scheme 3 Polymerization of 1 with a pentacoordinated alkynylsilicate prepared by the reaction of 3 and potassium t-butoxide. | ||
Next, we demonstrated the anionic “living” character of this polymerization end by applying it to the synthesis of a well-defined block copolymer (Scheme 4). After polymerization of 1 with a catalytic amount of TBAF, part of the mixture was studied to analyze the MW of the first block, and then a solution of 5 was added to the reaction mixture to give block copolymer 6.18 As shown in Fig. 2, the SEC curve of the polymer obtained at the end of the reaction was shifted toward a higher MW compared to that of the first polymerization. The first/second block ratio (n/m) of 6 was found to be 0.98 by 1H NMR analysis (Fig. S6 in the ESI†).
 | ||
| Scheme 4 Synthesis of block copolymer 6. | ||
 | ||
| Fig. 2 SEC curves (RI) obtained for the first polymerization of 1 (polymer 2, dotted line) and following sequential addition of 5 (polymer 6, solid line) after the first polymerization of 1. | ||
We also extended our method to the polymerization of a variety of aromatic monomers, 7–9, which possess trimethylsilylethynyl and pentafluorophenyl groups (Scheme 5). The reaction of 2-pentafluorophenyl-5-[(trimethylsilyl)ethynyl]thiophene 7 with 5 mol% TBAF gave polymer 10 with a Mn of 11200 and a PDI of 2.09 in a moderate yield. Fluorene-incorporated 8 was polymerized by addition of 5 mol% TBAF to give polymer 11 with a Mn of 10600 and a PDI of 1.99 in a good yield. A phenyleneethynylene monomer 9 was also polymerized to afford polymer 12 with a Mn of 11300 and a PDI of 1.85. The structures of these polymers are highly ordered, as demonstrated by NMR analyses (Fig. S7–S9 in the ESI†), indicating that the polymerizations also proceed with high regioselectivities.
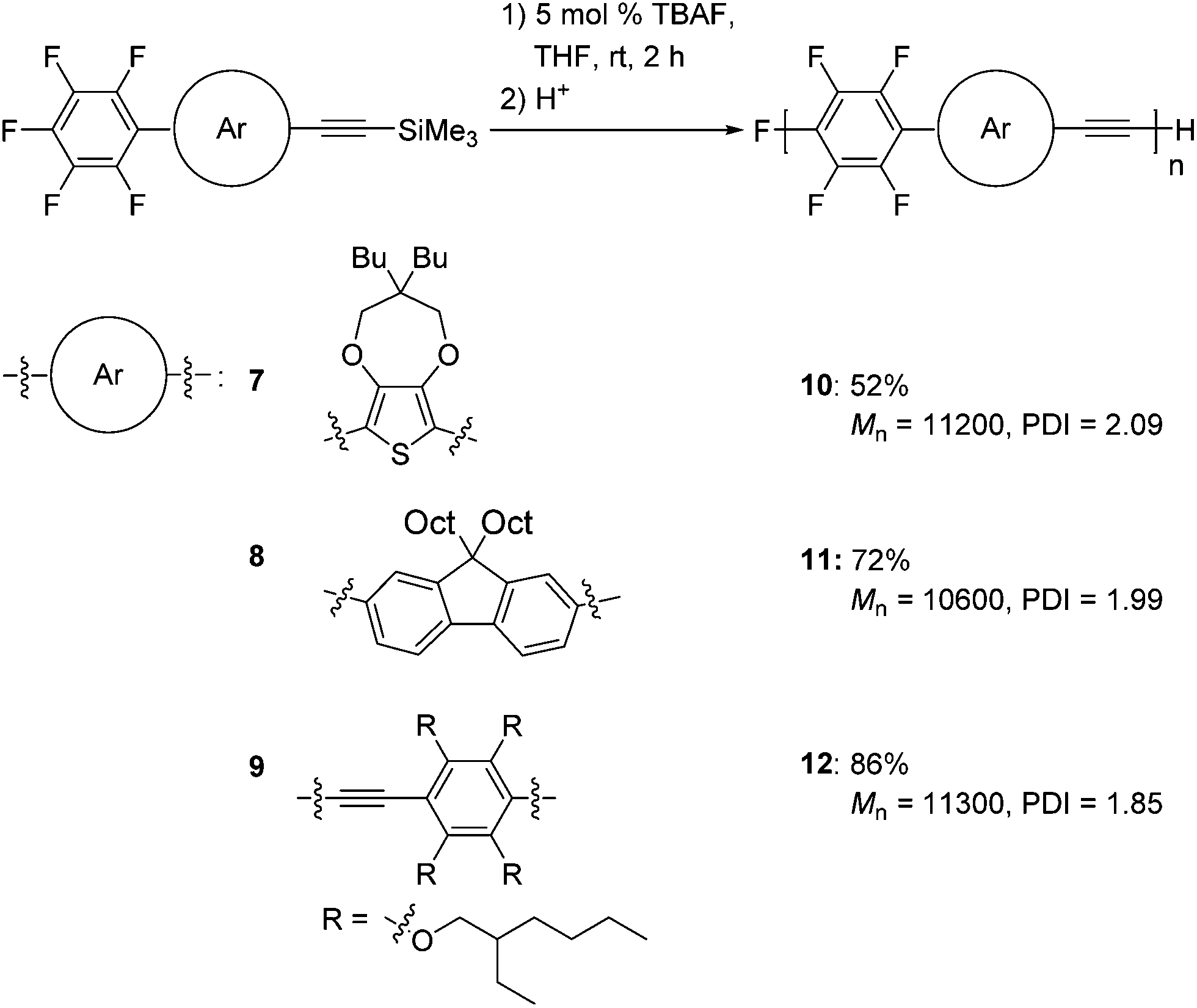 | ||
| Scheme 5 Polymerizations of 7–9. | ||
Next, we turned our attention to exploring the surface-terminated polymerization of 1. Au surfaces of nanometer-sized particles are an ideal substrate for this study because of their well-established chemistry and their features which could be exploited for molecular device applications.19 Surface-terminated polymerization was accomplished by the addition of a surface-enhanced Raman scattering (SERS)-active Au nanoparticle (ϕ 15 nm) array20 to a polymerization mixture of 1 (Scheme 6). After standing for 1 h, the plate was washed with THF and then analyzed by Raman spectroscopy. SEC analysis of the polymerization mixture shows that the resulting polymer, 13, has a Mn of 4700 and a PDI of 1.79. The νCC stretching region in the SERS spectra is particularly informative. The Raman spectrum of 2 shows a signal at around ∼2200 cm−1 owing to the ethynyl groups in the polymer main chain (Fig. 3). On the other hand, in the Raman spectrum of 13, a broad and downshifted signal is observed at 2050 cm−1. The red-shift of ∼150 cm−1 indicates a strong interaction with the Au substrate, which is in agreement with previous reports.21 Thus, the ethynyl group of the polymer end is attached to the gold substrate via covalent Au–CC bonds. Although this is a preliminarily demonstration, it represents a possible step for molecular device applications. Further details will be reported.
 | ||
| Scheme 6 Surface termination with a Au nanoparticle array for the polymerization of 1. | ||
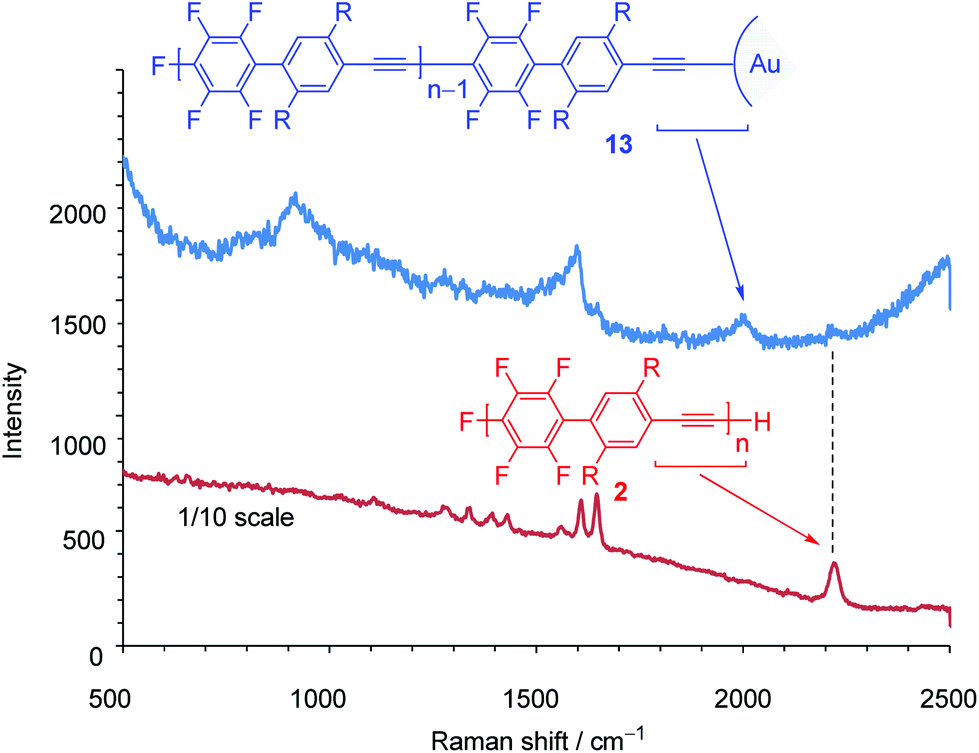 | ||
| Fig. 3 Raman spectra of 2 (film, red) and 13 (blue). | ||
Conclusions
We have developed a transition-metal-free controlled synthesis of PAEs promoted by fluoride anions. The polymerization proceeds in an anionic chain-growth-like manner to afford PAEs with controlled MWs and relatively low PDIs. The polymerization end is active and affords a block copolymer. We also demonstrated the synthesis of a surface-terminated PAE on a Au nanoparticle array. We expect that this concept can be extended to prepare other well-controlled conjugated polymers, providing new opportunities in optoelectronics and other applications. Further study along this line is currently in progress.Notes and references
- (a) A. C. Grimsdale, C. K. Leok, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS PubMed; (b) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed; (c) S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS PubMed; (d) D. L. Allara, J. J. Arnold, L. A. Bumm, T. P. Burgin, M. T. Cygan, T. D. Dunbar, L. Jones II, J. M. Tour and P. S. Weiss, Science, 1996, 271, 1705 Search PubMed.
- (a) U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS PubMed; (b) B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493 CrossRef CAS PubMed.
- U. H. F. Bunz, Acc. Chem. Res., 2001, 34, 998 CrossRef CAS PubMed.
- For recent reviews, see: (a) T. Yokozawa and A. Yokoyama, Chem. Rev., 2009, 109, 5595 CrossRef CAS PubMed; (b) T. Yokozawa and Y. Ohta, Chem. Commun., 2013, 49, 8281 RSC; (c) K. Okamoto and C. K. Luscombe, Polym. Chem., 2011, 2, 2424 RSC.
- J. M. Tour, Chem. Rev., 1996, 96, 537 CrossRef CAS PubMed.
- For reviews, see: (a) C. Chuit, R. J. P. Corriu, C. Reye and J. C. Young, Chem. Rev., 1993, 93, 1371 CrossRef CAS; (b) R. R. Holmes, Chem. Rev., 1996, 96, 927 CrossRef CAS PubMed; (c) G. K. S. Prakash and A. K. Yudin, Chem. Rev., 1997, 97, 757 CrossRef CAS PubMed; (d) M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley-Interscience, New York, 1999 Search PubMed.
- For example: (a) E. Nakamura and I. Kuwajima, Angew. Chem., Int. Ed. Engl., 1976, 15, 498 CrossRef; (b) T. Hiyama, M. Obayashi, I. Mori and H. Nozaki, J. Org. Chem., 1983, 48, 912 CrossRef CAS; (c) S. E. Denmark and J. Fu, J. Am. Chem. Soc., 2001, 123, 9488 CrossRef CAS PubMed; (d) T. Kitazawa, T. Minowa and T. Mukaiyama, Chem. Lett., 2006, 35, 1002 CrossRef CAS.
- (a) G. M. Brooke, J. Fluorine Chem., 1997, 86, 1 CrossRef; (b) F. Terrier, Modern Nucleophilic Aromatic Substitution, Wiley-VCH, Weinheim, 2013 Search PubMed.
- (a) J.-P. Kim, W.-Y. Lee, J.-W. Kang, S.-K. Kwon, J.-J. Kim and J.-S. Lee, Macromolecules, 2001, 34, 7817 CrossRef CAS; (b) K. B. Woody, J. E. Bullock, S. R. Parkin and M. D. Watson, Macromolecules, 2007, 40, 4470 CrossRef CAS; (c) P. A. Deck and C. R. Maiorana, Macromolecules, 2001, 34, 9 CrossRef CAS; (d) Y. Wang and M. D. Watson, J. Am. Chem. Soc., 2006, 128, 2536 CrossRef CAS PubMed.
- T. Sanji and T. Iyoda, J. Am. Chem. Soc., 2014, 136, 10238 CrossRef CAS PubMed.
- (a) T. Dutta, K. B. Woody and M. D. Watson, J. Am. Chem. Soc., 2008, 130, 452 CrossRef CAS PubMed; (b) T. Dutta, K. B. Woody, S. R. Parkin, M. D. Watson and J. Gierschner, J. Am. Chem. Soc., 2009, 131, 17321 CrossRef CAS PubMed.
- S. Kang, S. R. J. Ono and C. W. Bielawski, J. Am. Chem. Soc., 2013, 135, 4984 CrossRef CAS PubMed.
- A THF solution of TBAF (1 M) containing 5 wt% H2O was used in this study.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2002, 41, 3389 CrossRef.
- R. B. Lattan II and K. A. Scheidt, Org. Lett., 2005, 7, 3227 CrossRef PubMed.
- When pentacoordinated trimethyl silicates, which were prepared in situ by the reaction of 1-phenyl-2-(trimethylsilyl)acetylene with potassium t-butoxide or TBAF as a Lewis base, were used as an initiator, the polymerizations were not initiated effectively at the initiator (the introduction ratio of the initiator unit ≈10–50%, see Fig. S5 in the ESI†). This is because the reaction of the trimethylsilyl groups of the 1-phenyl-2-(trimethylsilyl)acetylene and 1 with the Lewis bases is in equilibrium and the initiation did not occur preferentially at the initiator units.
- In the 19F NMR, however, besides the two strong signals observed for the 1,4-tetrafluorophenylene units in the main chain, three weak signals which are assignable to the pentafluorophenyl group at the polymer end were found. This indicates that not all of the polymerizations were initiated at the initiator, but that the introduction ratio of the initiator unit is ≈80% (see ESI†).
- The polymerization of 5 with 5 mol% TBAF gave a polymer with a Mn of 4000 and a PDI of 1.42 in a 60% yield.
- (a) L. Sun, Y. A. Diaz-Fernandez, T. A. Gschneidtner, F. Westerlund, S. Lara-Avila and K. Moth-Poulsen, Chem. Soc. Rev., 2014, 43, 7378 RSC; (b) M. A. Reed, C. Zhou, C. J. Muller, T. P. Burgin and J. M. Tour, Science, 1997, 278, 252 CrossRef CAS.
- (a) W. Zou, Y. Wang, Z. Wang, A. Zhou, J. Li, A. Chang, Q. Wang, M. Komura, K. Ito and T. Iyoda, Nanotechnology, 2011, 22, 335301 CrossRef PubMed; (b) S. Hadano, H. Handa, K. Nagai, T. Iyoda, J. Li and S. Watanabe, Chem. Lett., 2013, 42, 71 CrossRef CAS.
- W. Hong, H. Li, S. X. Liu, Y. Fu, J. Li, V. Kaliginedi, S. Decurtins and T. Wandlowski, J. Am. Chem. Soc., 2012, 134, 19425 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterizations of new compounds and polymers. See DOI: 10.1039/c4sc02872d |
| This journal is © The Royal Society of Chemistry 2015 |