
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNMR and TRLFS studies of Ln(III) and An(III) C5-BPP complexes†
Christian
Adam
*ab,
Björn B.
Beele
ab,
Andreas
Geist
a,
Udo
Müllich
a,
Peter
Kaden
a and
Petra J.
Panak
ab
aKarlsruhe Institute of Technology (KIT), Institute for Nuclear Waste Disposal (INE), P.O. Box 3640, 76021 Karlsruhe, Germany. E-mail: christian.adam@kit.edu; Fax: +49 721 608 23927
bUniversity of Heidelberg, Institute of Physical Chemistry, Im Neuenheimer Feld 253, 69120 Heidelberg, Germany
First published on 19th December 2014
Abstract
C5-BPP is a highly efficient N-donor ligand for the separation of trivalent actinides, An(III), from trivalent lanthanides, Ln(III). The molecular origin of the selectivity of C5-BPP and many other N-donor ligands of the BTP-type is still not entirely understood. We present here the first NMR studies on C5-BPP Ln(III) and An(III) complexes. C5-BPP is synthesized with 10% 15N labeling and characterized by NMR and LIFDI-MS methods. 15N NMR spectroscopy gives a detailed insight into the bonding of C5-BPP with lanthanides and Am(III) as a representative for trivalent actinide cations, revealing significant differences in 15N chemical shift for coordinating nitrogen atoms compared to Ln(III) complexes. The temperature dependence of NMR chemical shifts observed for the Am(III) complex indicates a weak paramagnetism. This as well as the observed large chemical shift for coordinating nitrogen atoms show that metal–ligand bonding in Am(C5-BPP)3 has a larger share of covalence than in lanthanide complexes, confirming earlier studies. The Am(C5-BPP)3 NMR sample is furthermore spiked with Cm(III) and characterized by time-resolved laser fluorescence spectroscopy (TRLFS), yielding important information on the speciation of trace amounts of minor complex species.
Introduction
In 2010 about 13% of the world's electricity is supplied by nuclear power plants,1 producing 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 tons of spent nuclear fuel annually.2 Among the major challenges of used nuclear fuel are the long-term radiotoxicity and long-term thermal power that are dominated by plutonium and the minor actinides (MA = Np, Am, and Cm).
500 tons of spent nuclear fuel annually.2 Among the major challenges of used nuclear fuel are the long-term radiotoxicity and long-term thermal power that are dominated by plutonium and the minor actinides (MA = Np, Am, and Cm).
Both problems are addressed by the Partitioning and Transmutation strategy (P&T)3 that could have a beneficial impact on the design of a safe final repository.3,4 It involves separating plutonium and the minor actinides from the used fuel and converting them into shorter-lived fission products by neutron-induced nuclear reactions. In this context the separation of trivalent actinides An(III) from fission lanthanides Ln(III) is the key step, as some lanthanides have high neutron cross sections, consequently diminishing the efficiency of the transmutation step. Due to the similarity of An(III) and Ln(III) both in chemical properties and ionic radii, highly selective extracting agents are needed to achieve a reasonable separation.5
It has been shown that extractants containing either soft sulfur or soft nitrogen donor atoms exhibit the required selectivity.6 Heterocyclic N-donor ligands derived from the terpyridine motif have shown higher complex strengths towards trivalent actinides than towards trivalent lanthanides.7 Among these, heteroaromatic nitrogen donor ligands 2,6-bis(1,2,4-triazine-3-yl)pyridines (BTPs) were the first extractants to achieve separation factors for Am(III) over Eu(III) higher than 100 from nitric acid solutions.7,8 They show good solubility in a range of organic diluents and form stable and isostructural 1:3 complexes with lanthanides and actinides.9–16 Furthermore, they co-extract nitrate anions from the aqueous phase and, unlike other similar extracting agents, do not need additional lipophilic anion sources such as 2-bromocarboxylic acid.17–19 In order to attain a fundamental understanding of the BTP-type ligands' selectivity on a molecular level, the tridentate N-donor ligand C5-BPP was synthesized and tested for its extraction behavior.20 It was found that C5-BPP serves as a useful extracting agent with separation factors for Am(III) over Eu(III) over 100. However, it does not co-extract nitrate anions from the aqueous phase and is thus dependent on a lipophilic anion source. The ability to form stable 1:3 complexes and the different extraction behavior made C5-BPP an interesting target for investigations on the reason of the observed selectivity, especially in comparison to recent studies with nPrBTP.21
The molecular reason for the observed selectivity of some N-donor ligands is still largely unclear. A larger degree of covalence in the actinide–ligand bond, compared to lanthanide complexes, has been assumed to account for the observed extraction behavior.22–24 A more covalent bonding might result from a better overlap of the soft nitrogen lone pair with the diffuse 5f-orbitals of the actinide ions. In this case, the ratio of covalent to dative electrostatic bonding in actinide–N-donor complexes is expected to be larger than in isostructural lanthanide compounds. Results from K-edge XAS spectroscopy on An(III) complexes with ligands containing sulfur,25 oxygen and chlorine26 seem to support this explanation.
Actinide compounds are a challenge for quantum chemistry due to various reasons, like for example the inclusion of relativistic effects. So far, prediction of bonding modes and NMR shifts is limited to simple systems and hardly implemented in commercial software packages. As an example for these problems, quantum chemical treatment of Am(III) and Eu(III) complexes with Cyanex 301 seemed to show a more covalent bonding in the actinide case, based on consideration of the bond length as a marker for covalence.27 Yet this produces misleading results, as calculated lanthanide–ligand bond lengths are too long in comparison to experimental data and bond lengths calculated by more sophisticated quantum-chemical methods.28
Recently we were able to obtain the first NMR spectroscopic proof of a fundamentally different binding mode in Am(III) complexes with N-donor ligands.21
In general, NMR is an excellent spectroscopic method for the investigation of bonding interactions: the electrons of soft donor ligands can interact with positively charged cations either by sharing electron density in overlapping frontier orbitals or by electrostatic interactions. Both mechanisms lead to a rearrangement of electron density, which is monitored very precisely as the chemical shift in NMR spectroscopy. NMR focusing on the paramagnetism of the compounds allows the separation of the overall chemical shift into a part that is due to delocalization of electron spin density through covalent bonds (Fermi contact shift, FCS) and a distance- and angle-dependent part due to interaction of the anisotropic electron magnetic moment, assumed to be located at the metal ion, and the nuclear magnetic moment of ligand nuclei (pseudo contact shift, PCS).29–35 Currently, several methods for this separation of FCS and PCS32,36 are under investigation regarding their applicability to actinide complexes.
The scope of the work presented in this paper is to generate a reliable base of NMR spectra of diamagnetic and weakly paramagnetic C5-BPP lanthanide complexes and of the Am(III) complex. With these data, we aim to elucidate the bonding mode and potential bonding differences in lanthanide and actinide C5-BPP complexes, as this is expected to be the driving force of the ligand's selectivity for actinide over lanthanide extraction.
Theoretical and NMR background
The chemical shift – and thus the electron distribution – of the coordinating nitrogen atoms are of particular interest for the investigation of bonding interactions. The effect of covalent bonding is especially pronounced here, since transferred electron density can normally only be detected over a few covalent bonds. Only in some cases nuclei more than three bonds away from coordinating atoms are influenced by FCS. Unfortunately, obtaining resonance signals in one-dimensional direct excitation spectra from 15N atoms at natural abundance is impossible in a time-effective manner. This is due to the fact that 15N has a low natural abundance of 0.364% and a low negative gyromagnetic ratio (γ = −0.28), resulting in low receptivity of the nucleus (about 1% of the 13C receptivity at natural abundance).37 Furthermore, a negative gyromagnetic ratio means that the Nuclear Overhauser Effect will decrease the signal intensity for 15N if 1H broadband decoupling is used.In paramagnetic coordination compounds the overall experienced chemical shift δtot has several contributions:
| δtot = δdia + δcon + δpc + δanion | (1) |
δ dia is the diamagnetic (or orbital) shift of the compound, δcon represents the Fermi contact shift, a through-bond effect, δpc is the pseudo contact shift that originates from coupling of the electron magnetic moment on the metal ion and the ligand nuclei spins and δanion is the influence of the counter-anion. All published methods for the separation of these terms have in common that they rely on an isostructural diamagnetic analog to the paramagnetic complexes. The purely paramagnetic shift δpara = δcon + δpc is calculated by simply subtracting the chemical shift values of the diamagnetic reference compound from the measured chemical shifts of the paramagnetic complexes (eqn (2)). If reference and paramagnetic complexes have the same counter-anion, δanioncancels out.
| δpara = δtot − δdia | (2) |
In the lanthanide series, La(III) and Lu(III) are diamagnetic ions and their complexes are generally used as diamagnetic reference compounds. Furthermore, Y(III) often forms complexes which are isostructural to the lanthanide compounds and can also be used as a reference.
In principle, paramagnetic compounds provide a detailed insight into the bonding mode via the separation of the observed paramagnetic chemical shift δpara into FCS and PCS. For this task, several methods have been proposed in the literature. Methods based on the chemical shift dependence on the temperature38,39 have been a matter of controversy and their application has to be evaluated very carefully.40 Currently, the standard procedure is the evaluation of the purely paramagnetic shift throughout the complete lanthanide series vs. tabulated lanthanide-depending constants, i.e. spin-expectation values 〈Sz〉 and Bleaney parameters CLn.29,31,32,35,41 Lanthanide shift reagents42–45 and lanthanide probes for protein structure determination31,46–48 have been widely used in NMR spectroscopy and thus lanthanide magnetic properties are quite well understood. This is not the case for magnetic properties of elements of the actinide series, thus Bleaney parameters CLn and spin expectation values 〈Sz〉 are unknown for these cations.
So far, only a small number of proton spectra and a few heteronuclear spectra of actinide containing compounds are available. These are largely limited to uranium in several oxidation states and hence there is a paucity of NMR studies on organic complexes with transuranium elements.
The magnetic properties of the free Am(III) ion are still a matter of debate in literature, as deviations from the expected diamagnetism arising from a predicted J = 0 ground state have been found. Optical spectroscopy and DFT calculations show that the first non-diamagnetic excited states are some thousand wavenumbers higher in energy and thus thermally not populated and mixing of the states is not expected.49 This was also confirmed by experimental work on an [Am(H2O)9](CF3SO3)3 crystal in solid state, which exhibited a magnetic susceptibility curve that can be interpreted as non-magnetic behavior.50 On the other hand, surprisingly large magnetic susceptibilities and magnetic moments have been reported for different Am(III) compounds in the solid state, indicating that Am(III) is not purely diamagnetic.51–54 Recently, the magnetic susceptibility of Am(III) in perchloric acid solution was studied using the Evans NMR method.55 Results show a significant deviation from the expected magnetic behavior for Am(III) and Cf(III). Magnetic susceptibilities for both ions were found to be higher than expected.56
In a recent publication, the effect of spin–orbit coupling on the alignment of spins in a magnetic field and the applicability of the Russel–Saunders coupling scheme was discussed.57 Spin expectation values for different configurations of Am(III) were calculated. The authors show that the expected J = 0 ground state has a spin expectation value 〈Sz〉 = 0 and contains the expected Hund's multiplet to 63%. The energy difference to the J = 1 state with 〈Sz〉 = 0.5 is only 0.24 eV, which is significantly lower than the expected value of 3.0 for six unpaired electrons. The authors conclude that there is a significant deviation from the multiplets expected from Hund's rules, but that pure j–j-coupling cannot describe the electronic states as well.
In the case of the 243Am(nPrBTP)3(NO3)3 complexes, we were able to acquire one- and two-dimensional 1H, 13C and 15N spectra in good quality.21 The observed linewidths in the spectra and the range of chemical shifts indicate that Am(III) has only a weak paramagnetism, with effects even smaller than observed for Sm(III). These results encouraged us to expand our NMR studies to the Am(III) complexes of C5-BPP.
Synthesis
To compensate the unfavourable NMR spectroscopic properties of 15N, we synthesized a 15N enriched C5-BPP ligand, {15N}C5-BPP, in accordance to the already published 15N enriched nPrBTP ligand.21 The synthesis pathway is shown in Scheme 1. Successful labeling was confirmed by NMR spectroscopy and LIFDI-MS58,59 (cf. ESI†).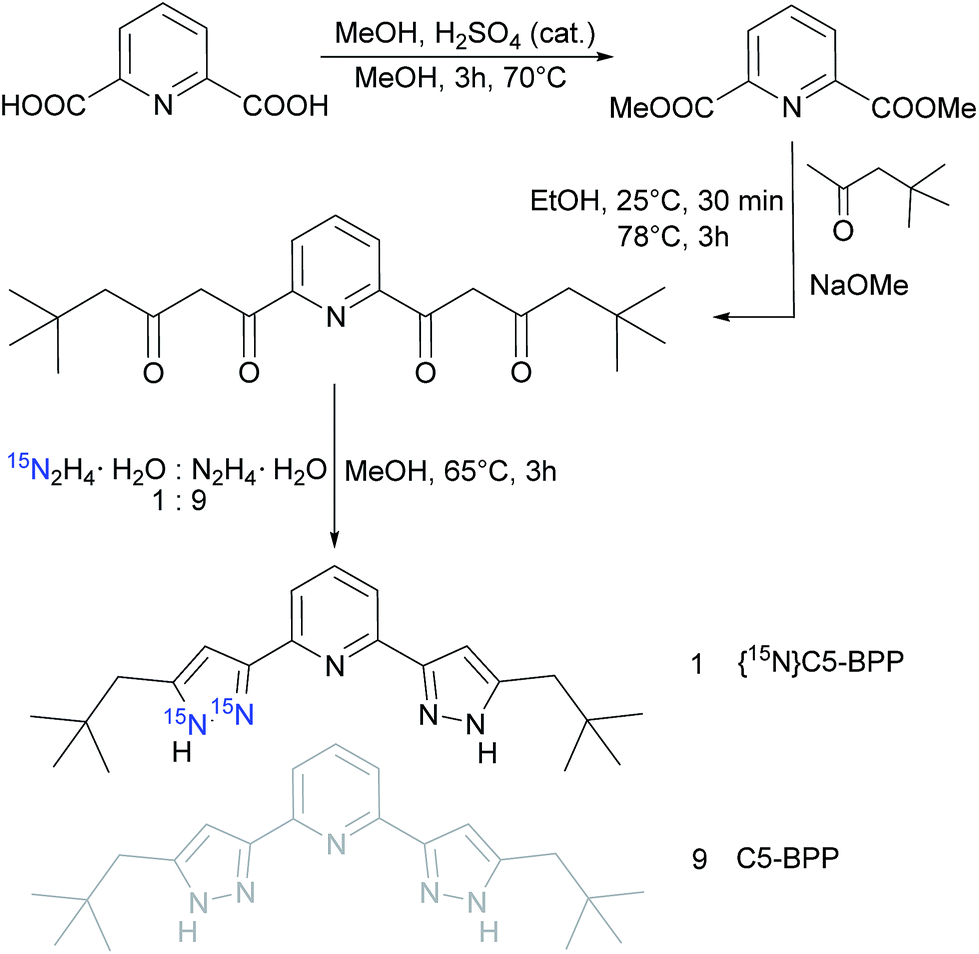 | ||
| Scheme 1 Synthesis and labeling of the pyrazole moiety in C5-BPP with 10% 15N; adapted from the synthesis protocol in ref. 20. | ||
Using {15N}C5-BPP, 1:3 complexes with lanthanides (La(III), Sm(III), Yb(III), Lu(III)) and Y(III) were prepared. In order to compare these complexes to a 1:3 complex with a trivalent actinide we also prepared a {15N}C5-BPP complex with 243Am (Fig. 1).
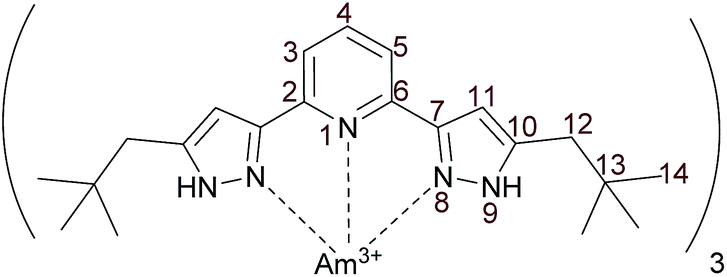 | ||
| Fig. 1 Molecular structure and numbering scheme of the [243Am(C5-BPP)3]3+ complex. | ||
All complexes were prepared in deuterated methanol. Earlier studies on crystal structures of the Ln(III) complexes report that C5-BPP does not displace all nitrate anions from the inner coordination sphere of the central metal ions during crystallization.20 In our case 1H NMR spectroscopy on Ln(III) complexes showed that more than one complex species was formed in samples in which nitrate anions were present. Diffusion-ordered NMR spectroscopy (DOSY)60–62 proved that several complex species with varying diffusion coefficients were present. This is due to the fact that nitrate anions are strongly complexing ligands in pure organic solvents. The formation of numerous different complex species was overcome by using triflate salts (OTf−, CF3SO3−) for which the counter-anion has been shown to be non-coordinating. Indeed, in NMR spectra of C5-BPP lanthanide triflate complexes, only the desired 1:3 complex and, occasionally, small traces of a 1:2 complex, were found.
In order to perform NMR investigations using complexes with the same counter-anion, Am(OTf)3 was prepared from an Am(NO3)3 stock solution. Subsequently 15N labeled and unlabeled C5-BPP was used to synthesize [Am(C5-BPP)3](OTf)3. To avoid potential magnetic impurities due to radiolysis of the solvent and impurities from radioactive decay products we used the long-lived isotope 243Am (t1/2 = 7370 a).
Results and discussion
Diamagnetic Ln(III)–(C5-BPP)3 complexes
As a first step in the investigation of bonding modes in C5-BPP complexes of lanthanide and actinide ions, we focused on diamagnetic or nearly diamagnetic compounds. A comparison of spectra of diamagnetic compounds is straightforward, as significant changes between isostructural complexes can be attributed to a change in binding mode.In our studies with nPrBTP we used the Lu(III) complex as a diamagnetic reference, since spectra of [La(nPrBTP)3](NO3)3 showed broadened spectral lines.21 This is due to a relatively weak coordination of nPrBTP to the large La(III) ion which decreases the complex symmetry and thus results in broad spectral lines. The bigger bite angle of the pyrazole nitrogen lone pairs in C5-BPP should enable this ligand to form structurally rigid complexes even with slightly larger cations. Indeed, we found that C5-BPP forms stable complexes with La(III), resulting in well-resolved NMR spectra with sharp lines.
Comparison of the three diamagnetic C5-BPP complexes (Y(III), La(III) and Lu(III)) shows that although all three metal ions are diamagnetic, there are significant differences in 1H, 13C and 15N NMR chemical shifts. These differences are strongest in close proximity to the metal ion, and only very weak at the aliphatic side chain. Differences between proton spectra of Y(III) and Lu(III) complexes are small, with a maximum of 0.01 ppm at the H4 triplet. The maximal discrepancy between proton signals of the La(III) and Lu(III) complexes is found for the signals of H3/5 with 0.04 ppm. The differences are more pronounced in 13C spectra. Again the spectra of Lu(III) and Y(III) complexes strongly resemble each other. Only for C2/6 (Δδ = 0.3 ppm) and C10 (Δδ = 0.28 ppm) significant discrepancies are observed. Differences between the La(III) and Lu(III) complexes are stronger in particular for the quaternary carbon atoms C7 (Δδ = 0.73 ppm), C2/6 (Δδ = 1.70 ppm), and C10 (Δδ = 1.20 ppm).
As the influence of the central metal ion seems to be strongly dependent on the distance to the observed nucleus it should be even more pronounced on the nitrogen atoms. In 15N spectra we observe only weak shift differences for the non-coordinating N9 (Y/Lu (Δδ = 0 ppm), La/Lu (Δδ = 1.2 ppm)). The coordinating nitrogen shifts show a stronger dependence on the central metal ion. The shift differences for N1 (Y/Lu (Δδ = 1.0 ppm), La/Lu (Δδ = 4.0 ppm) from 1H,15N-gHMQC spectra) are smaller than for N8 (Y/Lu (Δδ = 1.7 ppm), La/Lu (Δδ = 7.2 ppm)). These results coincide with the differences in ionic radii, which are quite similar for Y(III) (90.0 pm) and Lu(III) (86.1 pm), whereas La(III) is significantly larger (103.2 pm).63 Changes in the complex geometry and subsequently changed interaction between the metal ion and the ligand can explain the observed behavior.
These results clearly show that the diamagnetic reference compound needs to be chosen carefully, as the shift differences between La(III) and Lu(III) compounds are significant and several orders of magnitude larger than the spectral resolution. Based on our results we assume that La(III) is a good diamagnetic reference compound for the lighter part of the lanthanide series. The smaller metal ions Lu(III) and Y(III), which both have closed shells, are better suited as reference compounds for the heavier lanthanides. The error inferred from the reference compound on the determination of the purely paramagnetic chemical shift in strongly paramagnetic systems, where shifts of several hundred ppm can occur, are limited. However, the influence on weakly paramagnetic systems should not be underestimated.
It should be noted that in all diamagnetic lanthanide C5-BPP complexes and in the Y(III) C5-BPP complex, resonance signals for the coordinating nitrogen atoms N1 and N8 are generally found in a 12 ppm range around 266 ppm. For the non-coordinating nitrogen atom N9,resonance signals are found in a narrow 2 ppm range around 206 ppm (cf.Table 1 and Fig. 2). In comparison to the free ligand, the coordination of C5-BPP to a M(III) cation hardly influences the chemical shift of N9. N8 on the other hand is shifted approximately 30 ppm upfield. This is due to the rearrangement of electron density upon complex formation. Unfortunately, in the free ligand no resonance signal is observed for N1. Nevertheless, based on the diamagnetic lanthanide compound spectra, we would expect the resonance signal for N1 in the same shift range as N8. The same problems were encountered when we measured 13C spectra of the free ligand. We found that resonance signals for the quaternary carbon atoms C2/6, C7, and C10 are severely broadened and sometimes, as in the free 15N enriched ligand, unobservable in 1D spectra. So far we do not have a clear explanation for this behavior.
 | ||
| Fig. 2 15N direct excitation spectra in MeOD-d4 of all C5-BPP complexes in this study. N9 signals are labeled with green circles, N8 signals with red circles. | ||
Paramagnetic Ln(III)–(C5-BPP)3 complexes
In the following we studied the influence of a weakly and a strongly paramagnetic central metal cation on the NMR spectra of the C5-BPP complexes. We used [Sm(C5-BPP)3](OTf)3 as a representative for a weakly paramagnetic ion (Sm3+: μeff = 0.85μB) and [Yb(C5-BPP)3](OTf)3 as a strongly paramagnetic ion (Yb3+: μeff = 4.54μB).30,43 With the 15N labeled ligand in hand, our focus was on the influence of paramagnetism on the resonance signals of the coordinating nitrogen atoms. In the Sm(III) complex, the N9 resonance signal is observed at 205 ppm, i.e. without additional shift compared to the diamagnetic compounds. In contrast to the non-coordinating nitrogen, a larger shift is found for the coordinating nitrogen atoms. Compared to the La(III) complex, N1 is shifted 45 ppm upfield and N8 is shifted 48 ppm upfield. These values are in good agreement with observed shifts for nPrBTP complexes.21 Yb(III) complexes usually show the expected strong paramagnetic shifts, but paramagnetic relaxation enhancement for Yb(III) is still weak enough that spectral lines are not too broad to be observed and most multi-dimensional NMR experiments yield good results. Thus, unambiguous assignment of most signals is possible by heteronuclear correlation spectroscopy. However, due to the enhanced relaxation, the 15N signals for N8 and N9 in Yb(C5-BPP)3(OTf)3 are only found after 15N-labeling. N9 shows a notable shift of −10 ppm compared to the diamagnetic references, which can be attributed to the stronger PCS. The coordinating N8 is shifted by approximately −200 ppm to 20 ppm.A comparison of 15N direct excitation spectra of all investigated 1:3 M(III) C5-BPP complexes is shown in Fig. 2. The N9 signals (green circles) for the diamagnetic metal ions (Y(III), La(III), Lu(III)) show almost identical chemical shift values (green dotted line), while for N8 (red circles) there is a notable difference (red dotted line). Furthermore, the chemical shift for the non-coordinating N9 remains nearly constant even for the paramagnetic ions, while N8 shows a strong dependency on the paramagnetism of the ion.
The larger shift in the Yb(III) and the Sm(III) cases can be attributed to a stronger PCS (especially for Yb, which predominantly exhibits PCS) but as well a non-negligible FCS. Heteronuclei directly bonded to paramagnetic cations have only scarcely been investigated with respect to the different contributions to the experienced paramagnetic shift. Most research is limited to protons in close proximity to the metal ion center. However, although the influence of FCS decreases rapidly along covalent bonds, it often cannot be neglected.35 A strong impact of the FCS on directly coordinated nuclei can thus be expected, and, as in our case, might even contribute to a larger than expected share. Further research into this topic is necessary and currently under way in our group.
NMR-spectroscopy on Am(III)–(C5-BPP)3
The spectra of the Am(III)–C5-BPP complexes with and without 15N labeling show that more than one complex species was formed. Upon addition of further ligand solution two of the complex species could be assigned to the free ligand and the 1:2 complex. Signals from the 1:3 complex, which forms the major species present in the sample, increase in intensity with increasing ligand-to-metal ratio. However, during titration another minor complex species that contains only one ligand molecule and a so far unknown contaminant not visible by NMR spectroscopy is formed. However, the NMR signals of the 1:3 complex as the major species could easily be identified and unambiguously assigned. To further elucidate the composition of the complex speciation we studied the samples by further NMR spectroscopic methods and time-resolved laser fluorescence spectroscopy (TRLFS, see below).
For a sample containing several different components, diffusion-ordered NMR spectroscopy (DOSY) is a versatile method. 1H DOSY spectra show three well separated complex species. 19F direct excitation spectra only show one signal at −79.97 ppm, which corresponds to the triflate anion. 19F DOSY spectra yield one diffusion coefficient for this peak which differs from the coefficients for the complex species calculated from 1H DOSY measurements. Thus a coordinated triflate anion or exchange between a bound and a free form can be excluded. All 1D spectra are well-resolved, and unambiguous assignment of the signals of the 1:3 Am(III) C5-BPP complex is possible. The complex is fully characterized by 1H, 13C and 15N direct excitation spectroscopy at different temperatures as well as a range of 2D heteronuclear correlation spectroscopy methods.
Information about magnetic properties and the bonding situation can be deduced by comparison of the Am(III) complex spectra and those of a diamagnetic reference compound. Unfortunately, the diamagnetic actinides Ac(III) and Lr(III) have short half-lifes (t1/2(227Ac) = 21.8 a, t1/2(262Lr) = 3.6 h) and are not available in sufficient amounts. As we lack a diamagnetic actinide reference compound, we have to compare the Am(III) complex's chemical shifts to those of Ln(III) complexes. This comparison is displayed in Fig. 3 for [Am({15N}C5-BPP)3](OTf)3 and [Sm({15N}C5-BPP)3](OTf)3 compared to [La({15N}C5-BPP)3](OTf).
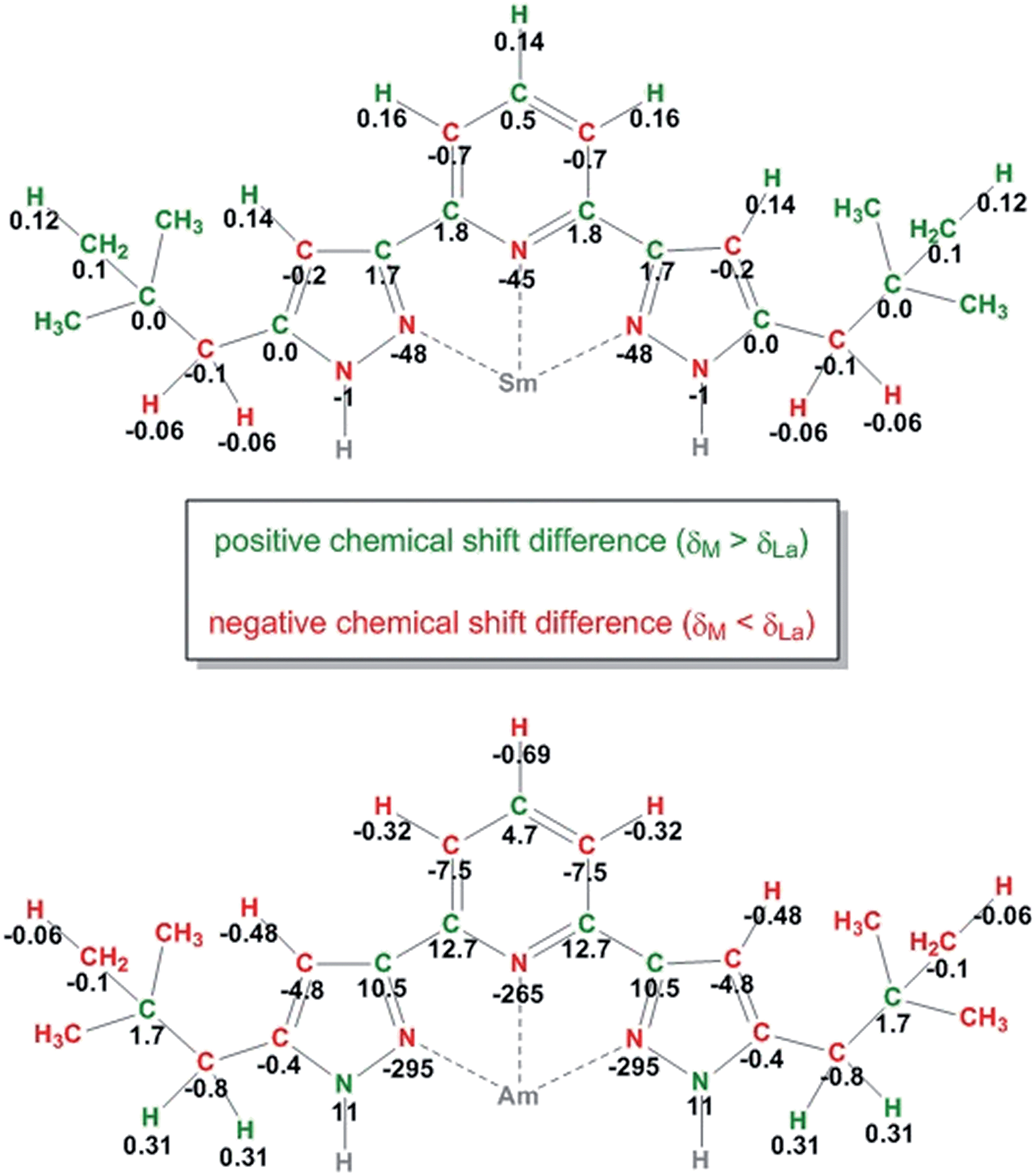 | ||
| Fig. 3 Chemical shift differences between the NMR signals in the Sm(III) (top) and Am(III) (bottom) complexes compared to the La(III) complex for all nuclei. All values are given in ppm. | ||
For most nuclei the effect of the Am(III) cation on the chemical shift is approximately ten times stronger than that of the Sm(III) cation. For the weakly paramagnetic Sm(III) a magnetic moment of μeff = 0.85μB is known.30,63 Measurements using the Evans method yield a magnetic moment of μeff = 1.64μB for Am(III).56 Recently, work on the influence of radioactive decay and radiolysis product formation on the accuracy of the Evans method has been published, suggesting a reduced magnetic moment of approximately μeff = 1.42μB.64 We therefore expect the paramagnetic influence of Am(III) to be stronger than the influence of Sm(III), but both should produce paramagnetic chemical shift effects in the same order of magnitude.
The large differences in the chemical shifts cannot be explained by the difference in the magnetic moments of the cations, but point to a fundamental change in the binding mode. Fig. 4 shows a 1H,15N-gHMQC spectrum of [Am({15N}C5-BPP)3](OTf)3. Indicated with the red boxes are the chemical shifts for the coordinating nitrogen atoms as expected from the free ligand and diamagnetic Ln(III) compounds. In the red circles are the measured values that differ vastly from the expectations. For the Am(III) complex, the immense shift differences of the coordinating nitrogen atoms (N1: −256 ppm, N8: −295 ppm) are noteworthy. Shifts of a comparable magnitude have only been found for a Yb(III) C5-BPP complex which has a considerably stronger effective magnetic moment (μeff = 4.54μB).32
 | ||
| Fig. 4 1H,15N-gHMQC spectrum of Am[({15N}C5-BPP)3](OTf)3 in MeOD-d4, optimized for a coupling constant JHN = 5 Hz. The expected value range is taken from similar experiments with the diamagnetic lanthanide complexes. The correlation signals in the gray circles originate from an additional minor complex species. | ||
Furthermore, carbon atoms in both the Sm(II) and the Am(III) complexes show alternating positive and negative chemicals shift differences along the carbon backbones of the ligands (see Fig. 3). This phenomenon is indicative of the simultaneous existence of spin polarization and spin delocalization at the ligand (polarized spin density delocalization).64-67 This spin delocalization would be due to a Fermi contact interaction between metal cation and N-donor ligands and thus to a share of covalence in the bonding. The pattern of the shift differences suggests that a part of the delocalized spin electron density resides in pz orbitals of the sp2 hybridized carbon atoms. However, if this is true we would expect the signs of the chemical shift difference of the protons to be inverse to the attached carbons' shift differences (two spins that are coupled electronically over one bond will have opposite signs). We find that for Sm(III) and Am(III) all pyridine proton shift differences have the same signs. In both cases H4 is shifted more towards deeper fields than the H3/5 protons. This behavior suggests that two (or even more) different mechanisms take part in the delocalization of electron spin density, showing that the bonding between Am(III) and the soft N-donor ligand is a very complicated matter. An explanation for the downfield shift of H4 could be that unpaired electron spin density is also transferred through σ bonds in the aromatic ring or the conjugated double bonds, respectively.
To gain insight into magnetic and bonding behavior, we acquired NMR spectra at different temperatures between 185 K and 335 K (cf. ESI†). In [La({15N}C5-BPP)3](OTf)3, N8 shows a temperature-dependent shift of 0.3 ppm. The N9 signal shows strong line broadening (FWHM 18.54 ± 0.28 Hz) even at low temperatures. Thus, N9 is only observable up to 315 K. In the monitored 130 K temperature range, the N9 signal shows a temperature-dependent shift of −1 ppm.
In the case of the Y(III) C5-BPP complex, N8 experiences a 0.5 ppm downfield shift between 185 K and 335 K, while N9 (FWHM at 185 K: 19.10 ± 0.18 Hz) shows a −1.4 ppm upfield shift.
The temperature-dependent chemical shift of the Am(III) complex shows a different behaviour (Fig. 5). The non-coordinating N9 (FWHM at 285 K: 20.04 ± 0.19 Hz) shows an upfield shift of approximately 1 ppm at 275 K with increasing broadening of the resonance signal. This signal is not observable above 300 K, while a new doublet appears 0.8 ppm downfield of the last broad signal. Up to a temperature of 335 K this signal is again shifted upfield by 0.5 ppm. In total, the temperature-dependent shift of N9 is 0.6 ppm. The coordinating N8, however, shows a continuous 11.3 ppm downfield shift. This temperature-dependent shift is approximately ten times the shift measured for diamagnetic reference compounds, which is another distinct piece of evidence showing that Am(III) is not diamagnetic.
 | ||
| Fig. 5 Sections of the 15N direct excitation spectra of [Am({15N}C5-BPP)3](OTf)3 in MeOD-d4 at increasing temperatures (N9 left side, N8 right side). All spectra are referenced to the internal standard TMS by the lock signal. | ||
The observed chemical shift of the coordinating nitrogen atoms in the Am(III) complex cannot be explained by the different strength of the ions' magnetic moments alone, as a comparison to the temperature dependent NMR spectra of the strongly paramagnetic [Yb({15N}C5-BPP)3](OTf)3 shows. As the chemical shift of N8 at room temperature is +20 ppm and thus close to the observed shift of the Am(III) complex, one could assume that the two ion's paramagnetism were in the same order of magnitude. However, the complex with the more paramagnetic Yb(III) cation shows a larger chemical shift range upon temperature change: In the monitored temperature range, N8 shows a 152 ppm shift, for the non-coordinating N9 the shift is still −8 ppm. Furthermore, even in the weakly paramagnetic Sm(III) complex, N8 shows a temperature-dependent shift of −162 ppm in the observed temperature range. Thus it is clear that the paramagnetism of the Am(III) ion is considerably weaker than at least in the Yb(III) ion and cannot satisfactorily explain the observed highfield shift of the N8 signal in [Am({15N}C5-BPP)3](OTf)3. The smaller temperature-dependent shift in the Am(III) complex, compared to Sm(III), could be due to a different ratio of covalent and dipolar bonding: FCS is transmitted through covalent bonds and has a linear temperature dependency. PCS, which can be associated to dipolar interactions, has a T−2 dependency. However, as long as no clear separation of the chemical shift contribution can be performed, this has to be seen as indicative of a more covalent bond, but not yet as a proof.
The observed behavior of alternating chemical shift effects in the carbon backbone, but not on the protons in the ligand, points towards a combination of direct spin delocalization and polarized spin density delocalization. Both rely on a Fermi contact interaction arising from covalent bonding between the trivalent metal cation and the nitrogen atoms of the ligands. From comparison of the observed chemical shift differences in the slightly paramagnetic Sm(III) complex and the Am(III) complex, which cannot be explained by paramagnetism alone. We interpret this fact as indicative of an higher share of covalence in the actinide compound, which is consistent with recently reported XAS and EXAFS studies.68 Another effect that might explain the shift differences between Am(III) and the Ln(III) complexes is the existence of spin-orbit coupling effects on the metal ion which influences the shift of the nitrogen atom.69–71 Spin–orbit coupling is strongly dependent on the atomic number of the nucleus and is thus considerably stronger in the actinide series than for lanthanides. Spin polarization from spin–orbit coupling resembles spin-spin coupling effects in NMR spectroscopy that are mediated by s-type orbitals.71 This is another mechanism that could explain the substantial shifts on the nitrogen atoms and why the shift differences cannot be observed on neighboring atoms. As a consequence, both paramagnetic effects in the form of FCS and spin–orbit coupling seem to play an important role in the observed chemical shifts. Fermi-contact interactions and thus the existence of a certain covalence compared to the lanthanide compounds could thus explain the observed shifts on the nitrogen atoms of the americium complex.
Cm(III) TRLFS studies to identify the minor complex species
As shown above minor Am(III) complex species are formed in addition to the prevailing 1:3 Am(III) complex. Small amounts of impurities or minor complex species cannot be characterized using NMR spectroscopy. Hence we used a different spectroscopic method to elucidate the composition of the minor complex species. Addition of a trace amount (6.6 × 10−8 mol × L−1) of Cm(III) to the [243Am(C5-BPP)3](OTf)3 NMR sample enabled us to make use of the excellent fluorescence properties of Cm(III). Furthermore, the chemical properties of Am(III) and Cm(III) are highly comparable which is for example reflected in very similar M(III)–N bond lengths in 1:3 complexes in solution9 (Am(nPr-BTP)3:Am(III)–N = 256 pm; Cm(nPr-BTP)3:Cm(III)–N = 257 pm).13,14
After addition of Cm(III) to the NMR sample the Cm(III) emission bands are recorded with increasing amounts of C5-BPP ligand. Upon addition of ligand solution to the Cm(III) spiked NMR sample the initial Am(III) and Cm(III) metal concentrations are diluted, and hence the ligand-to-metal ratio stepwise increases. The development of the Cm(III) fluorescence emission resulting from the 6D′7/2 → 8S′7/2 transition as function of the ligand-to-Am(III) ratio are shown in Fig. 6. The spectra are normalized to the same peak area for better comparison.
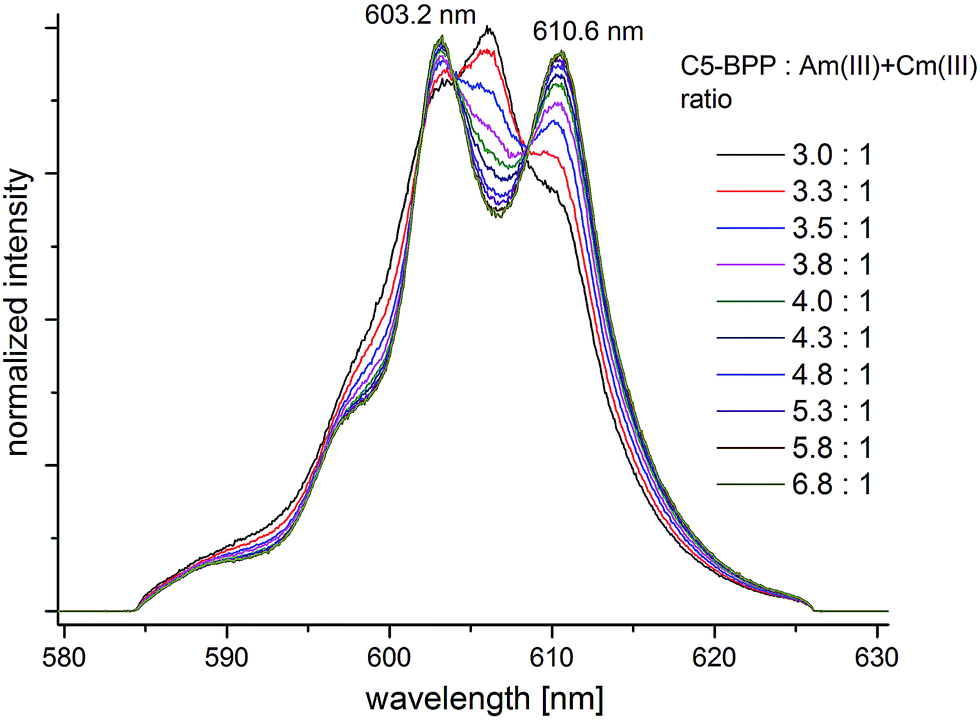 | ||
| Fig. 6 Normalized fluorescence spectra of Cm(III) in MeOD-d4 with increasing amount of C5-BPP. [Cm(III)]ini = 6.6 × 10−8 mol L−1, [Am(III)]ini = 6.0 × 10−6 mol L−1. | ||
At an initial C5-BPP-to-Am(III) + Cm(III) ratio of 3:1 (corresponding to a ligand-to-Cm(III) ratio of 4.5 × 105: 1) an emission band with a maximum at λmax = 606.0 nm and two weak shoulders at λmax = 603.2 nm and λmax = 610.3 nm are observed.
With increasing ligand-to-metal ratio the intensity of the emission band at λmax = 606.0 nm decreases significantly while both shoulders gain in intensity. At a final ligand-to-Am(III) + Cm(III) ratio of 6.8:1 two distinct emission bands with maxima at λmax = 603.2 nm and λmax = 610.6 nm are observed with an intensity ratio of approximately 1:1.
In earlier studies the emission bands of the Cm(III)–C5-BPP 1:1, 1:2 and 1:3 complex species in methanol were observed at λmax = 603.7 nm, λmax = 607.7 nm and λmax = 611.6 nm, respectively.20 Hence, the observed emission bands at λmax = 606.0 nm and λmax = 610.6 nm are attributed to a Cm(III)–C5-BPP 1:2 and a Cm(III)–C5-BPP 1:3 complex species. The hypsochromic shift of 1.0 nm in comparison to the literature known 1:2 and 1:3 complex species are assigned to the use of a deuterated solvent and the high concentration of triflate anions.
With increasing ligand-to-metal ratio a decreasing ratio of the 1:2 complex species and an increasing ratio of the 1:3 complex species are observed, showing a stepwise complexation of Cm(III). The emission band at λmax = 603.2 nm also gains in intensity upon increasing amount of C5-BPP, which confirms that does not result from the 1:1 Cm(III)–C5-BPP complex, and is attributed to a Cm(III) complex species with a minor impurity from the C5-BPP synthesis. At significantly higher metal concentrations used for NMR studies these minor complex species do not play an important role and all signals of the 1:3 complex species can be assigned unambiguously (see above).
Conclusions
We present the first NMR study on a series of 1:3 complexes of Ln(III) and Am(III) with the tridentate N-donor ligand C5-BPP. A key step in our investigations was the synthesis of a C5-BPP molecule with 15N enrichment in the pyrazole moieties.
Using {15N}C5-BPP we prepared 1:3 complexes with trivalent lanthanide ions (La, Sm, Yb, Lu and Y) and Am(III) as a representative of the trivalent actinides. In diamagnetic complexes, signals of the non-coordinating N9 are observed in a small chemical shift range between 195 ppm and 206 ppm. At room temperature, the coordinating N8 signals are found in a chemical shift range between 224 ppm and 275 ppm. Comparing the three diamagnetic complexes Y(III), La(III), and Lu(III), we found significant differences in 1H, 13C and 15N spectra. This shows that diamagnetic reference compounds for the extraction of purely paramagnetic shifts δpara have to be chosen with care. We conclude that La(III) serves as good diamagnetic reference for the lighter part of the lanthanide series and Lu(III) and Y(III) are better suited for the heavier lanthanides.
We furthermore prepared the [Am({15N}C5-BPP)3](OTf)3 complex and showed that NMR resonance signals for this complex have a stronger temperature dependence than signals of complexes with diamagnetic Ln(III), but weaker than for paramagnetic Yb(III) and Sm(III). This indicates a weak paramagnetism of the Am(III) complex, similar to earlier findings for BTP complexes.
In comparison to the diamagnetic lanthanide complexes, the coordinating N8 experiences a significant upfield shift to −20 ppm, which is in excellent agreement with data from earlier studies with the Am(III)–nPrBTP complex. As comparison to the Sm(III) and Yb(III) complexes shows, this extraordinary upfield shift cannot be explained as paramagnetic effects known from studies of similar lanthanide complexes, since shifts of the coordinating N8 in the same order of magnitude have only been found for the Yb(III) complex, which has a bigger magnetic moment. Explanations for this behavior are transfer of electron spin density to the nitrogen atoms by several possible mechanisms and spin–orbit coupling effects from Am(III). All transfer mechanisms rely on the existence of a Fermi contact interaction, which is mediated by covalent bonding through s-orbital containing binding orbitals.
Our results are an important contribution within current research efforts to identify the origin of selectivity of N-donor ligands in actinide–lanthanide separation. They show that NMR spectroscopy is a versatile and sensitive tool in the elucidation of fundamental bonding mechanisms especially for actinide compounds. Important insights into the metal–ligand bonding were obtained, which reveal valuable information for an optimized design of future extractants for the separation of actinides from lanthanides.
Further temperature dependent NMR experiments with paramagnetic cations of the entire lanthanide series and further transuranium element cations are in progress. Moreover, we endeavor to investigate the contributions to the chemical shift using quantum chemical calculations. The obtained experimental chemical shift values for all nuclei in the complexes are important benchmarks for those calculations.
Experimental section
General
All NMR spectra were recorded at T = 300 K on a Bruker Avance III 400 spectrometer operating at 400.18 MHz for 1H, 100.63 MHz for 13C and 40.56 MHz for 15N. The spectrometer was equipped with a z-gradient broadband observe probe optimized for x-magnetization detection. Chemical shifts are referenced internally to TMS (δ(TMS) = 0 ppm). 15N chemical shifts are referenced to 15NH4Cl with δ(NH4Cl) = 0 ppm. For all direct excitation and correlation spectra, standard Bruker pulse sequences were used. DOSY spectra were acquired using one-shot sequences.61 All 1D spectra for diamagnetic complexes and Am(III) were recorded with 32k data points and are zero filled to 64k points. 15N spectra were recorded at lower spectral resolution if necessary, allowing fast pulsing and high repetition rates to compensate the paramagnetic relaxation enhancement. The reported chemical shifts are taken from 1D spectra unless stated otherwise. 15N data at natural abundances are obtained from 1H,15N-HMQC spectra. Deuterated solvents were purchased from Euriso-Top GmbH. Chemicals for synthesis were purchased from VWR International and used as-is. 15N-labeled hydrazine hydrate (98% 15N) was purchased from Sigma-Aldrich and used as-is. Mass spectra using LIFDI and EI ionization methods were recorded using a JEOL JMS-700 magnetic sector instrument. Mass spectra using ESI ionization methods were recorded using a Bruker ApexQe FT-ICR instrument. All mass spectra were recorded at the mass spec facility of the Institute of Organic Chemistry at the University of Heidelberg. All mass spectra of 15N-labeled compounds were acquired using LIFDI-MS technology.58,59 Melting points were measured using a Stuart SMP30 melting point apparatus.TRLFS setup
All compounds for TRLFS experiments were used as received. Methanol (absolute) was purchased from Merck and stored over molecular sieves. The concentration of Cm(III) was set to 6.6 × 10−8 mol × L−1 by adding an aliquot of a stock solution [Cm(III)] = 6.7 × 10−6 mol L−1 in HClO4 (1.0 × 10−2 mol L−1) to the [243Am({15N}C5-BPP)3](OTf)3 NMR sample. The isotopic mass distribution of the Cm(III) solution was 89.7% 248Cm, 9.4% 246Cm, <0.5% 243Cm, 244Cm, 245Cm, and 247Cm, determined by alpha spectroscopy and ICP-MS. TRLFS measurements were performed using a Nd:YAG-pumped dye laser system [Surelite II laser (Continuum), NARROWscan D-R dye laser (Radiant Dyes Laser Accessories)]. For Cm(III) excitation a wavelength of 396.6 nm was used. The emission spectra were recorded at an angle of 90° to the exciting laser beam. A Shamrock 303i spectrograph (ANDOR), equipped with a 300, 900 and 1200 lines per mm grating turret was used for spectral decomposition. Fluorescence spectra were recorded in the 575–635 nm range using the 1200 lines per mm grating of the spectrograph. The fluorescence emission was detected by an ICCD camera [iStar Gen III, A-DH 720 18F-63 (ANDOR)]. Rayleigh scattering and shortlived fluorescence of organic ligands was discriminated by a delay time of 1.0 μs before the fluorescence light is recorded. The quartz cuvette was temperature controlled at T = 25 °C.TRLFS sample preparation
The [243Am(C5-BPP)3](OTf)3 NMR sample in 600 μL MeOD-d4 was transferred from a J. Young-type NMR tube into a quartz cuvette. 6 μL of an aqueous Cm(ClO4)3 stock solution (1.0 × 10−2 mol L−1 HClO4, [Cm(III)] = 6.7 × 10−6 mol L−1) was added and carefully shaken. The change in volume was limited to 1.0% (vol). Titrations were performed by stepwise addition of a C5-BPP solution (3.0 × 10−2 mol L−1) in MeOD. After each addition of the ligand solution the sample was carefully shaken and a Cm(III) fluorescence spectrum was recorded.Dimethyl 2,6-pyridinedicarboxylate
The preparation of dimethyl 2,6-pyridinedicarboxylate was carried out by a modification of a previously published method.72 2,6-Dipicolinic acid (10.0 g, 59.8 mmol) and 2.0 mL sulphuric acid (conc.) were refluxed in 40 mL methanol for 3 h. After the solution was cooled to room temperature the solution was neutralized with 1.5 g (14.2 mmol) Na2CO3. The resulting white precipitate was separated by filtration and washed three times with 20 mL portions of cold water. The solid was dried for 2 h at 60 °C in high vacuum yielding the desired product (10.65 g, 54.6 mmol, 91%) as a white solid.
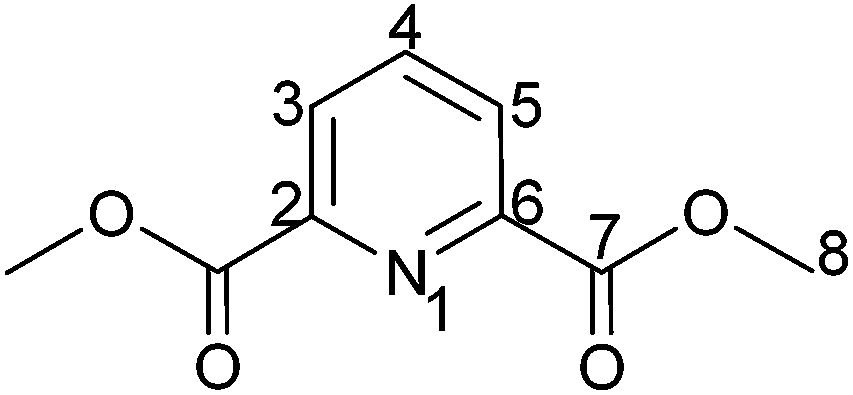
1 H-NMR (400.18 MHz, CDCl3): δ(ppm) = 8.26 (d, 2H, H3/5, 3J = 7.8 Hz), 7.99 (t, 1H, H4, 3J = 7.8 Hz), 3.98 (s, 6H, H8).
13 C-NMR (100.63 MHz, CDCl3): δ(ppm) = 164.9 (Cq, C7), 148.1 (Cq, C2/6), 138.3 (Ct, C4), 127.9 (Ct, C3/5), 53.1 (Cp, C8).
1,1′-(Pyridine-2,6-diyl)bis(5,5-dimethylhexane-1,3-dione)
1,1′-(Pyridine-2,6-diyl)bis(5,5-dimethylhexane-1,3-dione) was prepared from dimethyl 2,6-pyridinedicarboxylate and 4,4-dimethylpentan-2-one in an adapted literature procedure. 5.0 mL sodium methanolate (30% in methanol, 28.6 mmol) were added to 3.3 mL (22.9 mmol) 4,4-dimethylpentane-2-one and stirred in an argon atmosphere for 30 min at room temperature. Subsequently, a solution of 2.1 g (10.7 mmol) dimethyl 2,6-pyridinedicarboxylate in 20 mL diethyl ether (abs.) was added dropwise, and the reaction mixture was refluxed for 5 h. Subsequently, the reaction mixture was cooled to room temperature and neutralized with glacial acetic acid. The organic phase was washed three times with 30 mL portions of cold water, dried with sodium sulfate and concentrated in vacuo. The product was obtained in 79% yield as a yellowish crystalline solid. mp: 109.6 °C.
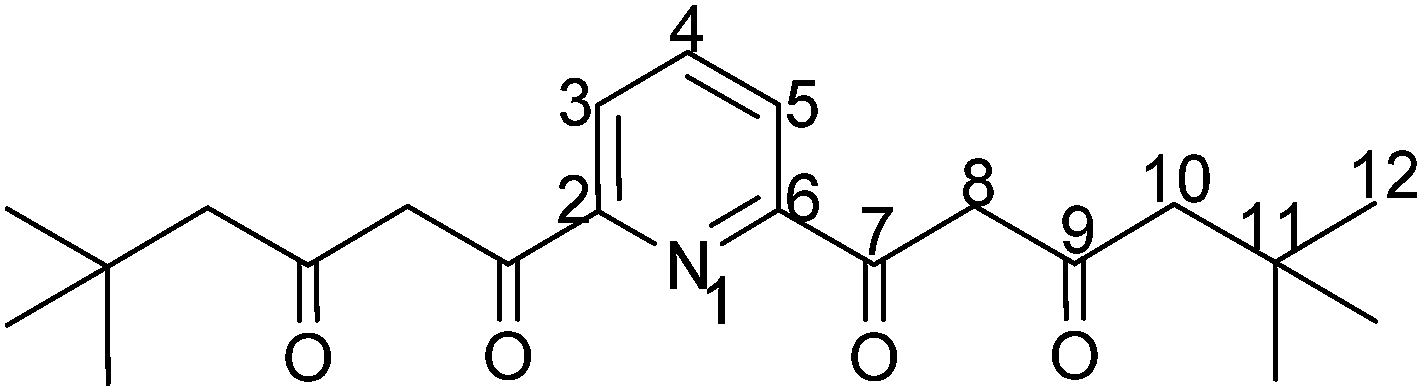
1 H-NMR (400.18 MHz, MeOD-d4, T = 328 K) keto form: δ(ppm) = 8.19 (d, 2H, H3/5), 8.08 (dd, 1H, H4), 6.87 (s, 4H, H8), 2.37 (s, 4H, H10), 1.08 (s, 18H, H12); enol form: (12% according to 1H-NMR) δ(ppm) = 8.27 (dd, 2H, H3/5)*, 6.80 (s, 4H, H8), 4.56 (s, OH), 2.59 (s, 4H, H10), 1.03 (s, 18H, H12). * Value for H4 could not be assigned unambiguously.
13 C-NMR (100.63 MHz, MeOD-d4, T = 328 K) keto form: δ(ppm) = 195.6 (Cq, C9), 183.7 (Cq, C7), 153.5 (Cq, C2/6), 139.7 (Ct, C4), 125.3 (Cs, C3/5), 99.6 (Cs, C8), 53.1 (Cs, C10), 32.7 (Cq, C11), 30.4 (Cp, C12); enol form: δ(ppm) = 195.5 (Cq, C9), 183.2 (Cq, C7), 153.4 (Cq, C2/6), 139.9 (Ct, C4), 126.5 (Cs, C3/5), 99.8 (Cs, C8), 56.6 (Cs, C10), 31.7 (Cq, C11), 30.0 (Cp, C12).
HR-MS (EI) calculated for C21H29NO4 [M]+ 359.2097; found: 359.2114; calculated for C20H26NO4 [M − CH3]+ 344.1862, found: 344.1841; calculated for C17H21NO4 [M − C4H8]+ 303.1471, found: 303.1492; calculated for C16H18NO4 [M − C5H11]+ 288.1236, found: 288.1275; calculated for C16H21NO3 [M − C5H8O]+ 275.1521, found: 275.1534; calculated for C15H18NO3 [M − C6H11O]+ 260.1287, found: 260.1283; calculated for C14H19NO2 [M − C7H10O2]+ 233.1416, found: 233.1416; calculated for C12H14NO3 204.1025, found: 204.0657; calculated for C11H12NO 190.0868, found: 190.0488; calculated for C6H11O 99.0810, found: 99.0791.
2,6-Bis(5-(2,2-dimethylpropyl)1H-pyrazol)-3-yl-pyridine (C5-BPP)
8.0 mL (129 mmol) N2H4·H2O (80% in H2O) were added to a solution of 540 mg (1.5 mmol) 1,1′-(pyridine-2,6-diyl)bis(4,4-dimethylhexane-1,3-dione) in 40 mL methanol (abs.) and refluxed for 3 h. After the solution was cooled to room temperature the resulting white precipitate was collected and washed three times each with 30 mL water and 30 mL diethyl ether. The desired product was obtained by drying in high vacuum (0.415 g, 1.18 mmol, 79%) as a white, crystalline solid. mp: 266.5 °C.
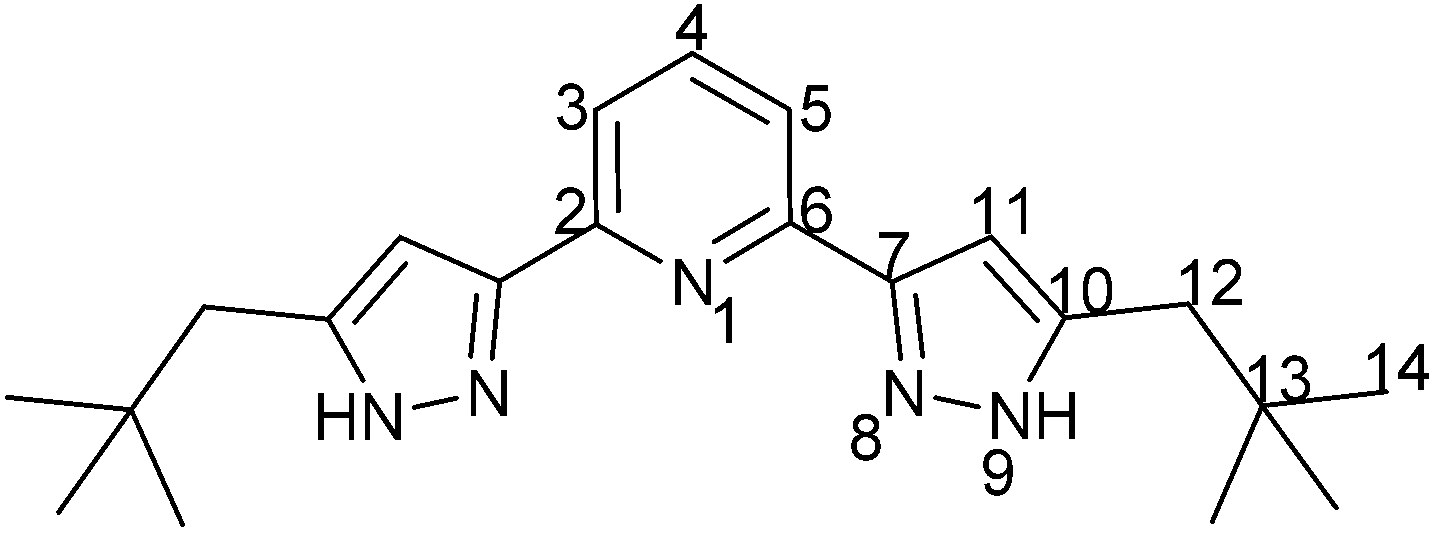
1 H-NMR (400.18 MHz, MeOD-d4): δ(ppm) = 7.84 (t, 1H, H4), 7.69 (s, 2H, H3/5), 6.72 (s, 2H, H11), 2.60 (s, 4H, H12), 1.00 (s, 18H, H14).
13 C-NMR (100.63 MHz, MeOD-d4): δ(ppm) = 153.1 (Cq, C2/6), 149.3 (Cq, C7), 144.2 (Cq, C10), 139.0 (Ct, C4), 119.7 (Ct, C3/5), 104.9 (Ct, C11), 43.3 (Cs, C12), 32.1 (Cq, C13), 29.8 (Cp, C14).
LIFDI-MS (CH3OH) calculated for C21H30N5 [M + H]+: 352.25, found: 352.21; calculated for C21H29N5 [M]+: 351.24, found: 351.22.
15N labeled 2,6-bis(5-(2,2-dimethylpropyl)1H-pyrazol)-3-yl-pyridine ({15N}C5-BPP)
100 mg (1.9 mmol) 15N2H4·H2O and 1.06 mL (17.3 mmol) 14N2H4·H2O were added to a mixture of 690 mg (1.9 mmol) 1,1′-(pyridine-2,6-diyl)bis(4,4-dimethylhexane-1,3-dione) and 10 mL methanol (abs.) and refluxed for 3 h. After the solution was cooled to room temperature the resulting white precipitate was collected and washed three times each with 20 mL water and 20 mL diethyl ether. The desired product was obtained by drying in high vacuum (0.618 g, 1.76 mmol, 92%) as a white, crystalline solid. mp: 266.5 °C.1 H-NMR (400.18 MHz, MeOD-d4): δ(ppm) = 7.82 (t, 1H, H4, 3J = 7.8 Hz), 7.69 (d, 2H, H3/5, 3J = 7.7 Hz), 6.71 (s, 2H, H11), 2.58 (s, 4H, H12), 0.98 (s, 18H, H14).
13 C-NMR (100.63 MHz, MeOD-d4): δ(ppm) = 153 (Cq, br. s., C7)*, 152 (Cq, C2/6)*, 143 (Cq, br. s., C10)*, 138.9 (Ct, C4), 119.8 (Ct, C3/5), 105.0 (Ct, C11), 41.7 (Cs, br. s., C12), 32.1 (Cq, C13), 29.8 (Cp, C14). * Value taken from a 1H,13C-HMBC spectrum.
15 N-NMR (40.56 MHz, MeOD-d4): δ(ppm) = 287 (N8)*, 206 (N9)*. * Value taken from an 1H,15N-HMQC spectrum.
LIFDI-MS (CH3OH) calculated for C21H30N315N2 [M + H]+: 354.24, found: 354.25; calculated for C21H29N315N2 [M]+: 353.25, found: 353.28.
Syntheses of lanthanide complexes
6 μmol of Ln(OTf)3 were weighted in a screw-cap glass. 18 μmol C5-BPP or {15N}C5-BPP, respectively, were dissolved in 600 μL MeOD-d4 with traces of TMS. The C5-BPP or {15N}C5-BPP ligand solution was added to the metal salt. After mixing the complex solution was transferred into an NMR tube. The sample was degassed by three freeze–pump–thaw cycles and subsequently flame-sealed. Complexes with the labeled and unlabeled ligand were prepared the same way. The chemical shift values for the unlabeled complex are equal to those of the labeled complexes and are not stated here for brevity. However, N1 chemical shifts could only be determined from 1H,15N-gHSQC spectra and are labeled accordingly (†).Synthesis of [243Am(C5-BPP)3](OTf)3
1.0 mL of a solution containing 4 mg mL−1 243Am in HNO3 (0.5 mol L−1) were transferred into a screw-cap glass. A total of 280 μL NaOH (2.0 mol L−1) was added in portions, resulting in precipitation of americium hydroxide. After 20 μL of additional NaOH (2.0 mol L−1) were added, the solution was centrifuged at 6000 rpm for 3 min. Additional 10 μL NaOH solution (2.0 mol L−1) were added, the solution was centrifuged again (6000 rpm, 2 min) and the supernatant was removed. Following this procedure, the precipitate was washed three times with 1.0 mL portions of NaOH (0.01 mol L−1) and once with 1.0 mL water. The americium hydroxide was dissolved in 1.0 mL H2O and 10 μL trifluoromethanesulfonic acid, forming Am(OTf)3. For complexation of Am(OTf)3 with C5-BPP or {15N}C5-BPP, respectively, 420 μL Am(OTf)3 solution were heated to dryness at about 100 °C on a heating plate. The obtained pale-pink solid was subsequently washed with 250 μL D2O and heated to dryness. The ligand solution (18 μmol in 600 μL MeOD-d4) was added to the Am(OTf)3, carefully mixed and transferred into a J. Young-type NMR tube.[Y({15N}C5-BPP)3](OTf)3
1 H-NMR (400.18 MHz, MeOD-d4): δ(ppm) = 8.15 (t, 1H, H4, 3J = 7.9 Hz), 7.93 (d, 2H, H3/5, 3J = 7.9 Hz), 6.77 (s, 2H, H11), 2.45 (d, 2H, H12, 2J = 14.0 Hz), 2.39 (d, 2H, H12, 2J = 14.0 Hz), 0.70 (s, 18H, H14).13 C-NMR (100.63 MHz, MeOD-d4): δ(ppm) = 153.6 (Cq, C7), 150.2 (Cq, C2, C6), 149.2 (Cq, C10), 143.0 (Ct, C4), 123.3 (Ct, C3, C5), 106.2 (Ct, C11), 39.5 (Cd, C12), 32.2 (Cq, C13), 29.7 (Cp, C14).
15 N-NMR (40.56 MHz, MeOD-d4): δ(ppm) = 264 (N1)†, 263 (t, N8, 1J = 9.4 Hz), 205 (s, N9).
ESI-MS (CH3OH) calculated for C65H87F6N15O6S2Y [Y(C5-BPP)3(OTf)2]+: 1440.5367, found: 1440.5516; calculated for C65H88F6N15O6S2NaK [3C5-BPP + HOTf + OTf + Na + K]+: 1414.5922, found: 1414.5880; calculated for C64H86F3N15O3SY [Y(C5-BPP)3(OTf)1 − H]+: 1290.5770, found: 1290.5872; calculated for C64H88F3N15O3SK [3C5-BPP + HOTf + K]+: 1242.6504, found: 1242.6491; calculated for C63H85N15Y [Y(C5-BPP)3 − 2H]+: 1140.6171, found: 1140.6252; calculated for C44H58F6N10O6S2Y [Y(C5-BPP)2(OTf)2]+: 1089.2945, found: 1089.3011; calculated for C43H57F3N10O3SY [Y(C5-BPP)2(OTf)1 − H]+: 939.3346, found: 939.3423; calculated for C42H58N10Na [2C5-BPP + Na]+: 725.4744, found: 725.4744; calculated for C21H30N5 [C5-BPP + H]+: 352.2501, found: 352.2500.
[La({15N}C5-BPP)3](OTf)3
1 H-NMR (400.18 MHz, MeOD-d4): δ(ppm) = 8.16 (t, 1H, H4, 3J = 7.9 Hz), 7.96 (d, 2H, H3/5, 3J = 7.9 Hz), 6.77 (s, 2H, H11), 2.41 (s, 4H, H12), 0.70 (s, 18H, H14).13 C-NMR (100.63 MHz, MeOD-d4): δ(ppm) = 154.3 (Cq, C7), 151.6 (Cq, C2, C6), 148.3 (Cq, C10), 142.9 (Ct, C4), 123.7 (Ct, C3, C5), 106.4 (Ct, C11), 39.5 (Cd, C12), 32.2 (Cq, C13), 29.7 (Cp, C14).
15 N-NMR (40.56 MHz, MeOD-d4): δ(ppm) = 272 (d, N81J = 9.9 Hz), 206 (m, N9).
ESI-MS (CH3OH) calculated for C67H92F10N15O10S3La [La(C5-BPP)3(OTf)3 + CH3OH + HF]+: 1692.5296, found: 1692.5454; calculated for C65H87F6N15O6S2La [La(C5-BPP)3(OTf)2]+: 1490.5373, found: 1490.5228; calculated for C64H86F3N15O3SLa [La(C5-BPP)3(OTf)1 − H]+: 1340.5774, found: 1340.5868; calculated for C63H87N15La [La(C5-BPP)3]+: 1192.6332, found: 1192.6338; calculated for C63H85N15La [La(C5-BPP)3 − 2H]+: 1190.6176, found: 1190.6127; calculated for C44H58F6N10O6S2La [La(C5-BPP)2(OTf)2]+: 1139.2950, found: 1139.3026; calculated for C42H58N10Na [2C5-BPP + Na]+: 725.4744, found: 725.4803; calculated for C64H87F3N15O3SLa [La(C5-BPP)3(OTf)1]2+: 670.7926, found: 670.7924; calculated for C63H86N15La [La(C5-BPP)3 − H]2+: 595.8127, found: 595.8125; calculated for C63H87N15La [La(C5-BPP)3]3+: 397.5444, found: 397.5463; calculated for C21H30N5 [C5-BPP + H]+: 352.2501, found: 352.2511.
[Lu({15N}C5-BPP)3](OTf)3
1 H-NMR (400.18 MHz, MeOD-d4): δ(ppm) = 8.15 (t, 1H, H4, 3J = 7.9 Hz), 7.93 (d, 2H, H3/5, 3J = 7.9 Hz), 6.77 (s, 2H, H11), 2.44 (d, 2H, H12, 2J = 14.0 Hz), 2.40 (d, 2H, H12, 2J = 14.0 Hz), 0.71 (s, 18H, H14).13 C-NMR (100.63 MHz, MeOD-d4): δ(ppm) = 153.6 (Cq, C7), 149.9 (Cq, C2, C6), 149.5 (Cq, C10), 142.9 (Ct, C4), 123.2 (Ct, C3, C5), 106.2 (Ct, C11), 39.6 (Cd, C12), 32.2 (Cq, C13), 29.7 (Cp, C14).
15 N-NMR (40.56 MHz, MeOD-d4): δ(ppm) = 267 (N1)†, 265 (d, N8, 1J = 9.4 Hz), 205 (m, N9).
ESI-MS (CH3OH) calculated for C65H87F6N15O6S2Lu [Lu(C5-BPP)3(OTf)2]+: 1526.5717, found: 1526.5793; calculated for C64H86F3N15O3SLu [Lu(C5-BPP)3(OTf)1 − H]+: 1376.6119, found: 1376.6199; calculated for C63H85N15Lu [Lu(C5-BPP)3 − 2H]+: 1226.6520, found: 1226.6662; calculated for C44H58F6N10O6S2Lu [Lu(C5-BPP)2(OTf)2]+: 1175.3294, found: 1175.3356; calculated for C43H57F3N10O3SLu [Lu(C5-BPP)2(OTf)1 − H]+: 1025.3696, found: 1025.3748; calculated for C43H59F3N10O3SK [2C5-BPP + HOTf + K]+: 891.4081, found: 891.4008; calculated for C64H87F3N15O3SLu [Lu(C5-BPP)3(OTf)1]2+: 688.8098, found: 688.8129; calculated for C21H30N5 [C5-BPP + H]+: 352.2501,; found: 352.2498.
[Sm({15N}C5-BPP)3](OTf)3
1 H-NMR (400.18 MHz, MeOD-d4): δ(ppm) = 8.30 (t, 1H, H4, 3J = 7.9 Hz), 8.12 (d, 2H, H3/5, 3J = 7.9 Hz), 6.90 (s, 2H, H11), 2.41 (s, 4H, H12), 0.81 (s, 18H, H14).13 C-NMR (100.63 MHz, MeOD-d4): δ(ppm) = 156.0 (Cq, C7), 153.4 (Cq, C2, C6), 148.4 (Cq, C10), 143.4 (Ct, C4), 122.9 (Ct, C3, C5), 106.2 (Ct, C11), 39.5 (Cd, C12), 32.3 (Cq, C13), 29.8 (Cp, C14).
15 N-NMR (40.56 MHz, MeOD-d4): δ(ppm) = 221 (s, N1)†, 224 (s, N8), 205 (s, N9).
ESI-MS (CH3OH) calculated for C65H88F6N15O6S2NaK [3C5-BPP + HOTf + OTf + Na + K]+: 1414.5922, found: 1414.5916; calculated for C64H89F3N15O3SSm [Sm(C5-BPP)3(OTf)1 + 2H]+: 1356.6143, found: 1356.6115; calculated for C63H87N15KSm [Sm(C5-BPP)3 + K]+: 1244.6103, found: 1244.6114; calculated for C64H88F3N15O3SK [3C5-BPP + HOTf + K]+: 1242.6504, found: 1242.6508; calculated for C63H88N15Sm [Sm(C5-BPP)3 + H]+: 1206.6545, found: 1206.6468; calculated for C63H87N15Sm [Sm(C5-BPP)3]+: 1205.6466, found: 1205.6438; calculated for C43H59F3N10O3SK [2C5-BPP + HOTf + K]+: 891.4081, found: 891.4008; calculated for C42H58N10Na [2C5-BPP + Na]+: 725.4744, found: 725.4783; calculated for C63H87N15Sm [Sm(C5-BPP)3]2+: 602.8233, found: 602.8167; calculated for C21H29N5Na [C5-BPP + Na]+: 374.2320, found: 374.2323; calculated for C21H30N5 [C5-BPP + H]+: 352.2501, found: 352.2514.
[Yb({15N}C5-BPP)3](OTf)3
1 H-NMR (400.18 MHz, MeOD-d4): δ(ppm) = 7.20 (br. s., 1H, H4), 6.61 (br. s., 2H, H3/5), 5.17 (s, 2H, H11), 2.83 (s, 4H, H12), 0.41 (s, 18-H, H14).13 C-NMR (100.63 MHz, MeOD-d4): δ(ppm) = 149.1 (Cq, C7), 147.8 (Cq, C2, C6), 144.1 (Cq, C10), 141.9 (Ct, C4), 118.4 (Ct, C3, C5), 101.4 (Ct, C11), 38.9 (Cd, C12), 32.1 (Cq, C13), 28.7 (Cp, C14).
15 N-NMR (40.56 MHz, MeOD-d4): δ(ppm) = 20 (s, N8), 194 (s, N9).
ESI-MS (CH3OH) calculated for C63H87N15NaYb [Yb(C5-BPP)3 + Na]+: 1250.6555, found: 1250.6565; calculated for C64H88F3N15O3SK [3C5-BPP + HOTf + K]+: 1242.6504, found: 1242.6489; calculated for C63H88N15Yb [Yb(C5-BPP)3 + H]+: 1228.6736, found: 1228.6736; calculated for C63H87N15K [3C5-BPP + K]+: 1092.6906, found: 1092.6861; calculated for C43H58F3N10O3SNa [2C5-BPP + OTf + Na]+: 874.4264, found: 874.4351; calculated for C42H58N10Na [2C5-BPP + Na]+: 725.4744, found: 725.4754; calculated for C63H88N15K [3C5-BPP + H + K]2+: 546.8492, found: 546.8461; calculated for C63H84N15Yb [Yb(C5-BPP)3 − 3H]3+: 408.2141, found: 408.2091; calculated for C21H29N5Na [C5-BPP + Na]+: 374.2320, found: 374.2321.
[Am(C5-BPP)3](OTf)3
1 H-NMR (400.18 MHz, MeOD-d4): δ(ppm) = 7.64 (d, 2H, H3/5, 3J = 7.9 Hz), 7.47 (t, 1H, H4, 3J = 7.9 Hz), 6.29 (s, 2H, H11), 2.91 (d, 2H, H12, 2J = 13.9 Hz), 2.55 (d, 2H, H12, 2J = 13.9 Hz), 0.64 (s, 18H, H14).13 C-NMR (100.63 MHz, MeOD-d4): δ(ppm) = 164.8 (Cq, C7), 164.2 (Cq, C2, C6), 147.9 (Ct, C4), 147.7 (Cq, C10), 116.3 (Ct, C3, C5), 101.7 (Ct, C11), 38.7 (Cd, C12), 33.9 (Cq, C13), 29.6 (Cp, C14).
15 N-NMR (40.56 MHz, MeOD-d4): δ(ppm) = 216 (N9)*, 1 (N1)†, −22 (N8)*. * Value taken from an 1H,15N-HMQC spectrum.
19 F-NMR (376.54 MHz, MeOD-d4): δ(ppm) = −80.00 (s, CF3SO3−).
[Am({15N}C5-BPP)3](OTf)3
1 H-NMR (400.18 MHz, MeOD-d4): δ(ppm) = 7.64 (d, 2H, H3/5, 3J = 7.9 Hz), 7.47 (tr, 1H, H4, 3J = 7.9 Hz), 6.28 (s, 2H, H11), 2.91 (d, 2H, H12, 2J = 13.9 Hz), 2.55 (d, 2H, H12, 2J = 13.9 Hz), 0.64 (s, 18H, H14).13 C-NMR (100.63 MHz, MeOD-d4): δ(ppm) = 164.8 (Cq, C7), 164.2 (Cq, C2, C6), 147.9 (Ct, C4), 147.7 (Cq, C10), 116.2 (Ct, C3, C5), 101.6 (Ct, C11), 38.7 (Cd, C12), 33.9 (Cq, C13), 29.6 (Cp, C14).
15 N-NMR (40.56 MHz, MeOD-d4): δ(ppm) = 217 (s, N9), 1 (N1)†, −23 (d, N8, 1J = 9.6 Hz). * Value taken from an 1H,15N-HMQC spectrum.
19 F-NMR (376.54 MHz, MeOD-d4): δ(ppm) = −80.00 (s, CF3SO3−).
Acknowledgements
This work was supported by the German Federal Ministry of Education and Research (BMBF) under contract numbers 02NUK020A and 02NUK020D. The authors wish to thank Lisa Böringer and Julia Schäfer (KIT-INE) for support with the labwork. We are grateful to Prof. Dr Frank Breher (KIT-AC, Karlsruhe) for cooperation during these funded projects involving the NMR spectrometer and associated equipment. We thank Dr Jürgen H. Gross, Doris Lang and Norbert Nieth (Institute of Organic Chemistry, University of Heidelberg) for the LIFDI and ESI MS measurements.Notes and references
- IEA, Key World Energy Statistics 2012, International Energy Agency (IEA), Vienna, 2012 Search PubMed.
- K. Gompper, A. Geist and H. Geckeis, Nachr. Chem., 2010, 58, 1015–1019 CrossRef CAS.
- M. Salvatores and G. Palmiotti, Prog. Part. Nucl. Phys., 2011, 66, 144–166 CrossRef CAS PubMed.
- OECD Report NEA No. 6894 Potential Benefits and Impacts of Advanced Nuclear Fuel Cycles with Actinide Partitioning and Transmutation, OECD, Nuclear Energy Agency (NEA), Paris, 2011.
- P. J. Panak and A. Geist, Chem. Rev., 2013, 113, 1199–1236 CrossRef CAS PubMed.
- C. Musikas, P. Vitorge and D. Pattee, Proc. - Int. Solvent Extr. Conf., 1983 ISEC'83, 1983, 6 Search PubMed.
- Z. Kolarik, Chem. Rev., 2008, 108, 4208–4252 CrossRef CAS PubMed.
- Z. Kolarik, U. Müllich and F. Gassner, Solvent Extr. Ion Exch., 1999, 17, 1155–1170 CrossRef CAS.
- N. L. Banik, M. A. Denecke, A. Geist, G. Modolo, P. J. Panak and J. Rothe, Inorg. Chem. Commun., 2013, 29, 172–174 CrossRef CAS PubMed.
- N. L. Banik, B. Schimmelpfennig, C. M. Marquardt, B. Brendebach, A. Geist and M. A. Denecke, Dalton Trans., 2010, 5117–5122 RSC.
- M. G. B. Drew, D. Guillaneux, M. J. Hudson, P. B. Iveson, M. L. Russell and C. Madic, Inorg. Chem. Commun., 2001, 4, 12–15 CrossRef CAS.
- P. B. Iveson, C. Riviere, D. Guillaneux, M. Nierlich, P. Thuery, M. Ephritikhine and C. Madic, Chem. Commun., 2001, 1512–1513 RSC.
- M. A. Denecke, A. Rossberg, P. J. Panak, M. Weigl, B. Schimmelpfennig and A. Geist, Inorg. Chem., 2005, 44, 8418–8425 CrossRef CAS PubMed.
- M. A. Denecke, P. J. Panak, F. Burdet, M. Weigl, A. Geist, R. Klenze, M. Mazzanti and K. Gompper, C. R. Chim., 2007, 10, 872–882 CrossRef CAS PubMed.
- M. J. Hudson, C. E. Boucher, D. Braekers, J. F. Desreux, M. G. B. Drew, M. R. S. Foreman, L. M. Harwood, C. Hill, C. Madic, F. Marken and T. G. A. Youngs, New J. Chem., 2006, 30, 1171–1183 RSC.
- J.-C. Berthet, Y. Miquel, P. B. Iveson, M. Nierlich, P. Thuery, C. Madic and M. Ephritikhine, J. Chem. Soc., Dalton Trans., 2002, 3265–3272 RSC.
- C. Ekberg, A. Fermvik, T. Retegan, G. Skarnemark, M. R. S. Foreman, M. J. Hudson, S. Englund and M. Nilsson, Radiochim. Acta, 2008, 96, 225–233 CrossRef CAS.
- A. Bremer, A. Geist and P. J. Panak, Dalton Trans., 2012, 7582–7589 RSC.
- A. Bremer, A. Geist and P. J. Panak, Radiochim. Acta, 2013, 101, 285–291 CrossRef CAS.
- A. Bremer, C. M. Ruff, D. Girnt, U. Müllich, J. Rothe, P. W. Roesky, P. J. Panak, A. Karpov, T. J. J. Müller, M. A. Denecke and A. Geist, Inorg. Chem., 2012, 51, 5199–5207 CrossRef CAS PubMed.
- C. Adam, P. Kaden, B. B. Beele, U. Müllich, S. Trumm, A. Geist, P. J. Panak and M. A. Denecke, Dalton Trans., 2013, 14068–14074 RSC.
- S. Trumm, P. J. Panak, A. Geist and T. Fanghänel, Eur. J. Inorg. Chem., 2010, 3022–3028 CrossRef CAS.
- S. Trumm, G. Lieser, M. R. S. J. Foreman, P. J. Panak, A. Geist and T. Fanghänel, Dalton Trans., 2010, 923–929 RSC.
- G. R. Choppin, J. Alloys Compd., 1995, 223, 174–179 CrossRef CAS.
- E. I. Solomon, B. Hedman, K. O. Hodgson, A. Dey and R. K. Szilagyi, Coord. Chem. Rev., 2005, 249, 97–129 CrossRef CAS PubMed.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS PubMed.
- A. Bhattacharyya, T. K. Ghanty, P. K. Mohapatra and V. K. Manchanda, Inorg. Chem., 2011, 50, 3913–3921 CrossRef CAS PubMed.
- M. Dolg, X. Cao and J. Ciupka, J. Electron Spectrosc. Relat. Phenom., 2014, 194, 8–13 CrossRef CAS PubMed.
- R. Golding and M. Halton, Aust. J. Chem., 1972, 25, 2577–2581 CrossRef CAS.
- B. Bleaney, R. J. P. Williams, A. V. Xavier, R. B. Martin, B. A. Levine and C. M. Dobson, J. Chem. Soc., Chem. Commun., 1972, 791–793 RSC.
- G. Pintacuda, M. John, X. C. Su and G. Otting, Acc. Chem. Res., 2007, 40, 206–212 CrossRef CAS PubMed.
- C. Piguet and C. F. G. C. Geraldes, in Handbook on the Physics and Chemistry of Rare Earths, ed. J. K.A. Gschneidner, J. C. G. Bünzli and V. K. Pecharsky, Elsevier, 2003, vol. 33, pp. 353–463 Search PubMed.
- J. A. Peters, J. Huskens and D. J. Raber, Prog. Nucl. Magn. Reson. Spectrosc., 1996, 28, 283–350 CrossRef CAS.
- J. Reuben, J. Magn. Reson., 1982, 50, 233–236 CAS.
- S. Di Pietro, S. L. Piano and L. Di Bari, Coord. Chem. Rev., 2011, 255, 2810–2820 CrossRef CAS PubMed.
- A. G. Martynov, Y. G. Gorbunova and A. Y. Tsivadze, Dalton Trans., 2011, 7165–7171 RSC.
- H. Friebolin, Basic one- and two-dimensional NMR spectroscopy, Wiley-VCH, Weinheim, 5th edn, 2011 Search PubMed.
- J. F. Desreux and C. N. Reilley, J. Am. Chem. Soc., 1976, 98, 2105–2109 CrossRef CAS.
- C. N. Reilley and B. W. Good, Anal. Chem., 1975, 47, 2110–2116 CrossRef CAS.
- J. W. M. Deboer, P. J. D. Sakkers, C. W. Hilbers and E. Deboer, J. Magn. Reson., 1977, 25, 455–476 CAS.
- B. Bleaney, J. Magn. Reson., 1972, 8, 91–100 CAS.
- J. Reuben and G. A. Elgavish, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr and E. LeRoy, Elsevier, 1979, vol. 4, pp. 483–514 Search PubMed.
- R. von Ammon and R. D. Fischer, Angew. Chem., 1972, 84, 737–755 CrossRef.
- J. H. Forsberg, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr and E. LeRoy, Elsevier, 1996, vol. 23, pp. 1–68 Search PubMed.
- S. P. Babailov, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 52, 1–21 CrossRef CAS PubMed.
- I. Bertini, C. Luchinat, G. Parigi and R. Pierattelli, ChemBioChem, 2005, 6, 1536–1549 CrossRef CAS PubMed.
- J. Reuben, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr and E. LeRoy, Elsevier, 1979, vol. 4, pp. 515–552 Search PubMed.
- G. Otting, Annu. Rev. Biophys., 2010, 39, 387–405 CrossRef CAS PubMed.
- P. Hessler Jan and T. W. Carnall, in Lanthanide and Actinide Chemistry and Spectroscopy, American Chemical Society, 1980, vol. 131, ch. 17, pp. 349–368 Search PubMed.
- C. Apostolidis, B. Schimmelpfennig, N. Magnani, P. Lindqvist-Reis, O. Walter, R. Sykora, A. Morgenstern, E. Colineau, R. Caciuffo, R. Klenze, R. G. Haire, J. Rebizant, F. Bruchertseifer and T. Fanghänel, Angew. Chem., Int. Ed., 2010, 49, 6343–6347 CrossRef CAS PubMed.
- L. Soderholm, J. Less-Common Met., 1987, 133, 77–85 CrossRef CAS.
- L. Soderholm, N. Edelstein, L. R. Morss and G. V. Shalimoff, J. Magn. Magn. Mater., 1986, 54–7, 597–598 CrossRef.
- P. G. Huray, S. E. Nave and R. G. Haire, J. Less-Common Met., 1983, 93, 293–300 CrossRef CAS.
- S. E. Nave, R. G. Haire and P. G. Huray, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 2317–2327 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- T. F. Wall, S. Jan, M. Autillo, K. L. Nash, L. Guerin, C. L. Naour, P. Moisy and C. Berthon, Inorg. Chem., 2014, 2450–2459 CrossRef CAS PubMed.
- P. S. Bagus, E. S. Ilton, R. L. Martin, H. J. A. Jensen and S. Knecht, Chem. Phys. Lett., 2012, 546, 58–62 CrossRef CAS PubMed.
- H. B. Linden and J. H. Gross, J. Am. Soc. Mass Spectrom., 2011, 22, 2137–2144 CrossRef CAS PubMed.
- H. B. Linden and J. H. Gross, Rapid Commun. Mass Spectrom., 2012, 26, 336–344 CrossRef PubMed.
- G. A. Morris, Diffusion-Ordered Spectroscopy in eMagRes, John Wiley & Sons, Ltd, 2009 Search PubMed.
- M. D. Pelta, G. A. Morris, M. J. Stchedroff and S. J. Hammond, Magn. Reson. Chem., 2002, 40, S147–S152 CrossRef CAS.
- C. S. Johnson Jr, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203–256 CrossRef.
- S. Cotton, Lanthanide and actinide chemistry, Wiley, Hoboken, NJ, 2006 Search PubMed.
- M. Autillo, P. Kaden, A. Geist, L. Guerin, P. Moisy and C. Berthon, Phys. Chem. Chem. Phys., 2014, 8608–8614 RSC.
- I. Bertini, C. Luchinat and S. Aime, Coord. Chem. Rev., 1996, 150, 29–76 CrossRef.
- E. Ruiz, J. Cirera and S. Alvarez, Coord. Chem. Rev., 2005, 249, 2649–2660 CrossRef CAS PubMed.
- M. Enders, in Modeling of molecular properties, ed. P. Comba, Wiley-VCH, Weinheim, 2011, pp. 49–63 Search PubMed.
- M. A. Denecke, Dalton Trans., 2015 10.1039/c4dt02716g.
- P. Hrobárik, V. Hrobáriková, A. H. Greif and M. Kaupp, Angew. Chem., 2012, 124, 11042–11046 CrossRef.
- P. Hrobárik, V. Hrobáriková, A. H. Greif and M. Kaupp, Angew. Chem., Int. Ed., 2012, 51, 10674 CrossRef.
- M. Kaupp, O. L. Malkina, V. G. Malkin and P. Pyykkö, Chem.–Eur. J., 1998, 4, 118–126 CrossRef CAS.
- E. J. T. Chrystal, L. Couper and D. J. Robins, Tetrahedron, 1995, 51, 10241–10252 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: LIFDI-MS spectra and additional NMR spectra. See DOI: 10.1039/c4sc03103b |
| This journal is © The Royal Society of Chemistry 2015 |