
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal complexes as potential modulators of inflammatory and autoimmune responses
Chung-Hang
Leung
*a,
Sheng
Lin
b,
Hai-Jing
Zhong
a and
Dik-Lung
Ma
*b
aState Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macao, China. E-mail: duncanleung@umac.mo
bDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong, China. E-mail: edmondma@hkbu.edu.hk
First published on 7th November 2014
Abstract
Over the past few decades, the realm of inorganic medicinal chemistry has been dominated by the study of the anti-cancer properties of transition metal complexes, particularly those based on platinum or ruthenium. However, comparatively less attention has been focused on the development of metal complexes for the treatment of inflammatory or autoimmune diseases. Metal complexes possess a number of advantages that render them as attractive alternatives to organic small molecules for the development of therapeutic agents. In this perspective, we highlight recent examples in the development of transition metal complexes as modulators of inflammatory and autoimmune responses. The studies presented here serve to highlight the potential of transition metal complexes in modulating inflammatory or immune pathways in cells.
 Chung-Hang Leung | Chung-Hang Leung completed his PhD in 2002 at the City University of Hong Kong. After completing a five-year postdoctoral fellowship at the Department of Pharmacology, Yale University, he was appointed Research Assistant Professor at the University of Hong Kong and then at the Hong Kong Baptist University. He is currently an Associate Professor at the University of Macau. His primary research interests are in structure-based drug discovery and in the development of oligonucleotide-based drug screening methods. |
 Sheng Lin | Sheng Lin completed his BSc degree in Biotechnology at Beijing University of Chemical Technology, Beijing, China. He is currently undertaking his MPhil studies at the Department of Chemistry at the Hong Kong Baptist University under the supervision of Dr Dik-Lung Ma and Dr Chung-Hang Leung. His research interests include the development of luminescent oligonucleotide-based assays for biomolecules and metal ions. |
 Hai-Jing Zhong | Hai-Jing Zhong completed her BSc degree in 2011 at Jinan University, Guangzhou, China. She is currently undertaking her PhD studies at the University of Macau under the supervision of Dr Chung-Hang Leung and Dr Dik-Lung Ma. Her research interest lies in the development of screening platforms for drug discovery and development. |
 Dik-Lung Ma | Dik-Lung Ma completed his PhD in 2004 at the University of Hong Kong under the supervision of Prof. C.-M. Che. Between 2005 and 2009 he worked at the University of Hong Kong, the Hong Kong Polytechnic University, and The Scripps Research Institute with Prof. C.-M. Che, Prof. K.-Y. Wong, and Prof. R. Abagyan. His research mainly focuses on the luminescent sensing of biomolecules and metal ions, computer-aided drug discovery, and inorganic medicine. In 2010 he was appointed Assistant Professor at the Hong Kong Baptist University. |
1 Introduction
The past few decades have witnessed the emergence of inorganic medicinal chemistry as an independent field. Leading the forefront of this movement has been the identification and development of platinum anti-cancer drugs for the treatment of various types of cancer. For example, the introduction of cisplatin 1 (Fig. 1) as the frontline treatment for testicular cancer has seen survival rates for this previously deadly disease exceed 90%.1 More recently, transition metal complexes based on other metal ions such as ruthenium, iridium and rhodium have also attracted attention as anti-cancer agents. Notable examples of ruthenium agents that have reached clinical trials include the anti-metastatic agent indazolium trans-[tetrachloro-(S-dimethyl sulfoxide)(1H-imidazole)ruthenate(III)] (NAMI-A) 2 and the anti-neoplastic agent imidazolium trans-[tetrachlorobis(1H-indazole)-ruthenate(III)] (KP-1019) 3.2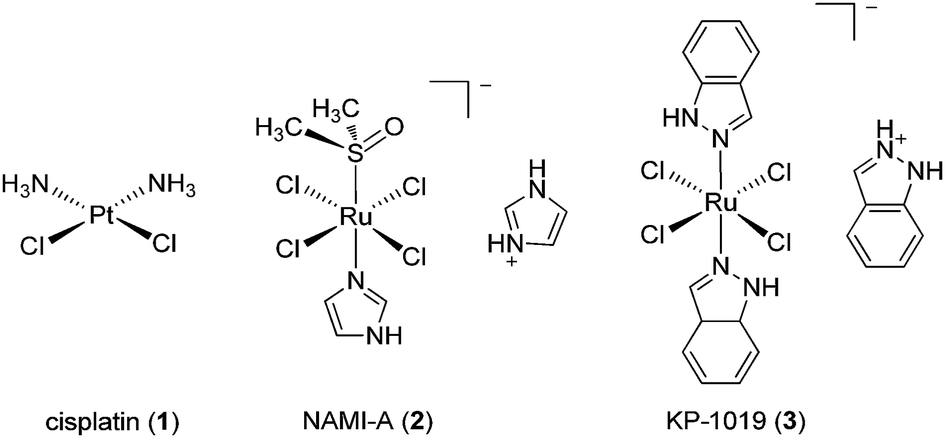 | ||
| Fig. 1 Chemical structures of cisplatin 1, NAMI-A 2 and KP-1019 3. | ||
Given the revolutionary success of cisplatin and its analogues, it is unsurprising that the field of inorganic medicinal chemistry has been dominated by studies on the anti-cancer activity of metal complexes. Recent review articles describing the application of transition metal complexes as anti-cancer agents have been published by leaders in the field, including Sadler,3 Meggers,4 Che,5 Barton,6 Alessio,7 Ott,8 Dyson,9–11 Sheldrick12 and others.13–19 However, relatively less attention has been devoted to the development of metal complexes for anti-inflammatory or anti-autoimmune applications. Auranofin 4 [triethylphosphine-(2,3,4,6-tetra-O-acetyl-β-D-thiopyranosato)-gold(I)], solganal/aurothioglucose [{(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)-oxane-2-thiolato}gold(I)] 5 and myochrysine/sodium aurothiomalate [sodium((2-carboxy-1-carboxylatoethyl)thiolato)gold(I), GST] 6 (Fig. 2) are gold compounds that are used for the treatment of rheumatoid arthritis. However, to our knowledge, no other metal-based drug has been approved for the treatment of inflammatory or autoimmune diseases.
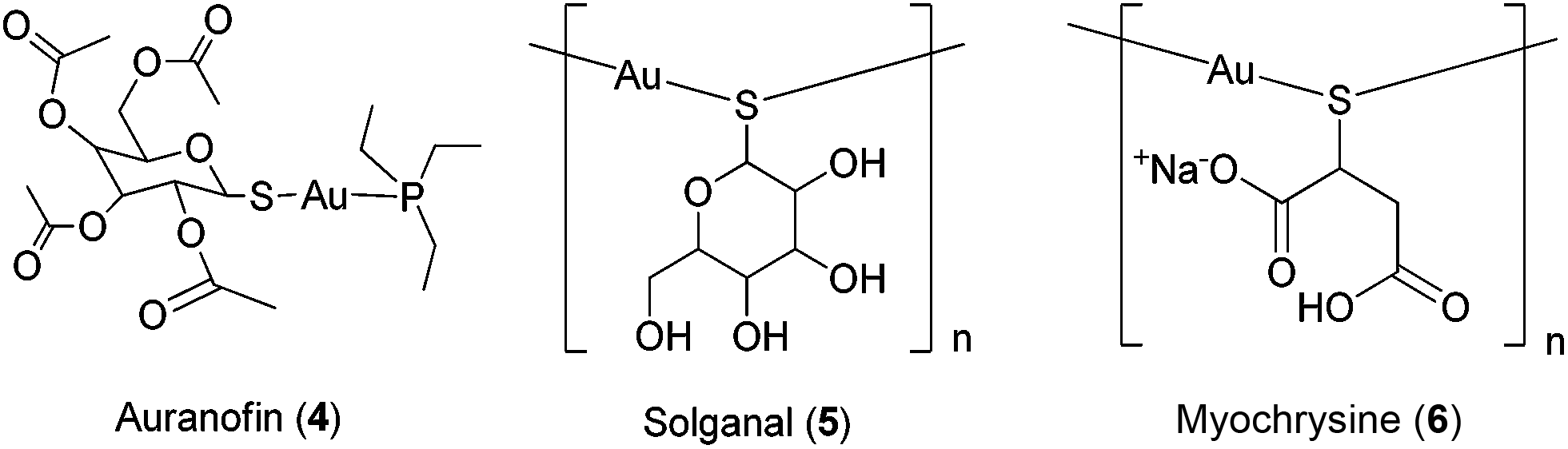 | ||
| Fig. 2 Chemical structures of auranofin 4, solganal 5 and myochrysine 6. | ||
Metal complexes possess several notable advantages that render them as attractive alternatives to organic small molecules for the development of therapeutic agents. The basic structure of a transition metal complex consists of a central metal ion bonded via dative covalent bonds to one or more organic or inorganic ligands. Metal complexes can adopt numerous geometries, including square-planar, square-pyramidal, trigonal-bipyramidal and octahedral, depending on the coordination number of the metal ion. Notably, many of these geometries are unavailable to purely organic molecules, which are limited to linear, trigonal planar or tetrahedral shapes because carbon cannot normally exceed a coordination number of 4. Extraordinarily, an octahedral metal complex that possesses six different ligands can adopt up to 30 different stereoisomeric configurations, whereas a tetrahedral carbon atom with four different groups generates only a maximum of two enantiomers. The diversity of molecular architectures afforded by transition metal complexes may therefore allow them to sample regions of the chemical space that are inaccessible to organic molecules. Additionally, the nature of the auxiliary ligands has a large influence on the thermodynamic and kinetic properties of the metal complexes. Together with the modular nature of inorganic synthesis, the pharmacological properties of metal complexes can often be readily fine-tuned by adjustment of the attached ligands without the need for lengthy synthetic protocols. Finally, metal complexes can undergo redox reactions and ligand-exchange reactions inside the body, allowing for unique mechanisms of action to take place. For example, the high oral bioavailability of satraplatin (bis-(acetato)-ammine dichloro-(cyclohexylamine)platinum(IV)) compared to classical platinum anti-cancer drugs has been attributed to the presence of its two acetate groups. Once inside the body, satraplatin is reduced to Pt(II) and behaves analogously with cisplatin, undergoing ligand-exchange reactions with nucleobases and generating intrastrand and interstrand DNA cross-links.20
This perspective aims to survey examples of metal complexes that been investigated for anti-inflammatory or anti-autoimmune activity that have been developed within the past five years. For ease of access, the examples chosen have been classified by the type of mechanism displayed: (i) metal complexes of anti-inflammatory compounds, (ii) metal complexes that covalently bind biomolecules, and (iii) kinetically inert metal complexes. This review intends not to be exhaustive, but aims to highlight some of the more interesting examples in this field that have been reported in the last few years.
2 Metal complexes of anti-inflammatory compounds
The coordination of bioactive molecules to metal ions is a common strategy to improve the therapeutic potency and/or to reduce the toxicity of drug molecules. The resulting metal complexes frequently possess superior lipophilicity profiles compared to the free ligands, allowing them to more easily pass through cell membranes to exert their biological effects. In an example from antibiotic drug discovery, a Pt(II) tetracycline complex was six times more potent against Escherichia coli compared to the free ligand.21 The following examples highlight studies in which bioactive ligands have been tethered to metal ions, generating metal complexes with anti-inflammatory and other biological activities.The anti-inflammatory effects of the classical non-steroidal anti-inflammatory drug (NSAID) aspirin (asp) 7 (Fig. 3) are achieved by inhibiting prostaglandin (PG) synthesis, via the irreversible inhibition of cyclo-oxygenases (COX). COX-1 is present in most tissues, including in the gastrointestinal tract where it plays an important role in maintaining the stomach lining, whereas COX-2 is primarily present at sites of inflammation.22 Therefore, COX-2-selective inhibitors such as celecoxib and rofecoxib are associated with less gastric irritation and ulceration.23,24
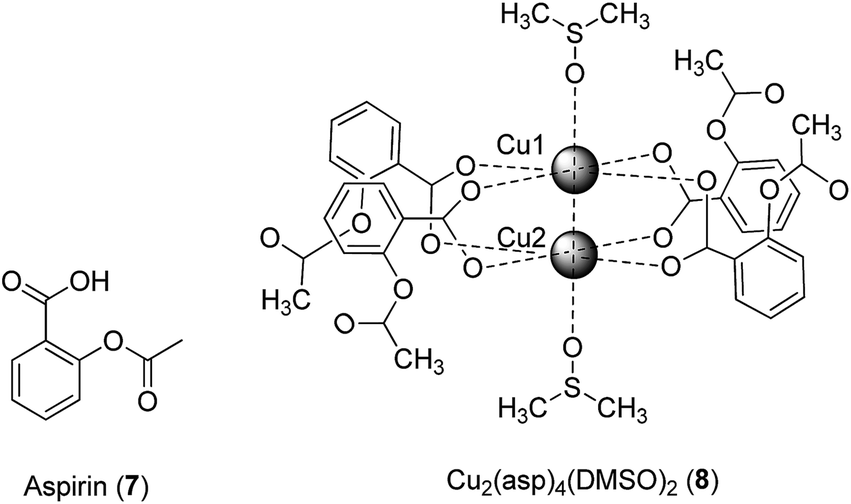 | ||
| Fig. 3 Chemical structure of aspirin 7 and crystal structure of Cu2(asp)4(DMSO)28. | ||
Previous studies have indicated that the copper–aspirin complex tetrakis-μ-acetylsalicylato-dicopper(II) (Cu–aspirin) exhibited more potent anti-inflammatory activity than aspirin 7 in rats or mice, with fewer adverse effects.25 The crystal structure of Cu2(asp)4 has been previously reported,26 though in aqueous solution the compound will most likely coordinate to water in a manner similar to that observed in the crystal structure of Cu2(asp)4(DMSO)28.27 To investigate the basis of the enhanced efficacy of Cu–aspirin, Shen and co-workers evaluated the selectivity of Cu–aspirin for COX-1 and COX-2.28 The results revealed that Cu–aspirin exhibited weaker inhibition of COX-1 in resting human umbilical vein endothelial cells compared to aspirin 7. In contrast, Cu–aspirin displayed significantly stronger inhibition of COX-2-mediated prostaglandin E2 (PGE2) synthesis in activated macrophages compared to aspirin 7. The selectivity index of Cu–aspirin for COX-2 over COX-1 was 3.33 compared to 0.42 for aspirin 7, indicating that Cu–aspirin was ca. 7-fold more selective for COX-2 compared to aspirin 7. This significant selectivity may be attributed to the steric properties of Cu–aspirin when it interacts with COX-1 and COX-2. Being a small molecule, aspirin 7 is able to fit into the pockets of both COX-1 and COX-2 to acetylate Ser530, leading to pan-COX inhibition. On the other hand, the COX-2-selective inhibitors celecoxib and rofecoxib possess a large sulfanilamide side chain, and are therefore unable to enter the side pocket of COX-1. The Cu–aspirin complex, being far larger than aspirin 7 alone, is likely to exhibit selectivity for COX-2 over COX-1 for the same reason. In a control experiment, the Cu–asp complex was found to be more potent against COX-2 compared to a mixture of CuSO4 and aspirin, with CuSO4 alone having no activity, indicating that the COX-2 inhibitory activity of asp was more than a simple cumulative effect of the drug and the metal ion. Moreover, the mixture of CuSO4 and aspirin showed improved COX-2 inhibitory activity compared with aspirin alone, suggesting that some Cu–asp could have been formed in situ.
Kale and co-workers investigated the effect of zinc complexation on the anti-inflammatory activity of the non-steroidal anti-inflammatory drug (NSAID) aceclofenac 9 (Fig. 4).29 Like a number of other NSAIDs with similar side effects, aceclofenac 9 promotes the formation of stomach ulcers with the mechanism of association in part due to its carboxylic acid functionality. In the stomach, aceclofenac 9 exists in its neutral form, and is able to pass into the cells of the gastric mucosa. However, the higher pH of the cells promotes in the intracellular dissociation of the acid, resulting in a constant backflow of protons into these cells with associated cellular injury. In the study, the authors hypothesized that the carboxyl group in aceclofenac 9 could be effectively masked by coordination to zinc. The zinc complex was more stable in HCl (pH 1.2) than in phosphate buffer (pH 7.4), suggesting that it would remain stable in the stomach. The results revealed that aceclofenac 9 and its corresponding zinc complex showed similar abilities to reduce inflammatory edema in the paws of rats challenged by carrageenan. Importantly, however, the zinc–aceclofenac complex induced fewer ulcers in rat stomachs compared to the parent drug 9. This suggests that the complexation of NSAIDs with zinc may be an effective strategy for limiting the adverse gastrointestinal side effects of these agents. The structure of the zinc complex was characterized by elemental analysis, but not by mass spectrometry or NMR spectrometry. While the authors proposed a two-coordinate zinc ion in the structure of the zinc–aceclofenac complex, we think it more likely that the zinc ion will adopt an octahedral geometry in aqueous solution, with two bidentate aceclofenac ligands and two water ligands.
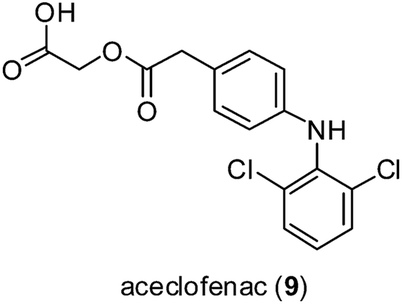 | ||
| Fig. 4 Chemical structure of the NSAID aceclofenac 9. | ||
Co(II) complexes bearing the NSAID mefenamic acid ligand have also been investigated as potential anti-inflammatory agents (Fig. 5).30 Physicochemical and spectroscopic data suggested that mefenamic acid acted as a deprotonated monodentate ligand, which coordinated to the Co(II) ion through its carboxylato oxygen atom, forming octahedral [Co(mef)2(MeOH)4] 10 or [Co(mef)2(MeOH)2(N^N)] 11a–c (where mef = mefenamic acid and N^N = 2,2′-bipyridine, 1,10-phenanthroline or (pyridine)2) complexes. In this study, the free radical scavenging abilities of the complexes against 2,2-diphenylpicrylhydrazyl (DPPH), 2,2′-azinobis [3-ethylbenzothiazoline-6-sulfonicacid]-diammonium (ABTS) and hydroxyl radicals were investigated. All of the complexes showed stronger DPPH free radical scavenging activity than mefenamic acid. [Co(mef)2(MeOH)2(phen)] 11b was the only complex to exhibit a lower scavenging activity than the parent ligand against hydroxyl radicals, whereas [Co(mef)2(MeOH)2(py)2] 11c showed the highest scavenging activity against both hydroxyl and ABTS radicals. The distorted octahedral geometry of the complexes was confirmed by X-ray crystallography, while EPR studies suggested that the complexes retain their structures in frozen solution. However, it is likely that the methanol ligands will be replaced by water in an aqueous environment. In later studies, research groups also reported Cu(II) complexes of mefenamic acid,31 naproxen, diclofenac,31 diflunisal,32 and flufenamic acid,33 Co(II) complexes of naproxen34 and tolfenamic acid,35 and Mn(II) complexes of tolfenamic acid36 that showed anti-inflammatory activity.
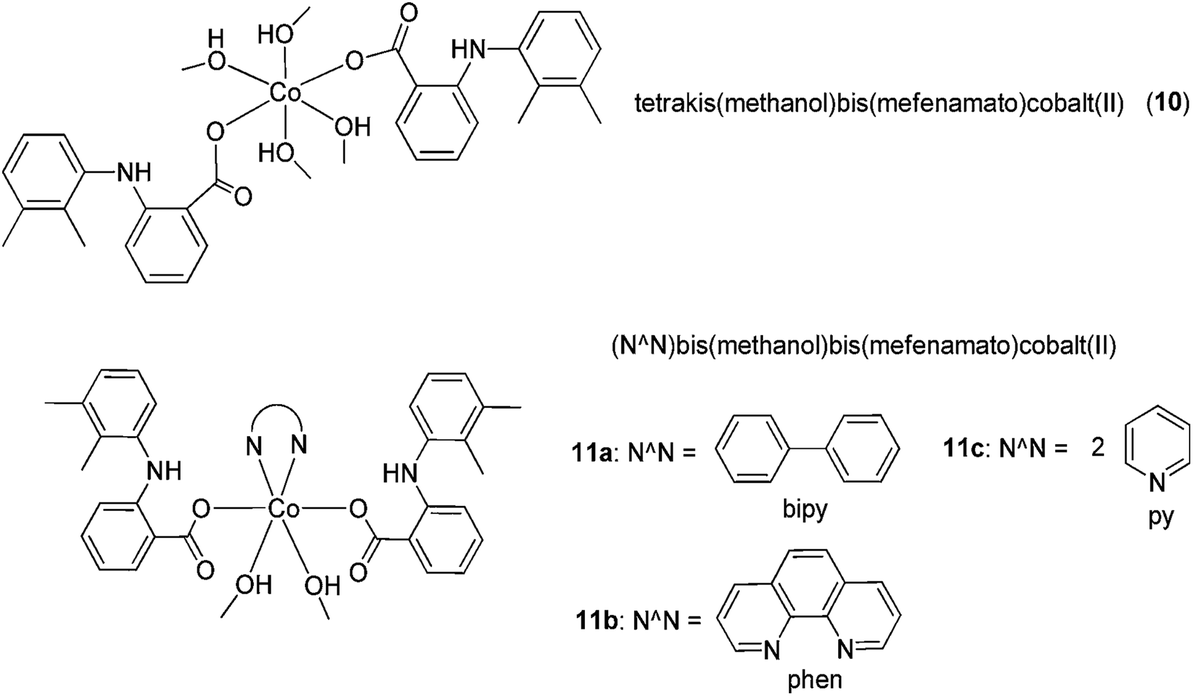 | ||
| Fig. 5 Chemical structures of [Co(mef)2(MeOH)4] 10 and [Co(mef)2(MeOH)2(N^N)] 11a–c (where mef = mefenamic acid and N^N = 2,2′-bipyridine, 1,10-phenanthroline or (pyridine)2) complexes. | ||
Anthranilic acid is a core precursor to a number of anti-inflammatory agents, such as mefenamic acid.37 From 2013, Iqbal and co-workers evaluated the anti-inflammatory and COX inhibitory activities of Mn(II) (12), Fe(II) (13), Co(II) (14), Ni(II) (15) and Zn(II) (16) complexes containing Schiff base ligands derived from anthranilic acid and aldoses (Fig. 6).38 The metal ions formed four-coordinate, ML2-type complexes with the bidentate Schiff base ligands, with spectral and magnetic data indicating a tetrahedral geometry for Mn(II) and Fe(II) complexes, and a square-planar geometry for the Co(II), Ni(II) and Zn(II) complexes. Oral administration of the Mn(II) and Zn(II) complexes reduced kaolin-induced paw edema in rats, indicating that the complexes possessed anti-inflammatory activity. Furthermore, all of the complexes displayed COX-2/COX-1 selectivity index values of 0.34–0.52, which were similar to that of aspirin 7 (0.41). Unfortunately, the ligands were found to be unstable and could not be isolated in the free state. Additionally, the stability of the complexes was not reported.
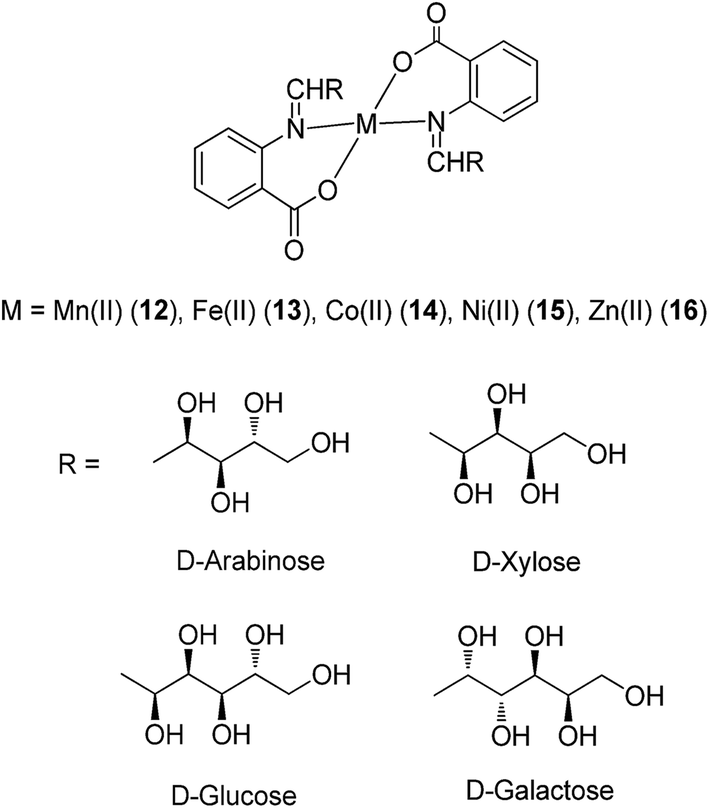 | ||
| Fig. 6 Chemical structures of the metal complexes 12–16 containing Schiff base ligands derived from anthranilic acid and aldoses. | ||
In 2011, Beraldo and co-workers reported the anti-inflammatory activities of salicylaldehyde benzoyl hydrazones 17 and 18 and their dimeric Zn(II) complexes (Fig. 7).39 The structures of the Zn(II) complexes were characterized by elemental analysis, but not by mass spectrometry. All compounds showed the ability to inhibit the migration of monocytes and neutrophils into the peritoneal cavity induced by zymosan with comparable or superior potency compared to indomethacin, a COX-1 selective inhibitor. Furthermore, the ability of the complexes to reduce formalin-induced pain in mice was investigated. Formalin-induced pain can be divided into two phases, known as the neurogenic (first) and inflammatory (second) phases of the nociceptive response. The results showed that all four compounds reduced the sensitivity of mice to pain induced by formalin. Interestingly, the analgesic ability of the 2-benzoyl derivative H2LASSBio-466 17 was enhanced upon complexation with Zn(II), whereas the reverse was true for the 4-benzyol derivative H2LASSBio-1064 18. H2LASSBio-466 17 was only able to inhibit the neurogenic phase of the formalin-induced nociceptive response, whereas its Zn(II) complex showed activity in both the first and second phases, indicating the potential of the complex to inhibit inflammatory pain. On the other hand, H2LASSBio-1064 18 was active in both neurogenic and inflammatory phases, but its effects were diminished upon complexation with Zn(II). The authors postulated that the different potencies of the Zn(II) complexes could be attributed to differences in the rate of the release of hydrazone. Alternatively, as [Zn(LASSBio-466)OH2]2 contains a di-anionic hydrazone moiety, whereas [Zn(HLASSBio-1064)Cl]2 contains a mono-anionic hydrazone moiety, the different electronic effects of the complexes could also affect their bioavailability profiles in different ways. However, the pharmacological targets of the complexes remain unknown.
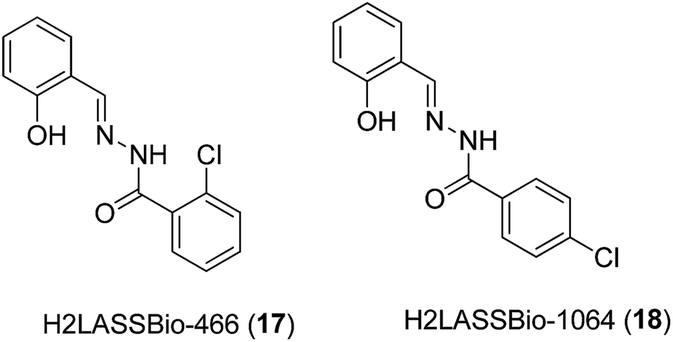 | ||
| Fig. 7 Chemical structures of the salicylaldehyde benzoyl hydrazones 17 and 18. | ||
Recently, Ali and co-workers investigated the anti-inflammatory properties of Ni(II) (19), Zn(II) (20) and Co(II) (21) metal complexes bearing Schiff base ligands derived from salicylaldehyde and glycine (Fig. 8).40 Oral administration of the complexes reduced inflammatory edema in rats challenged by carrageenan, with greater potency compared to the NSAID diclofenac. The ability of the compounds to inhibit paw edema in the second phase of inflammation (mediated by prostaglandin release) suggested that the compounds may possibly inhibit COX, in a manner similar to indomethacin. The finding demonstrated that the compounds showed moderate analgesic activity against acetic acid or thermal injury, but with lower potency compared to diclofenac. However, whether the peripheral anti-nociceptive activity of the compounds was mediated by interfering with the local reactions induced by the irritant or by inhibiting the synthesis, release and/or antagonizing the activity of pain regulators at the peripheral target sites was undetermined.
 | ||
| Fig. 8 Chemical structures of the transition metal complexes 19–21 bearing Schiff base ligands derived from salicylaldehyde and glycine. | ||
Naik and co-workers have investigated the anti-inflammatory activity of a series of Schiff bases derived from 2-mercapto-3-formyl quinoline/2-hydroxy-3-formyl quinoline with 2,6-diaminopyridine (DAP) and their corresponding Co(II) (22, 26), Ni(II) (23, 27), Cu(II) (24, 28) and Zn(II) (25, 29) complexes (Fig. 9).41 Most of the complexes significantly reduced inflammatory edema in rats challenged by carrageenan. Among the compounds tested, the Cu(II) complexes 24 and 28 showed the highest biological activities. Moreover, the complexes did not induce ataxia, tremors, convulsions, sedation, lacrimation or changes in motor activity in mice, and caused no significant toxicity to the stomach, intestines and liver of mice.
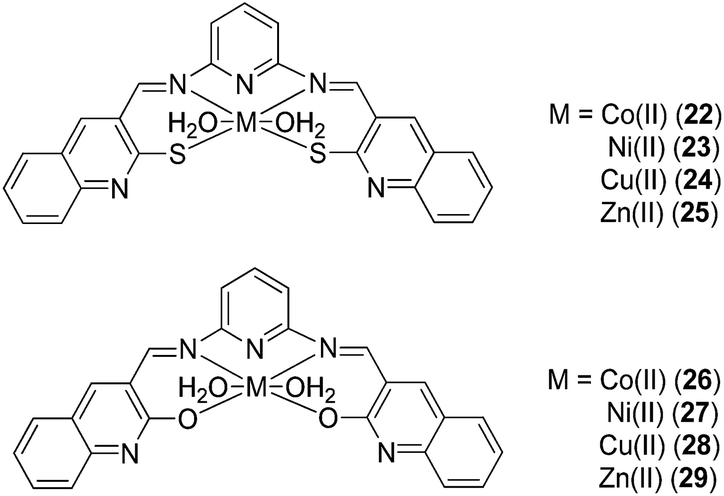 | ||
| Fig. 9 Chemical structures of the Co(II), Ni(II), Cu(II) and Zn(II) complexes 22–29 with Schiff bases derived from 2-mercapto-3-formyl quinoline or 2-hydroxy-3-formyl quinoline with 2,6-diaminopyridine (DAP). | ||
A series of Cu(II), Ni(II) and Co(II) metal complexes with the tridentate Schiff base ligand 2,4-bis(indolin-3-one-2-ylimino)-6-phenyl-1,3,5-triazine (BIPTZ) 30 (Fig. 10) has recently been reported by Tharmaraj and co-workers.42 Metal complexes were observed to adopt a square-pyramidal geometry for the [ML]-type complexes and an octahedral geometry for [ML2]-type complexes by electronic absorption spectra and magnetic susceptibility. Moreover, metal complexes and the free ligand were able to reduce inflammatory paw edema volume in rats challenged by carrageenan, indicating that they possessed anti-inflammatory activity.
A range of germ-line-encoded receptors, termed pattern-recognition receptors (PRRs), respond to conserved signatures on invading microorganisms, resulting in the transcriptional activation of pro-inflammatory responses.43 PRRs include the Toll-like receptors (TLRs), C-type lectins (CTLs), cytoplasmic RI GI-like receptors (RLRs), and nucleotide binding domain, leucine-rich repeat-containing family proteins (NLRs), which include the nucleotide-binding oligomerization domain (NOD) proteins NOD1 and NOD2. NOD2 is important in triggering inflammatory responses to bacterial infection because it recognizes muramyl dipeptide (MDP), which is common to all bacterial peptidoglycans. In 2010, Schmalz, Kufer and co-workers investigated the application of chromium-containing arene (arene–Cr(CO)3) complexes 31a–d (Fig. 11) as specific inhibitors of NOD2 signaling.44 The complexes contained an arene ligand that was structurally related to anti-inflammatory diterpenes produced by the sea whip Pseudopterogorgia elisabethae.45 Several of the Cr(0) complexes specifically interfered with NOD1/2-mediated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling in human embryonic kidney (HEK293T) cells and endogenous NOD activity in human acute monocytic leukemia (THP1) myeloid cells. The most active compound 31a probably inhibited NOD2 activation directly in a competitive manner. Further evidence for this mechanism stemmed from the observation that while 31a reduced NOD2/MDP-induced NF-κB activation, it did not inhibit NF-κB activation driven by overexpression of receptor-interacting protein kinase 2, which is a signaling molecule located directly downstream of NOD2 in the signaling cascade. Structure–activity analysis of a number of chromium derivatives revealed the structural features required for NOD2 specificity. The most efficacious compound 31a, specifically antagonized MDP-mediated NF-κB activation without inhibiting TLR2-, TLR4- or tumor necrosis factor (TNF)-mediated NF-κB activation. However, complexes that lacked a benzylamino or benzyloxy substituent at the benzylic position (C3) (e.g.31b and 31c) or a 2-methyl-2-propenyl side chain at C7 (e.g. 31d) were effective against all the tested pathways, indicating that those structures were important for NOD2 specificity. Additionally, the free ligand exhibited pronounced cytotoxicity but did not exhibit significant activity against NOD2- or TLR4-mediated NF-κB activation, demonstrating the importance of the Cr(0) metal center in determining biological activity. However, the precise mechanism by which the Cr(0) complexes antagonized NOD2 was not determined.
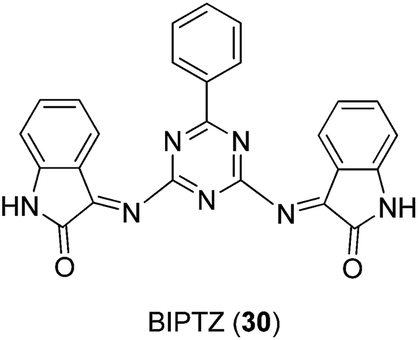 | ||
| Fig. 10 Chemical structure of BIPTZ 30. | ||
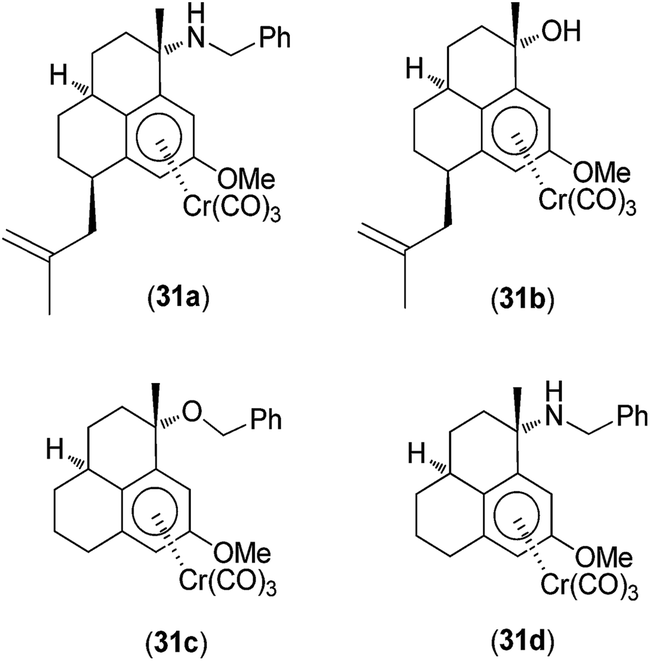 | ||
| Fig. 11 Chemical structures of the chromium-containing arene (arene–Cr(CO)3) complexes 31a–d. | ||
Gudasi and co-workers investigated the anti-inflammatory properties of Cu(II) (32), Zn(II) (33), Mn(II) (34) and Ni(II) (35) complexes of a 1,2-dihydroquinazolinone derivative (Fig. 12).46 Pyridine-2-ethyl-(3-carboxylideneamine)-3-(2-phenyl)-1,2-dihydroquinazolin-4(3H)-one behaves as a neutral, tridentate ligand, generating five-coordinated transition metal complexes with metal ions. The complexes exhibited a dose-dependent reduction of edema volume in rats challenged with carrageenan, with the Cu(II) complex 32 again exhibiting the most potent anti-inflammatory activity. Significantly, the anti-inflammatory activities of the metal complexes were superior to that of the parent ligand, which was attributed to their enhanced lipophilicities. Enoxacin (eno) 36 is a potent inhibitor of the bacterial enzyme DNA gyrase that exhibits anti-bacterial activity against both Gram-negative and Gram-positive bacteria and reduces gingival inflammation.47 In 2009, Arayne and co-workers investigated the anti-inflammatory properties of Cu(II) (37), Ni(II) (38), Mn(II) (39) or Fe(III) (40) complexes of enoxacin 36 (Fig. 13).48 Infrared spectroscopic analysis suggested that enoxacin 36 acted as a monoanionic bidentate ligand and coordinated to the metal ions via its carboxyl and carbonyl groups. The anti-inflammatory properties of the enoxacin complexes 37–39 were evaluated by measuring the levels of reactive free radicals released by activated phagocytic cells. The Mn(II) (39) and Cu(II) (37) enoxacin complexes were found to be the most active against free radical release, with IC50 values of 15.3 and 18.7 μg mL−1, respectively, whereas enoxacin and its Fe(III) (40) and Ni(II) (38) complexes were less potent (IC50 > 50 μg mL−1). However, the molecular mechanism governing the immunomodulatory effect of the enoxacin complexes was not determined.
 | ||
| Fig. 12 Chemical structures of the transition metal complexes 32–35 of pyridine-2-ethyl-(3-carboxylideneamine)-3-(2-phenyl)-1,2-dihydroquinazolin-4(3H)-one. | ||
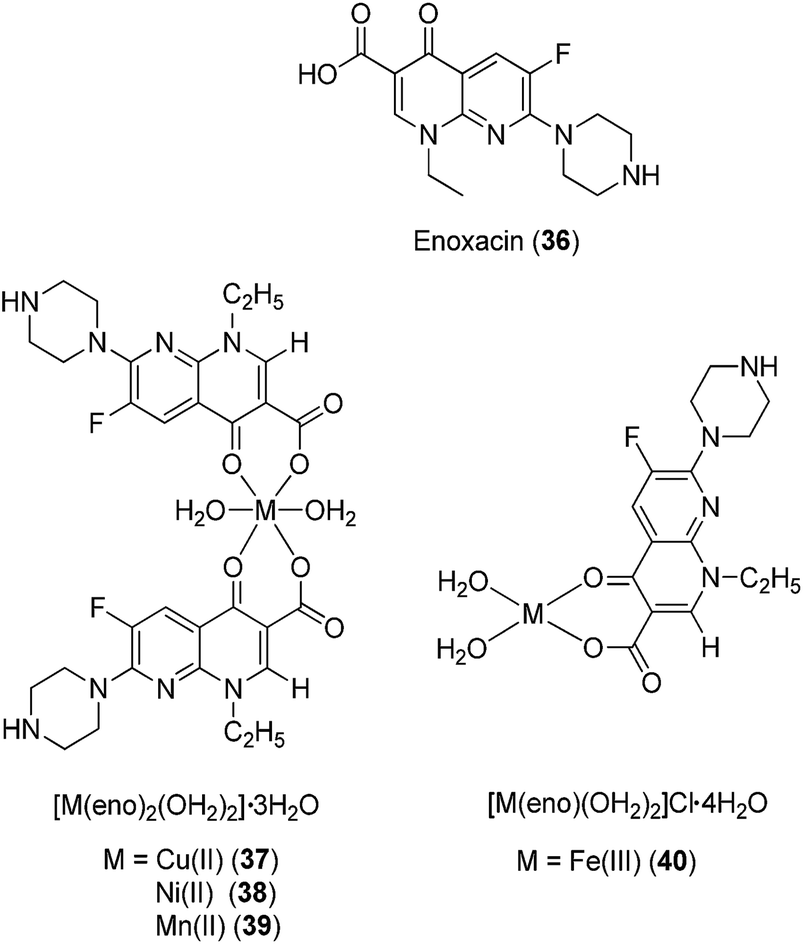 | ||
| Fig. 13 Chemical structures of the complexes of enoxacin (eno) 36, [M(eno)2(OH2)2]·3OH237–39 (where M = Cu(II), Ni(II) or Mn(II)) and [Fe(eno)(OH2)2]Cl·4OH240. | ||
The coagulation of blood and the inflammatory activation of platelets, endothelial cells and monocytes are events that can lead to the formation of an oncogenic inflammatory microenvironment in tissues. These events are regulated by a network of mediators, including thrombin and platelet-activating factor (PAF).49,50 Mitsopoulou and co-workers have recently reported the biological properties of fac-[Re(phendione)(CO)3Cl] 41 (where phendione = 1,10-phenanthroline-5,6-dione) as inhibitors of PAF-induced aggregation of washed rabbit platelets (WRPs) in vitro (Fig. 14).51 Additionally, complex 41 and [Re(CO5)Cl] showed activity against PAF-basic metabolic enzyme activities in rabbit leukocyte homogenates. In cellular proliferation studies, the free ligand phendione was found to be highly cytotoxic towards a range of cancer cell lines, but this toxicity was markedly reduced upon complexation to Re(I).
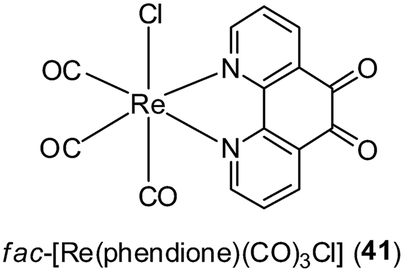 | ||
| Fig. 14 Chemical structure of fac-[Re(phendione)(CO)3Cl] 41 (where phendione = 1,10-phenanthroline-5,6-dione). | ||
One limitation of the studies presented in this section is that control experiments with the separate metal ions and the ligands were not always performed to assess whether or not the effects observed were not only a cumulative effect of the individual species. Several studies compared the potency of the metal complexes to the free ligand, but only one (Cu–aspirin) investigated the activity of the metal ion alone. Besides ruling out cumulative effects, such control experiments could also clarify the role of the metal ion in conferring enhanced biological activity to the metal complex over the free ligand. Additionally, the stabilities of many of the complexes in water were not reported. Given that potential drugs will eventually enter into an aqueous environment in the body, it would be important to investigate the possibility of metal complex hydrolysis to ascertain whether the intact complex and/or its derivatives are the biologically active species.
3 Metal complexes that covalently bind biomolecules
The ability of metal ions to form coordinative bonds with biomolecules has been harnessed in the development of numerous classes of metallodrugs. For example, cisplatin and its analogues act by covalently binding to nucleobases of DNA, generating intrastrand or interstrand DNA lesions that interfere with cellular replication or metabolic processes.52 In this section, we discuss examples of metal complexes that have been demonstrated to form covalent bonds to biomolecules, thereby exerting potential anti-inflammatory effects.The gold(I) salts auranofin 4, solganal 5, and myochrysine 6 have been approved for the treatment of rheumatoid arthritis. Despite their over two decades of use, however, their mechanism of action has not yet been fully elucidated. The gold compounds have been hypothesized to produce various metabolites in vivo, including gold(I),53 [Au(CN)2]−,54 or Au(III)55 that exert various biological effects. Proposed mechanisms of action of gold salts include the inhibition of selenium enzymes such as thioredoxin reductase,56 redox cycling to target mitochondria,57 the induction of heme oxygenase-1 (HO-1) expression,58 and the inhibition of signal transducer and activator of transcription 3 (STAT3),59 NF-κB,60 and TLR4 activation.61
The ability of gold(I) complexes to interact with thioredoxin reductase can be rationalised by Pearson's acid base concept,62 which suggests that the “soft” gold(I) ions preferentially bind to soft Lewis base ligands such as thiolato and selenolato ions, or phosphine derivatives. In the blood, the gold(I) complexes bind well to sulfhydryl-containing compounds, such as cysteine or glutathione, as well as to albumin or globulins via a ligand exchange mechanism. In the case of auranofin 4, the tetraacetylthioglucose is displaced first, followed by the replacement of the triethylphosphine ligand that is subsequently oxidized to triethylphosphine oxide. The cooperative effects of adjacent thiolato or selenolato ligands neighbouring the interaction site play an important role in the incorporation of gold into the active site of selenium-containing flavoreductases, such as thioredoxin reductase.
In 2012, Trávníček and co-workers investigated the anti-inflammatory activities of gold(I) complexes with the general formula [Au(L)(PPh3)]·xOH2 (42a–h; x = 0–1.5), where L represented the deprotonated form of benzyl-substituted derivatives of 6-benzylaminopurine (Fig. 15).63 The gold(I) complexes significantly reduced the production of pro-inflammatory cytokines such as TNF-α, interleukin-1 beta (IL-1β) and high-mobility group protein B1 (HMGB1) in lipopolysaccharide (LPS)-activated macrophages,64 but did not inhibit the secretion of the anti-inflammatory cytokine interleukin-1 receptor antagonist (IL-1Ra). Complexes 42a and 42f also reduced inflammatory edema volume in rats challenged by carrageenan. Notably, all of the tested gold(I) complexes exhibited comparable or superior activity compared to auranofin at equitoxic doses. Electrospray ionization mass spectrometry experiments revealed that the complexes could bind to cysteine via the displacement of the 6-benzylaminopurine N-donor ligand. Hence, these gold(I) complexes were considered to act as pro-drugs in a similar manner to auranofin 4. Moreover, certain complexes showed cytotoxic activity against THP1 cells. In a later study, Trávníček and co-workers studied the biological properties of related derivatives containing a 2-chloro substituent on the adenine moiety.65 These complexes inhibited TNF-α and IL-1β secretion in cells, but did not show anti-inflammatory activity in vivo. Electrospray ionization mass spectrometry analysis suggested that the 2-chloro complexes were relatively unstable, as revealed by the formation of [Au(PPh3)2]+ in all solutions. Taken together, these studies demonstrate that the anti-inflammatory activities of gold(I) complexes may be highly sensitive to structural variation in the auxiliary ligands.
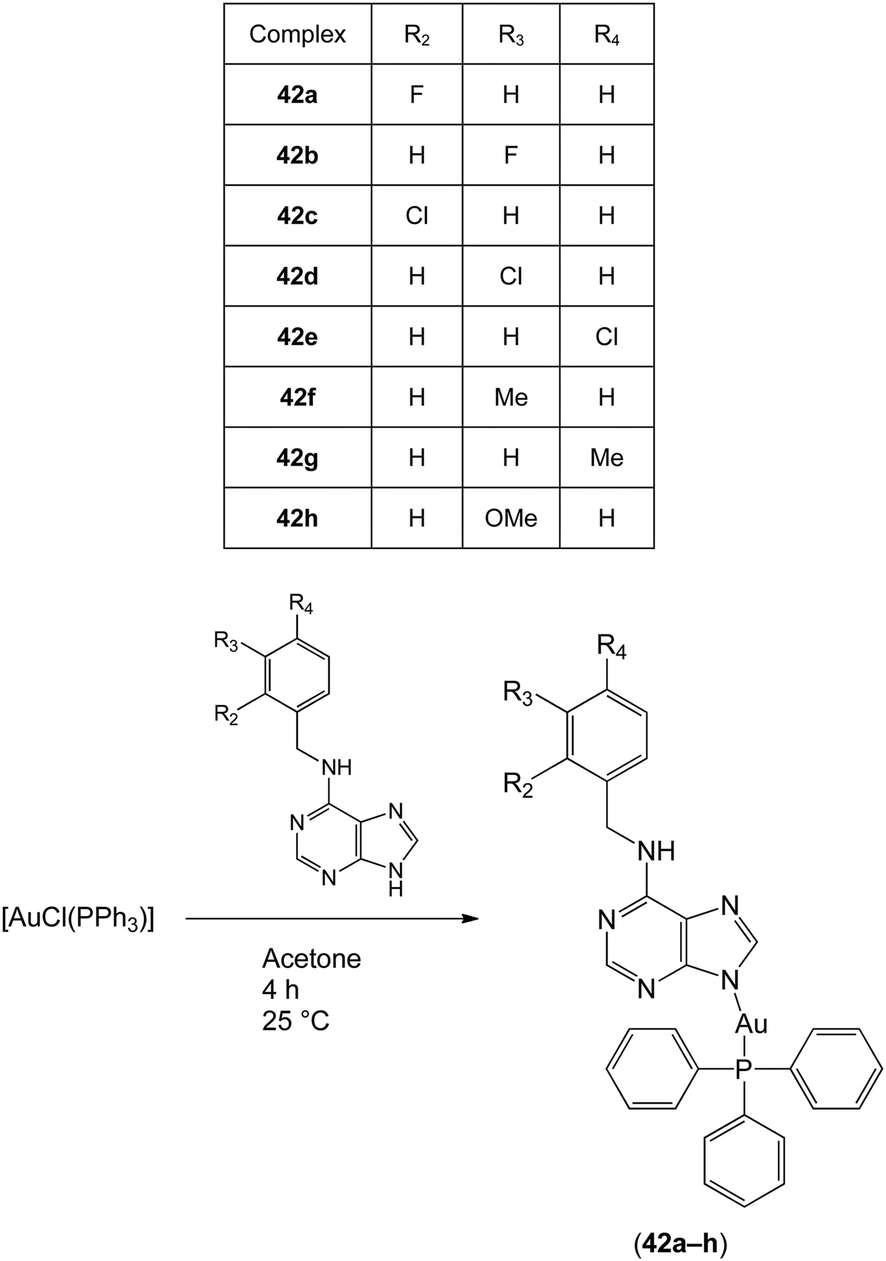 | ||
| Fig. 15 Synthesis and chemical structures of the gold(I) complexes with the general formula [Au(L)(PPh3)]·xOH2 (42a–h; x = 0–1.5). | ||
STAT proteins are responsible for regulating fundamental cellular processes, including cell growth and differentiation, development, apoptosis, inflammation and immunity.66,67 Constitutive STAT3 activity has been detected in a number of human cancers.68 The tumorigenic potential of dysregulated STAT3 has been linked to its effects on pro-oncogenic inflammatory pathways, including NF-κB and interleukin-6 (IL-6)-GP130-Janus kinase (JAK) pathways.69,70 Moreover, STAT3 has been found to play a critical role in both chronic inflammation and joint inflammation in rheumatoid arthritis.71 Alternatively, STAT3 has been reported to exert anti-inflammatory activities via interleukin-10 (IL-10) signaling.72
Turkson and co-workers have investigated the application of platinum compounds 43–46 (Fig. 16) as inhibitors of STAT3 signaling.73 The Pt(IV) compounds CPA-1 43, CPA-7 45 and Pt(IV)Cl446 blocked STAT3 DNA-binding activity and STAT3-driven activity in vitro, and inhibited cell growth and induced apoptosis in malignant cells harboring constitutively active STAT3. On the other hand, the Pt(II) compounds CPA-3 44 and cisplatin 1 showed little inhibition of STAT3 activity in cells, indicating the importance of the Pt(IV) center in achieving cellular activity. Based on kinetic experiments, the authors postulated that there could be two mechanisms by which the Pt(IV) complexes disrupted STAT3 activity in cells. The first mechanism involved the direct targeting of STAT3 protein by the Pt(IV) compounds, while the second mechanism involved a decrease in STAT3 phosphorylation levels mediated by the complexes. In a later study, the group identified IS3 295 47, a piperazine salt of hexachloroplatinate(IV) (Fig. 16), from the NCI 2000 diversity set as an inhibitor of STAT3 activity.74 Preliminary evidence suggested that IS3 295 47 interacted with cysteine residue(s) within STAT3, forming thiol conjugates that do not bind effectively to DNA.
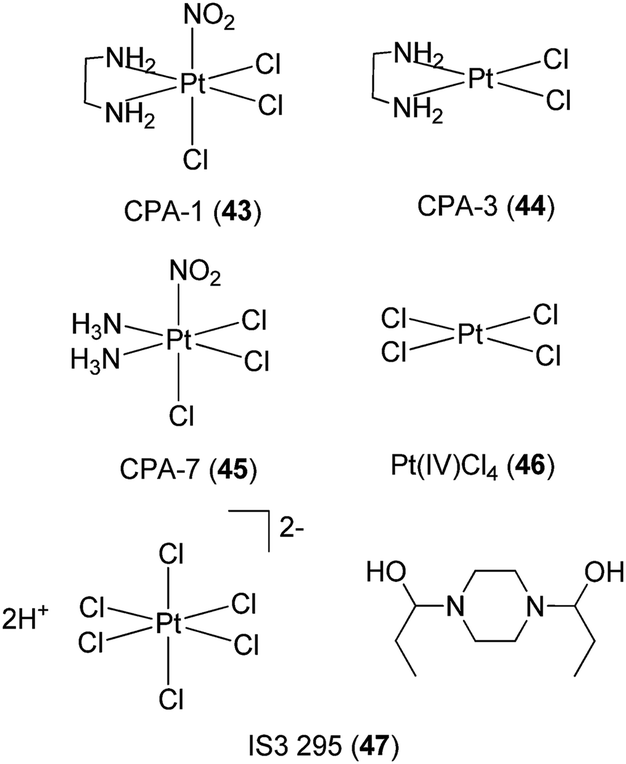 | ||
| Fig. 16 Chemical structures of CPA-1 43, CPA-3 44, CPA-7 45 and Pt(IV)Cl446 and IS3 295 47. | ||
4 Kinetically inert metal complexes
Meggers has pioneered the development of kinetically inert metal complexes as inhibitors of protein kinases.75 Remarkably, even though all kinases share highly similar active sites, the compounds have managed to achieve significant selectivity for specific kinases despite being limited to purely reversible interactions, and many of these compounds showed potent anti-cancer activity. This section highlights the application of kinetically inert metal complexes as inhibitors of inflammatory or autoimmune signaling molecules.Tumor necrosis factor-α (TNF-α) is a pro-inflammatory cytokine that plays an important role in regulating key biological processes, including inflammation, immunity and haematopoiesis.76 The dysregulation of TNF-α activity has been implicated in transplant rejection, viral replication, septic shock, diabetes, tumorigenesis, and autoinflammatory diseases such as rheumatoid arthritis, psoriatic arthritis, and Crohn's disease. In 2012, Leung, Ma and co-workers reported the cyclometalated Ir(III) complex [Ir(ppy)2(biq)]PF648 containing the bidentate 2-phenylpyridinato (ppy) ligand and the 2,2′-biquinoline (biq) ligand (Fig. 17) as the first iridium-based inhibitor of TNF-α.77
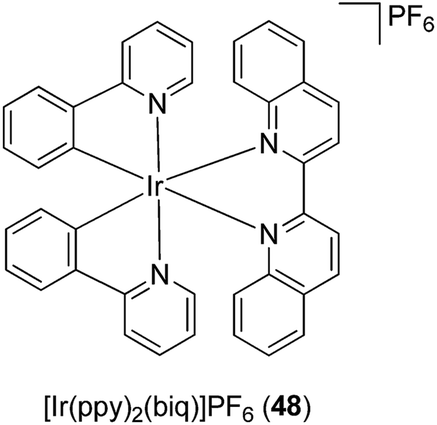 | ||
| Fig. 17 Chemical structure of the cyclometalated Ir(III) complex [Ir(ppy)2(biq)]PF648. | ||
The synthesis of the enantiopure complexes Δ-48 and Λ-48 was achieved using a novel asymmetric strategy reported by Meggers and co-workers in 2013,78 which employed (S)-4-tert-butyl-2-(2′-hydroxyphenyl)oxazoline as a chiral auxiliary (Fig. 17). In a cell-free assay, complexes Δ-48 and Λ-48 inhibited the TNF-α–TNFR interaction with a potency comparable to SPD304, a potent small-molecule inhibitor of TNF-α. Furthermore, complexes Δ-48 and Λ-48 inhibited TNF-α-induced NF-κB luciferase activity in human hepatocellular carcinoma (HepG2) cells with superior potency to SPD304.77 Intriguingly, the analogous Rh(III) complex showed markedly reduced inhibitory activity, indicating the importance of the metal center in determining the TNF-α inhibitory activity of the complexes.
Similar to most protein–protein interfaces, the binding site on the TNF-α dimer is mostly hydrophobic and featureless, primarily consisting of glycine, leucine, and tyrosine subunits. Consequently, the interaction between small molecules and the TNF-α dimer has been presumed to be mainly hydrophobic and shape-driven. The authors anticipated that the extended aromatic biq ligand of complex 48 could facilitate favorable hydrophobic interactions with the TNF-α dimer binding interface, thereby blocking the association of the third subunit and the formation of the active trimer complex. Molecular modeling analysis revealed that the biq ligand of Δ-48 contacted the β-strand of the A subunit of TNF-α, while the ppy units contacted the β-strand of the B subunit. On the other hand, the Λ-48 enantiomer was predicted to be situated in the same binding pocket, but with the biq ligand and one of the ppy ligands of Λ-48 contacting the β-strands from both subunits of the TNF-α dimer complex. The absence of hydrogen bonding or salt-bridge interactions in the models suggested that the interaction between the β-strands of the TNF-α dimer with Δ-48 or Λ-48 was primarily hydrophobic in nature, which was consistent with previous reports. This report demonstrates that kinetically inert organometallic compounds can be utilised for targeting protein–protein interactions, which are typically considered to be difficult to target with small molecules.
The cyclometallated rhodium(III) complex 49 (Fig. 18) has recently been reported as an inhibitor of lipopolysaccharide (LPS)-induced nitric oxide (NO) production in RAW264.7 macrophages.79 The generation of NO by inducible nitric oxide synthase (iNOS) can be triggered by various inflammatory stimuli, leading to the formation of reactive intermediates of NO that can promote pro-inflammatory responses.80 Complex 49 inhibited NO production as well as NF-κB transcriptional activity in LPS-induced RAW264.7 macrophages. Moreover, complex 49 antagonized vasculogenic activity in human umbilical vein endothelial cells, which was attributed, at least in part, to the inhibition of NF-κB activity and NO production in cellulo. However, the mechanism of action of this rhodium(III) complex was undetermined.
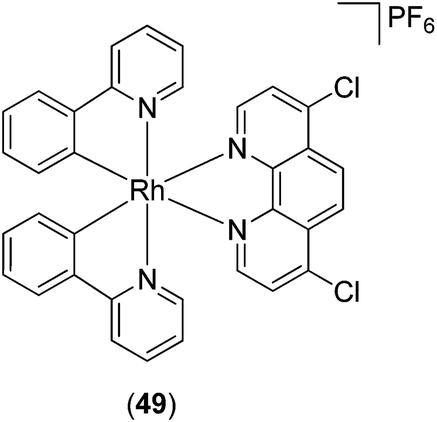 | ||
| Fig. 18 Chemical of structure of Rh(III) complex 49. | ||
As upstream activators of the STAT proteins, Janus kinase 2 (JAK2) has also been considered as a possible target for anti-inflammatory and anti-autoimmune applications.81–83 In 2012, two cyclometalated rhodium complexes rac-[Rh(ppy)2(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) NL)2]+50 and rac-[Rh(bzq)2(CNL)2]+51 (ppy = 2-phenylpyridine; bzq = benzoquinoline and CNL = 2-naphthylisocyanide) (Fig. 19) were developed as non-covalent JAK2 inhibitors.84 Results from biological experiments indicated that complexes 50 and 51 inhibited JAK2 enzyme phosphorylation activity and reduced JAK2 autophosphorylation in cellulo. Furthermore, the complexes exhibited micromolar cytotoxicity towards human erythroleukemia (HEL) cancer cells, which harbor constitutive JAK2 activity.
NL)2]+50 and rac-[Rh(bzq)2(CNL)2]+51 (ppy = 2-phenylpyridine; bzq = benzoquinoline and CNL = 2-naphthylisocyanide) (Fig. 19) were developed as non-covalent JAK2 inhibitors.84 Results from biological experiments indicated that complexes 50 and 51 inhibited JAK2 enzyme phosphorylation activity and reduced JAK2 autophosphorylation in cellulo. Furthermore, the complexes exhibited micromolar cytotoxicity towards human erythroleukemia (HEL) cancer cells, which harbor constitutive JAK2 activity.
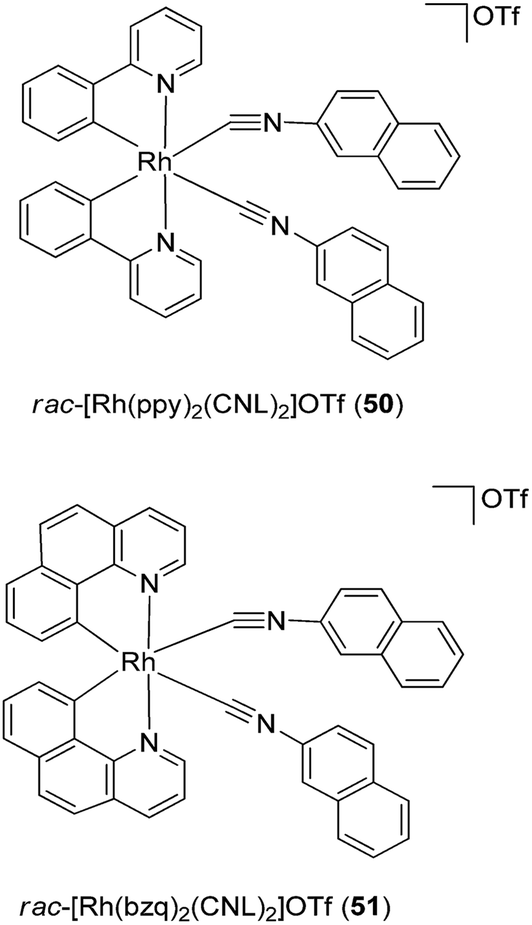 | ||
| Fig. 19 Chemical structures of rac-[Rh(ppy)2(CNL)2]OTf 50 and rac-[Rh(bzq)2(CNL)2]OTf 51 (ppy = 2-phenylpyridine; bzq = benzoquinoline and CNL = 2-naphthylisocyanide). | ||
Ma, Leung and co-workers have also reported a Rh(III) complex 52 (Fig. 20) as a kinetically inert metal-based inhibitor of STAT3 dimerisation.85 A fluorescence polarisation assay revealed that complex 52 targeted the SH2 domain of STAT3. Furthermore, complex 52 inhibited the STAT3 DNA-binding activity, phosphorylation, dimerization, and signaling activity in vitro. Whereas complex 52 was able to significantly reduce tumor size and angiogenesis in an in vivo mouse xenograft model, immunohistochemical experiments showed that the levels of COX-2 and iNOS in the tumor tissues were also reduced by complex 52. Since STAT3 promotes the expression of COX-286 and iNOS,87 this suggests a possible mechanism by which complex 52 may exert anti-inflammatory activity. Moreover, complex 52 induced no signs of gross toxicity or weight loss in treated rats, and did not significantly affect the mean weights of the heart, liver and kidney, demonstrating its relatively non-toxic nature.
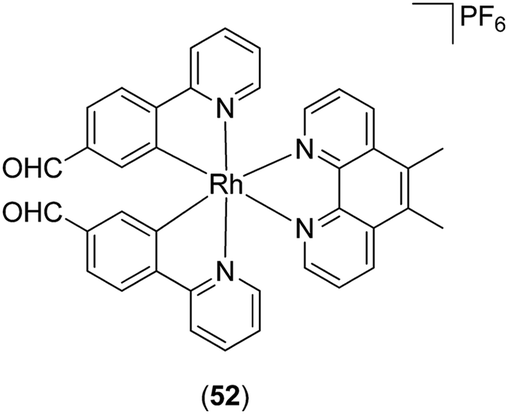 | ||
| Fig. 20 Chemical of structure of Rh(III) complex 52. | ||
Gunning and co-workers reported the first application of coordination complexes 53, 54 and 55 (Fig. 21) mimicking the Src homology 2 (SH2) phosphopeptide-binding domain.88 The authors postulated that the SH2 domain of STAT3 could be effectively mimicked by metal complexes, leading to the inhibition of SH2-phosphotyrosine interactions that are essential for STAT3 signaling. As a proof-of-concept, functionalized bis-dipicolylamine (BDPA) copper(II) complexes were developed that disrupted STAT3–STAT3 protein interactions. The Cu(II) complexes inhibited STAT3 homodimer DNA-binding activity in mouse fibroblast nuclear extracts containing constitutively activated STAT3. The inhibitory action of the Cu(II) complexes wAS attributed to their mimicry of the SH2 domain–phosphotyrosine binding function.
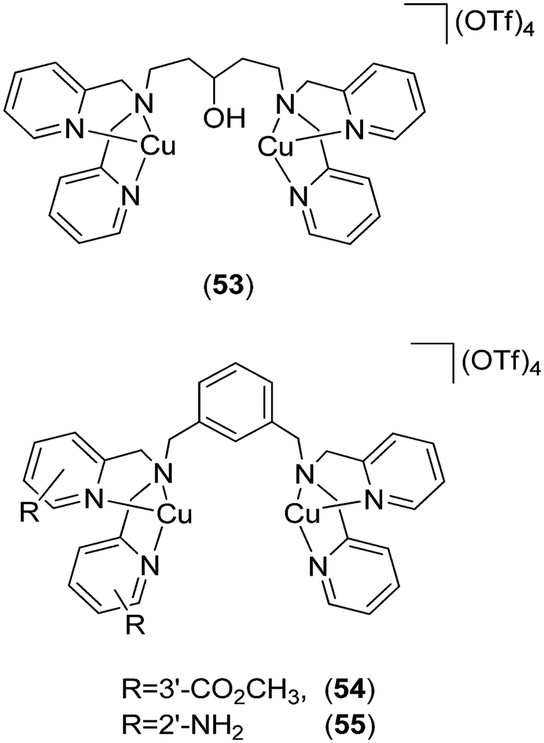 | ||
| Fig. 21 Chemical structures of the coordination complexes 53, 54 and 55 as functional proteomimetics of the Src homology 2 (SH2) phosphopeptide-binding domain. | ||
The mammalian target of rapamycin (mTOR) controls metabolism, growth, and cell survival, and has recently emerged as a key regulator of innate immune cell homeostasis.89 Inhibition of mTOR by rapamycin in human monocytes induces the production of IL-12, TNF-α, or IL-6 via the activation of transcription factor NF-κB, but attenuates the active release of IL-10 via STAT3 activation. Recently, a series of Rh(III) and Ir(III) complexes have been reported as inhibitors of mTOR activity.90 The Rh(III) complex 56 (Fig. 22) attenuated mTOR activity in a cell-free system and in mTOR immunoprecipitates from treated cells, with a potency comparable to that of rapamycin. Furthermore, the inhibition by complex 56 was found to be dependent on FK506-binding protein 12 (FKBP12). The authors postulated that complex 56 may first bind to FKBP12, creating an interface that is subsequently recognized by the mTOR FKBP12–rapamycin-binding domain. Therefore, complex 56 may be tentatively considered as a protein–protein interaction stabilizer of the mTOR–FKBP12 interaction, and could potentially be further developed as a modulator of the inflammatory response. Preliminary structure–activity relationship analysis with related complexes indicated that the size and hydrophobicity of the N^N donor ligand and the nature of the metal center were important determinants for mTOR inhibitory activity.
 | ||
| Fig. 22 Chemical structure of Rh(III) complex 56. | ||
A redox imbalance between endogenous reactive species, such as nitric oxide, superoxide, hydrogen peroxide, peroxynitrite, and endogenous antioxidants, can cause damage to biological molecules and impair signaling pathways. Oxidative stress has been linked to a wide range of human diseases, such as ischemia–reperfusion disorders, central nervous system disorders, cardiovascular disorders, inflammatory disorders, cancer and diabetes. Therefore, compounds capable of reducing the levels of reactive species can suppress oxidative damage to biological molecules as a consequence of primary oxidative insult. Moreover, certain transcription factors such as NF-κB require reactive species for activation. Hence, the removal of reactive species can also reduce general transcriptional activity, thereby suppressing excessive inflammation and leading to continuous oxidative stress and secondary oxidative damage of biomolecules.
Superoxide dismutase (SOD) is an endogenous enzyme that eliminates superoxide by catalyzing its dismutation into O2 and H2O2 (Fig. 23). A large range of Mn porphyrins,91–101 Mn cyclic polyamines102–105 and Mn salen derivatives106–111 have been developed as SOD mimics (Fig. 24). The redox cycle of the Mn SOD mimics involves catalyzing the oxidation of O2˙− to O2 by Mn(III)SOD, followed by reduction of O2˙− to H2O2 by Mn(II)SOD. Some Mn porphyrins have been reported to target the mitochondria, whereas others are able to cross the blood–brain barrier and were effective in stroke and hemorrhage models. Moreover, as reactive species are required for general transcriptional activity, some Mn SOD mimics have been found to be effective not only when delivered to the site of primary oxidative injury, but also when administered hours (as in stroke) or weeks (as in radiation exposure) after the injurious event, via the repair of damage caused by continuous inflammation. Excellent review articles on the application of Mn complexes as anti-oxidant and anti-inflammatory compounds have been published.111–114 Besides Mn compounds, other metallo SOD mimics such as Fe porphyrins or Cu porphyrins have been reported, though these have found more limited application.
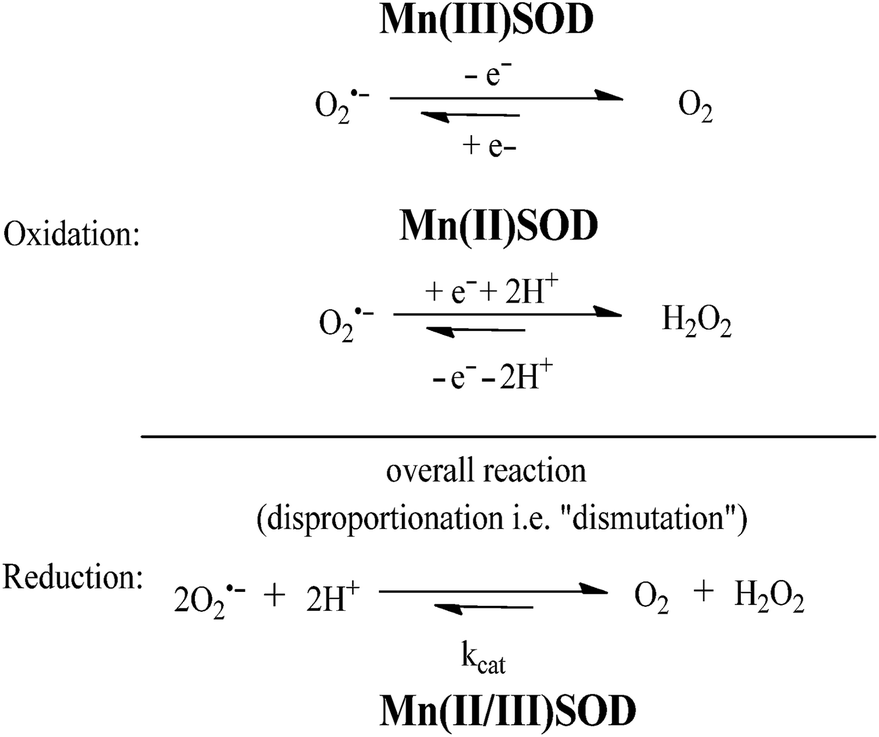 | ||
| Fig. 23 The dismutation of O2˙− catalyzed by MnSOD. | ||
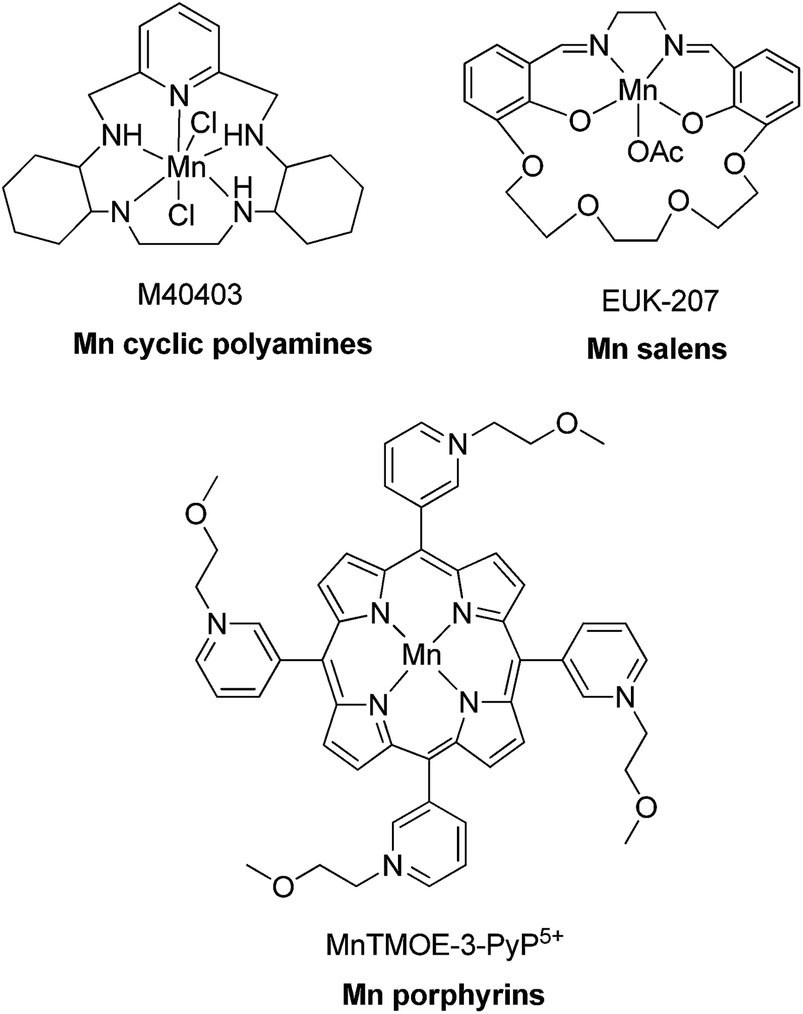 | ||
| Fig. 24 Chemical structures of Mn porphyrins, Mn cyclic polyamines and Mn salen derivatives. | ||
5 Conclusion and outlook
Compared to the comprehensive body of work established with metal anti-cancer compounds, the field of metal-based anti-inflammatory and anti-autoimmune agents is still immature. To date, the gold compounds auranofin 4, solganal 5 and myochrysine 6 used for the treatment of rheumatoid arthritis are among the only metal-based compounds that have been approved for the treatment of inflammatory or autoimmune diseases.This review has highlighted some recent examples of transition metal complexes that have been investigated for anti-inflammatory or anti-autoimmune activity. A common strategy that has been employed is the coordination of anti-inflammatory compounds or other bioactive molecules to metal ions, resulting in enhanced activity. For example, the anti-inflammatory activities of metal complexes 32–35 of a 1,2-dihydroquinazolinone derivative were superior to those of the free ligand.46 This was attributed to the higher lipophilicity of the metal complexes compared to the free ligand, potentially improving the ability of the complexes to pass through cellular membranes and into cells. In the case of the NSAID aceclofenac 9, complexation with zinc generated a zinc–aceclofenac complex that induced fewer stomach ulcers in rats, while having comparable anti-inflammatory activity to the parent drug.29 The authors postulated that the ulcerogenic carboxyl group in aceclofenac could be effectively masked by its coordination to zinc, resulting in less severe adverse side effects in the stomach.
The second strategy utilises metal complexes with labile ligands that can coordinate directly with biomolecules. Trávníček and co-workers demonstrated that gold(I) complexes 41 bearing substituted 6-benzylaminopurine and triphenylphosphine ligands displayed stronger anti-inflammatory activity in vitro and in vivo to auranofin at equitoxic doses.63 Interestingly, analogues containing a 2-chloro group on the adenine moiety were inactive in vivo, indicating that minor changes in the structure of these gold(I) complexes can have a significant effect on their anti-inflammatory properties.65 The third approach described in this review involves kinetically inert metal complexes that target specific biomolecules in a non-covalent fashion. For example, the cyclometalated Ir(III) complex [Ir(ppy)2(biq)]PF6 developed by Leung, Ma and co-workers represented the first metal-based inhibitor of TNF-α 48.77 Alternatively, Mn porphyrin, cyclic polyamine and salen derivatives are able to catalyse redox reactions that eliminate reactive species and suppress oxidative stress and inflammation.
One important aspect that has to be critically considered in the design and development of metal complexes as anti-inflammatory agents is their toxicity. Notable adverse side effects of gold drugs include oral ulcers, altered taste, skin rashes, skin pigmentation, anemia (thrombocytopenia) or renal toxicity (proteinuria, nephropathy). Additionally, toxic effects on the brain have also been reported. On the other hand, platinum compounds have been linked with nephrotoxicity, neurotoxicity, cardiotoxicity and myelosuppression,115 and their use in the body can also lead to the development of chemoresistance via multiple mechanisms.116 As the use of metal complexes as anti-inflammatory agents is still a relatively new and emerging topic, extensive studies on the toxicity of the compounds described in this review were infrequently performed. Nevertheless, as toxicity often represents one of the most common limits to the use of metal complexes in medicine, toxicity evaluation is an essential step that is needed to determine the potential pharmacological application of these metal complexes as anti-inflammatory agents.
Toward the future, we believe that further elucidation of the precise mechanism of action of anti-inflammatory activity of the bioactive metal complexes is necessary in order to facilitate the rational design of metal complex analogues that could display improved potency or selectivity, while minimizing toxicity. In particular, potential chemical transformations or reactions of the metal complexes in vivo should be more fully characterised. Additionally, we believe that the application of kinetically inert metal complexes to specifically target biomolecules in cells deserves further investigation. Members of cellular inflammatory or immune pathways interact with each other via protein–protein interfaces that are typically large and amorphous. Such protein–protein interactions could be potentially targeted by kinetically inert metal complexes that possess ligands of suitable shape and hydrophobicity for binding to the protein–protein interface. Finally, advances in molecular biology and interactomics may present novel targets for the development of new anti-inflammatory or anti-autoimmune metal complexes. Given the diverse variety of promising studies highlighted in this review, we envisage that it is only a matter of time before the next transition metal complexes are approved in the clinic for the treatment of inflammatory or autoimmune diseases.
Acknowledgements
This work is supported by Hong Kong Baptist University (FRG2/13-14/008), Centre for Cancer and Inflammation Research, School of Chinese Medicine (CCIR-SCM, HKBU), the Health and Medical Research Fund (HMRF/13121482), the Research Grants Council (HKBU/201811, HKBU/204612, and HKBU/201913), the French National Research Agency/Research Grants Council Joint Research Scheme (A-HKBU201/12), the Science and Technology Development Fund, Macao SAR (103/2012/A3) and the University of Macau (MYRG091(Y3-L2)-ICMS12-LCH, MYRG121(Y3-L2)-ICMS12-LCH and MRG023/LCH/2013/ICMS).References
- D. R. Feldman, G. J. Bosl, J. Sheinfeld and R. J. Motzer, JAMA, J. Am. Med. Assoc., 2008, 299, 672 CrossRef CAS PubMed.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817 RSC.
- Z. Liu and P. J. Sadler, Acc. Chem. Res., 2014, 47, 1174 CrossRef CAS PubMed.
- M. Dörr and E. Meggers, Curr. Opin. Chem. Biol., 2014, 19, 76 CrossRef PubMed.
- C.-M. Che and R. W.-Y. Sun, Chem. Commun., 2011, 47, 9554 RSC.
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617 RSC.
- J. Kljun, I. Bratsos, E. Alessio, G. Psomas, U. Repnik, M. Butinar, B. Turk and I. Turel, Inorg. Chem., 2013, 52, 9039 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3 CrossRef CAS PubMed.
- C. M. Clavel, E. Paunescu, P. Nowak-Sliwinska and P. J. Dyson, Chem. Sci., 2014, 5, 1097 RSC.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677 CrossRef CAS.
- G. Sava, A. Bergamo and P. J. Dyson, Dalton Trans., 2011, 40, 9069 RSC.
- Y. Geldmacher, M. Oleszak and W. S. Sheldrick, Inorg. Chim. Acta, 2012, 393, 84 CrossRef CAS PubMed.
- B. Kupcewicz, K. Sobiesiak, K. Malinowska, K. Koprowska, M. Czyz, B. Keppler and E. Budzisz, Med. Chem. Res., 2013, 22, 2395 CrossRef CAS PubMed.
- M. Groessl and C. G. Hartinger, Anal. Bioanal. Chem., 2013, 405, 1791 CrossRef CAS PubMed.
- C. J. Marmion, D. M. Griffith, J. Parker, Z. Ude, H. Nimir, M. Morgan, M. Brennan, B. Duff, D. Egan, A. Chubb, J. Kasparkova and V. Brabec, JBIC, J. Biol. Inorg. Chem., 2014, 19, S463 Search PubMed.
- G. Gasser and N. Metzler-Nolte, Curr. Opin. Chem. Biol., 2012, 16, 84 CrossRef CAS PubMed.
- A. Levina, A. Mitra and P. A. Lay, Metallomics, 2009, 1, 458 RSC.
- Y. Chen, M. Y. Qin, J. H. Wu, L. Wang, H. Chao, L. N. Ji and A. L. Xu, Eur. J. Med. Chem., 2013, 70, 120 CrossRef CAS PubMed.
- L. Y. Li, K. J. Du, Y. Wang, H. N. Jia, X. J. Hou, H. Chao and L. N. Ji, Dalton Trans., 2013, 42, 11576 RSC.
- H. Choy, C. Park and M. Yao, Clin. Cancer Res., 2008, 14, 1633 CrossRef CAS PubMed.
- E. Chartone-Souza, T. L. Loyola, M. Bucciarelli-Rodriguez, M. Ã. d. B. C. Menezes, N. A. Rey and E. C. Pereira-Maia, J. Inorg. Biochem., 2005, 99, 1001 CrossRef CAS PubMed.
- J. R. Vane, Y. S. Bakhle and R. M. Botting, Annu. Rev. Pharmacol. Toxicol., 1998, 38, 97 CrossRef CAS PubMed.
- L. J. Marnett, Curr. Opin. Chem. Biol., 2000, 4, 545 CrossRef CAS.
- J. J. Talley, Expert Opin. Ther. Pat., 1997, 7, 55 CrossRef CAS.
- Z. H. Chohan, M. S. Iqbal, H. S. Iqbal, A. Scozzafava and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2002, 17, 87 CrossRef CAS PubMed.
- L. Manojlovic-Muir, Acta Crystallogr., 1973, 29, 2033 CrossRef CAS.
- T. Fujimori, S. Yamada, H. Yasui, H. Sakurai, Y. In and T. Ishida, JBIC, J. Biol. Inorg. Chem., 2005, 10, 831 CrossRef CAS PubMed.
- Y. Yun, P. Chen, C. L. Zheng, Y. Yang, W. G. Duan, L. Wang, B. He, J. Q. Ma, D. H. Wang and Z. Q. Shen, Yakugaku Zasshi, 2007, 127, 1869 CrossRef CAS.
- M. A Kale, R. Shelke and R. B Nawale, Anti-Inflammatory Anti-Allergy Agents Med. Chem., 2014, 13, 36 CrossRef.
- F. Dimiza, A. N. Papadopoulos, V. Tangoulis, V. Psycharis, C. P. Raptopoulou, D. P. Kessissoglou and G. Psomas, Dalton Trans., 2010, 39, 4517 RSC.
- F. Dimiza, S. Fountoulaki, A. N. Papadopoulos, C. A. Kontogiorgis, V. Tangoulis, C. P. Raptopoulou, V. Psycharis, A. Terzis, D. P. Kessissoglou and G. Psomas, Dalton Trans., 2011, 40, 8555 RSC.
- S. Fountoulaki, F. Perdih, I. Turel, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2011, 105, 1645 CrossRef CAS PubMed.
- C. Tolia, A. N. Papadopoulos, C. P. Raptopoulou, V. Psycharis, C. Garino, L. Salassa and G. Psomas, J. Inorg. Biochem., 2013, 123, 53 CrossRef CAS PubMed.
- F. Dimiza, A. N. Papadopoulos, V. Tangoulis, V. Psycharis, C. P. Raptopoulou, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2012, 107, 54 CrossRef CAS PubMed.
- S. Tsiliou, L.-A. Kefala, F. Perdih, I. Turel, D. P. Kessissoglou and G. Psomas, Eur. J. Med. Chem., 2012, 48, 132 CrossRef CAS PubMed.
- M. Zampakou, N. Rizeq, V. Tangoulis, A. N. Papadopoulos, F. Perdih, I. Turel and G. Psomas, Inorg. Chem., 2014, 53, 2040 CrossRef CAS PubMed.
- S. Sharma, V. K. Srivastava and A. Kumar, Eur. J. Med. Chem., 2002, 37, 689 CrossRef CAS.
- M. S. Iqbal, S. J. Khurshid and B. Muhammad, Med. Chem. Res., 2013, 22, 861 CrossRef CAS.
- W. B. Júnior, M. S. Alexandre-Moreira, M. A. Alves, A. Perez-Rebolledo, G. L. Parrilha, E. E. Castellano, O. E. Piro, E. J. Barreiro, L. M. Lima and H. Beraldo, Molecules, 2011, 16, 6902 CrossRef PubMed.
- M. Jesmin, M. K. Islam and S. M. M. Ali, Int. Lett. Chem., Phys. Astron., 2014, 8, 64 Search PubMed.
- Narayanachar, S. D. Dhumwad, S. Mutalik, M. H. Hugar and P. N. Naik, Main Group Chem., 2013, 12, 87 CAS.
- R. Shanmugakala, P. Tharmaraj, C. Sheela and N. Chidambaranathan, Med. Chem. Res., 2014, 23, 329 CrossRef CAS.
- J. D. Hansen, L. N. Vojtech and K. J. Laing, Dev. Comp. Immunol., 2011, 35, 886 CrossRef CAS PubMed.
- H. Bielig, J. Velder, A. Saiai, M. Menning, S. Meemboor, W. Kalka-Moll, M. Krönke, H. G. Schmalz and T. A. Kufer, ChemMedChem, 2010, 5, 2065 CrossRef CAS PubMed.
- A. M. S. Mayer, P. B. Jacobson, W. Fenical, R. S. Jacobs and K. B. Glaser, Life Sci., 1998, 62, 401 CrossRef.
- R. S. Hoonur, B. R. Patil, D. S. Badiger, R. S. Vadavi, K. B. Gudasi, P. R. Dandawate, M. M. Ghaisas, S. B. Padhye and M. Nethaji, Eur. J. Med. Chem., 2010, 45, 2277 CrossRef CAS PubMed.
- M. J. Wood, Clin. Pharmacokinet., 1989, 16, 38 CrossRef PubMed.
- S. Arayne, N. Sultana, U. Haroon and M. A. Mesaik, Bioinorg. Chem. Appl., 2009, 2009, 914105 Search PubMed.
- A. Tsoupras, C. Iatrou, C. Frangia and C. Demopoulos, Infect. Disord.: Drug Targets, 2009, 9, 390 CrossRef CAS.
- V. Melnikova and M. Bar-Eli, Cancer Metastasis Rev., 2007, 26, 359 CrossRef CAS PubMed.
- M. Kaplanis, G. Stamatakis, V. D. Papakonstantinou, M. Paravatou-Petsotas, C. A. Demopoulos and C. A. Mitsopoulou, J. Inorg. Biochem., 2014, 135, 1 CrossRef CAS PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265 CrossRef CAS PubMed.
- A. J. Canumalla, N. Al-Zamil, M. Phillips, A. A. Isab and C. F. Shaw Iii, J. Inorg. Biochem., 2001, 85, 67 CrossRef CAS.
- G. G. Graham and A. J. Kettle, Biochem. Pharmacol., 1998, 56, 307 CrossRef CAS.
- J. Zou, Z. Guo, J. A. Parkinson, Y. Chen and P. J. Sadler, J. Inorg. Biochem., 1999, 74, 352 Search PubMed.
- V. Gandin, A. P. Fernandes, M. P. Rigobello, B. Dani, F. Sorrentino, F. Tisato, M. Björnstedt, A. Bindoli, A. Sturaro and R. Rella, Biochem. Pharmacol., 2010, 79, 90 CrossRef CAS PubMed.
- O. Rackham, S. J. Nichols, P. J. Leedman, S. J. Berners-Price and A. Filipovska, Biochem. Pharmacol., 2007, 74, 992 CrossRef CAS PubMed.
- N.-H. Kim, M.-K. Oh, H. J. Park and I.-S. Kim, J. Pharmacol. Sci., 2010, 113, 246 CrossRef CAS.
- N. H. Kim, M. Y. Lee, S. J. Park, J. S. Choi, M. K. Oh and I. S. Kim, Immunology, 2007, 122, 607 CrossRef CAS PubMed.
- K.-I. Jeon, M.-S. Byun and D.-M. Jue, Exp. Mol. Med., 2003, 35, 61 CrossRef CAS PubMed.
- H. S. Youn, J. Y. Lee, S. I. Saitoh, K. Miyake and D. H. Hwang, Biochem. Biophys. Res. Commun., 2006, 350, 866 CrossRef CAS PubMed.
- R. G. Pearson, J. Chem. Educ., 1987, 64, 561 CrossRef CAS.
- Z. Trávníček, P. Štarha, J. Vančo, T. Šilha, J. Hošek, P. Suchý and G. Pražanová, J. Med. Chem., 2012, 55, 4568 CrossRef PubMed.
- H. Fan and J. A. Cook, J. Endotoxin Res., 2004, 10, 71 CrossRef CAS PubMed.
- J. Hošek, J. Vančo, P. Štarha, L. Paráková and Z. Trávníček, PLoS One, 2013, 8, e82441 Search PubMed.
- J. Bromberg and J. E. Darnell Jr, Oncogene, 2000, 19, 2468 CrossRef CAS PubMed.
- J. E. Darnell, Science, 1997, 277, 1630 CrossRef CAS.
- P. A. Johnston and J. R. Grandis, Mol. Interventions, 2011, 11, 18 CrossRef CAS PubMed.
- Y. Fan, R. Mao and J. Yang, Protein Cell, 2013, 4, 176 CrossRef CAS PubMed.
- H. Yu, D. Pardoll and R. Jove, Nat. Rev. Cancer, 2009, 9, 798 CrossRef CAS PubMed.
- K. Pathak, Expert Opin. Ther. Targets, 2014, 18, 581 CrossRef CAS PubMed.
- P. Murray, Biochem. Soc. Trans., 2006, 34, 1028 CrossRef CAS PubMed.
- J. Turkson, S. Zhang, J. Palmer, H. Kay, J. Stanko, L. B. Mora, S. Sebti, H. Yu and R. Jove, Mol. Cancer Ther., 2004, 3, 1533 CAS.
- J. Turkson, S. Zhang, L. B. Mora, A. Burns, S. Sebti and R. Jove, J. Biol. Chem., 2005, 280, 32979 CrossRef CAS PubMed.
- E. Meggers, Chem. Commun., 2009, 1001 RSC.
- K. J. Tracey and A. Cerami, Annu. Rev. Med., 1994, 45, 491 CrossRef CAS PubMed.
- C.-H. Leung, H.-J. Zhong, H. Yang, Z. Cheng, D. S.-H. Chan, V. P.-Y. Ma, R. Abagyan, C.-Y. Wong and D.-L. Ma, Angew. Chem., Int. Ed., 2012, 51, 9010 CrossRef CAS PubMed.
- M. Helms, Z. Lin, L. Gong, K. Harms and E. Meggers, Eur. J. Inorg. Chem., 2013, 2013, 4164 CrossRef CAS.
- L.-J. Liu, S. Lin, D. S.-H. Chan, C. T. Vong, P. M. Hoi, C.-Y. Wong, D.-L. Ma and C.-H. Leung, J. Inorg. Biochem., 2014, 140, 23 CrossRef CAS PubMed.
- Y. C. Luiking, M. P. Engelen and N. E. Deutz, Curr. Opin. Clin. Nutr. Metab. Care, 2010, 13, 97 CrossRef CAS PubMed.
- B. Belli, D. Brigham, A. Dao, R. Nepomuceno, E. Setti, G. Liu, M. Hadd, S. Bhagwat, W. Wierenga and M. Holladay, Arthritis Rheum., 2010, 62, 269 Search PubMed.
- J. S. Fridman, P. A. Scherle, R. Collins, T. Burn, C. L. Neilan, D. Hertel, N. Contel, P. Haley, B. Thomas and J. Shi, J. Invest. Dermatol., 2011, 131, 1838 CrossRef CAS PubMed.
- X. Yang, G. He, Y. Hao, C. Chen, M. Li, Y. Wang, G. Zhang and Z. Yu, J. Neuroinflammation, 2010, 7, 54 CrossRef PubMed.
- C.-H. Leung, H. Yang, V. P.-Y. Ma, D. S.-H. Chan, H.-J. Zhong, Y.-W. Li, W.-F. Fong and D.-L. Ma, Med. Chem. Commun., 2012, 3, 696 RSC.
- D.-L. Ma, L.-J. Liu, K.-H. Leung, Y.-T. Chen, H.-J. Zhong, D. S.-H. Chan, H.-M. D. Wang and C.-H. Leung, Angew. Chem., Int. Ed., 2014, 53, 9178 CrossRef CAS PubMed.
- H.-W. Lo, X. Cao, H. Zhu and F. Ali-Osman, Mol. Cancer Res., 2010, 8, 232 CrossRef CAS PubMed.
- H.-W. Lo, S.-C. Hsu, M. Ali-Seyed, M. Gunduz, W. Xia, Y. Wei, G. Bartholomeusz, J.-Y. Shih and M.-C. Hung, Cancer Cell, 2005, 7, 575 CrossRef CAS PubMed.
- J. A. Drewry, S. Fletcher, P. Yue, D. Marushchak, W. Zhao, S. Sharmeen, X. Zhang, A. D. Schimmer, C. Gradinaru, J. Turkson and P. T. Gunning, Chem. Commun., 2010, 46, 892 RSC.
- M. Säemann, M. Haidinger, M. Hecking, W. Hörl and T. Weichhart, Am. J. Transplant., 2009, 9, 2655 CrossRef PubMed.
- H.-J. Zhong, K.-H. Leung, L.-J. Liu, L. Lu, D. S.-H. Chan, C.-H. Leung and D.-L. Ma, ChemPlusChem, 2014, 79, 508 CrossRef CAS.
- S. Rausaria, M. M. E. Ghaffari, A. Kamadulski, K. Rodgers, L. Bryant, Z. Chen, T. Doyle, M. J. Shaw, D. Salvemini and W. L. Neumann, J. Med. Chem., 2011, 54, 8658 CrossRef CAS PubMed.
- S. Asayama, E. Kawamura, S. Nagaoka and H. Kawakami, Mol. Pharmaceutics, 2006, 3, 468 CrossRef CAS PubMed.
- I. Batinić-Haberle, S. Cuzzocrea, J. S. Rebouças, G. Ferrer-Sueta, E. Mazzon, R. Di Paola, R. Radi, I. Spasojević, L. Benov and D. Salvemini, Free Radical Biol. Med., 2009, 46, 192 CrossRef PubMed.
- I. Batinic-Haberle, I. Spasojevic, M. T. Hubert, A. Tovmasyan, Z. Rajic, D. K. S. Clair, Z. Vujaskovic, M. W. Dewhirst and J. D. Piganelli, Amino Acids, 2012, 42, 95 CrossRef CAS PubMed.
- L. Benov and I. Batinic-Haberle, Free Radical Res., 2005, 39, 81 CrossRef CAS.
- B. Gauter-Fleckenstein, K. Fleckenstein, K. Owzar, C. Jiang, I. Batinic-Haberle and Z. Vujaskovic, Free Radical Biol. Med., 2008, 44, 982 CrossRef CAS PubMed.
- I. Kos, J. S. Rebouças, G. DeFreitas-Silva, D. Salvemini, Z. Vujaskovic, M. W. Dewhirst, I. Spasojević and I. Batinić-Haberle, Free Radical Biol. Med., 2009, 47, 72 CrossRef CAS PubMed.
- D. Lahaye, K. Muthukumaran, C.-H. Hung, D. Gryko, J. S. Rebouças, I. Spasojević, I. Batinić-Haberle and J. S. Lindsey, Bioorg. Med. Chem., 2007, 15, 7066 CrossRef CAS PubMed.
- J. S. Rebouças, I. Spasojević, D. H. Tjahjono, A. Richaud, F. Méndez, L. Benov and I. Batinić-Haberle, Dalton Trans., 2008, 1233 RSC.
- A. G. Tovmasyan, Z. Rajic, I. Spasojevic, J. S. Reboucas, X. Chen, D. Salvemini, H. Sheng, D. S. Warner, L. Benov and I. Batinic-Haberle, Dalton Trans., 2011, 40, 4111 RSC.
- I. Batinic-Haberle, L. Benov, I. Spasojevic and I. Fridovich, J. Biol. Chem., 1998, 273, 24521 CrossRef CAS PubMed.
- R. Shimanovich, S. Hannah, V. Lynch, N. Gerasimchuk, T. D. Mody, D. Magda, J. Sessler and J. T. Groves, J. Am. Chem. Soc., 2001, 123, 3613 CrossRef CAS.
- X. Zhang and D. H. Busch, J. Am. Chem. Soc., 2000, 122, 1229 CrossRef CAS.
- S. Rausaria, A. Kamadulski, N. P. Rath, L. Bryant, Z. Chen, D. Salvemini and W. L. Neumann, J. Am. Chem. Soc., 2011, 133, 4200 CrossRef CAS PubMed.
- P. Failli, D. Bani, A. Bencini, M. Cantore, L. Di Cesare Mannelli, C. Ghelardini, C. Giorgi, M. Innocenti, F. Rugi and A. Spepi, J. Med. Chem., 2009, 52, 7273 CrossRef CAS PubMed.
- S. J. Ophoven and G. Bauer, Anticancer Res., 2010, 30, 3967 CAS.
- S. R. Doctrow, K. Huffman, C. B. Marcus, G. Tocco, E. Malfroy, C. A. Adinolfi, H. Kruk, K. Baker, N. Lazarowych and J. Mascarenhas, J. Med. Chem., 2002, 45, 4549 CrossRef CAS PubMed.
- Y. G. Abashkin and S. K. Burt, Inorg. Chem., 2005, 44, 1425 CrossRef CAS PubMed.
- M. Sharpe, R. Ollosson, V. Stewart and J. Clark, Biochem. J., 2002, 366, 97 CAS.
- W. Park and D. Lim, Bioorg. Med. Chem. Lett., 2009, 19, 614 CrossRef CAS PubMed.
- S. J. Ophoven and G. bauer, Anticancer Res., 2010, 30, 3967 CAS.
- I. Batinić-Haberle, S. J. Rebouças and I. Spasojević, Antioxid. Redox Signaling, 2010, 13, 877 CrossRef PubMed.
- I. Batinic-Haberle, Z. Rajic, A. Tovmasyan, J. S. Reboucas, X. Ye, K. W. Leong, M. W. Dewhirst, Z. Vujaskovic, L. Benov and I. Spasojevic, Free Radical Biol. Med., 2011, 51, 1035 CrossRef CAS PubMed.
- D. Bani and A. Bencini, Curr. Med. Chem., 2012, 19, 4431 CrossRef CAS.
- J. T. Hartmann and H.-P. Lipp, Expert Opin. Pharmacother., 2003, 4, 889 CrossRef CAS PubMed.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |