DOI:
10.1039/C4SC03599B
(Edge Article)
Chem. Sci., 2015,
6, 5120-5127
NKT cell-dependent glycolipid–peptide vaccines with potent anti-tumour activity†
Received
20th November 2014
, Accepted 25th June 2015
First published on 25th June 2015
Abstract
It is known that T cells can eliminate tumour cells through recognition of unique or aberrantly expressed antigens presented as peptide epitopes by major histocompatibility complex (MHC) molecules on the tumour cell surface. With recent advances in defining tumour-associated antigens, it should now be possible to devise therapeutic vaccines that expand specific populations of anti-tumour T cells. However there remains a need to develop simpler efficacious synthetic vaccines that possess clinical utility. We present here the synthesis and analysis of vaccines based on conjugation of MHC-binding peptide epitopes to α-galactosylceramide, a glycolipid presented by the nonpolymorphic antigen-presenting molecule CD1d to provoke the stimulatory activity of type I natural killer T (NKT) cells. The chemical design incorporates an enzymatically cleavable linker that effects controlled release of the active components in vivo. Chemical and biological analysis of different linkages with different enzymatic targets enabled selection of a synthetic vaccine construct with potent therapeutic anti-tumour activity in mice, and marked in vitro activity in human blood.
Introduction
With the recent regulatory approval of a cancer vaccine for prostate cancer1 and the clinical success of antibodies that cause blockade of immune checkpoints in T cells,2 immunotherapies that stimulate T cell function are now emerging as credible treatment options for patients with advanced malignancies. However, despite cancer vaccines having the potential to induce T cell responses with high specificity for defined tumour-associated antigens, in general they have not yet realised this promise, and issues remain around potency, lack of clinical efficacy,3,4 manufacturing and cost. Therefore new scalable strategies for the production of efficacious cancer vaccines are still needed. Synthetic vaccines based on antigenic peptides are highly defined and easily manufactured. However, in order to generate a robust anti-tumour response it is important not only that peptide fragments are acquired by antigen presenting cells (APCs) and presented to T cells in the context of MHC molecules, but also that the APCs are in the correct activation state.5 This can be achieved by using adjuvants that target pattern-recognition receptors on APCs, such as the Toll-like receptors (TLRs)6 or selected C-type lectin receptors.7 Advancing this idea further, conjugation of antigens directly to TLR ligands has been shown to achieve appropriate targeting to APCs as well to induce their activation, leading to enhanced stimulation of peptide-specific cytotoxic T-lymphocytes (CTLs).8,9 A less explored approach to activating APCs is to specifically stimulate accessory cells in the local environment. In animal models it has been shown that compounds that stimulate innate-like T cells, a class of semi-activated T cells with an invariant T cell receptor (TCR), can serve as powerful immune adjuvants when delivered with antigens.10,11 The most studied innate-like T cells are the invariant natural killer T (NKT) cells, which recognise glycolipid antigens that bind to the lipid antigen-presenting molecule CD1d.12,13 A number of natural and synthetic CD1d ligands have been defined, of which the most studied is α-galactosylceramide (α-GalCer, KRN7000), a synthetic compound discovered through SAR studies on a class of glycolipid originally isolated from a marine sponge.14 Interestingly, similar compounds containing the α-galactosyl linkage have recently been found in mammalian tissue.15 When administered with peptide antigens, α-GalCer provokes NKT cell-dependent activation of APCs, mediated by CD40-CD40L interactions, which in turn licences the APCs to stimulate superior peptide-specific T cell responses.10 We, and others, hypothesised that stronger responses could be obtained by conjugation of peptides to α-GalCer, as this would provide focussed delivery of antigen and adjuvant to the same cell.16,17 Crucial to the strategy was inclusion of an enzymatically cleavable linker to provide controlled release of the active components in vivo. Having recently described the first example of a vaccine of this type with an esterase-cleavable linker,16 and its application in the suppression of allergic airway inflammation, we now report that the same type of construct can be used to promote an immune response against established tumours in mice. We describe the synthesis and biological analysis of an expanded series of conjugates with linkers that are susceptible to either esterase or protease activity. The aim was to select a vaccine construct that could be produced in a facile synthetic process and that has therapeutic anticancer activity. In addition, we examined whether these conjugates activate human NKT cells and enhance peptide-specific CTL responses in human blood in vitro.
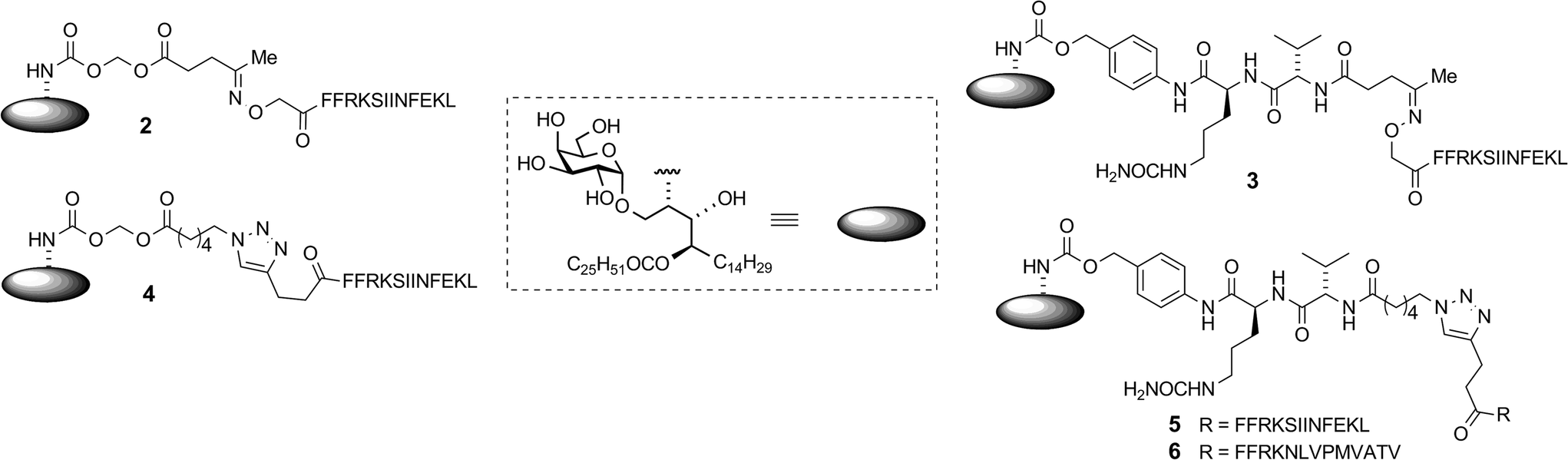
A key consideration in the construction of conjugates was the site of conjugation of the antigen component to α-GalCer. As detailed below, we used a rearranged α-GalCer derivative with a free amino group to provide a synthetically selective and stable point of attachment. Also of consideration in conjugate design was the type of self-immolative linker joining the two components, which would influence serum stability and the release of the active components in vivo. This would largely be influenced by the enzymatic activity to be exploited, which ideally would be enhanced within APCs. Of the many self-immolative linkers described in the literature or used in approved pharmaceuticals, we chose to examine esterase-sensitive acyloxymethyl carbamates18 and protease-sensitive valine–citrulline–p-aminobenzyl (VC–PAB) carbamates19 in this series of conjugates. We have previously shown that conjugates incorporating the acyloxymethyl carbamate group were able to focus activity on splenic CD8α+ dendritic cells (DCs), which are APCs that efficiently cross-present antigens to stimulate CTLs.16 The VC–PAB group is a known substrate20,21 for lysosomal proteases, including cathepsin B which is abundant in APCs.22 A third consideration in conjugate design was the choice of chemistry with which to attach the peptide moiety, as this has been shown to have an impact on the immune response.23 We investigated oxime ligation and copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC), both of which, although well-established, required considerable optimization in our system. For analysis of T cell responses, initial conjugates in this series contained an immunodominant CD8 epitope from ovalbumin (OVA257). This was flanked at the N-terminus by the cleavage sequence Phe–Phe–Arg–Lys, as the extra proteolytic step required for its removal has been shown to support cross-presentation of peptides onto MHC class I molecules for stimulation of CTLs.24 With these considerations in mind we undertook the synthesis of vaccine candidates 2–5 (Fig. 1).
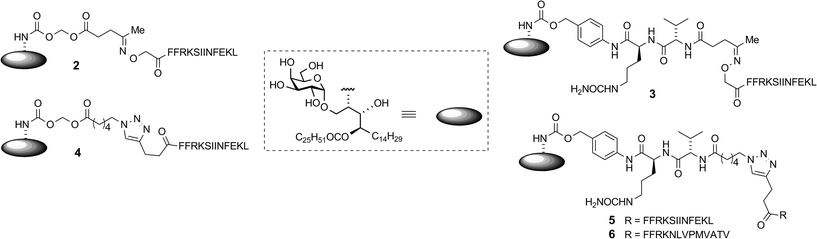 |
| | Fig. 1 Candidate vaccine structures. | |
Results and discussion
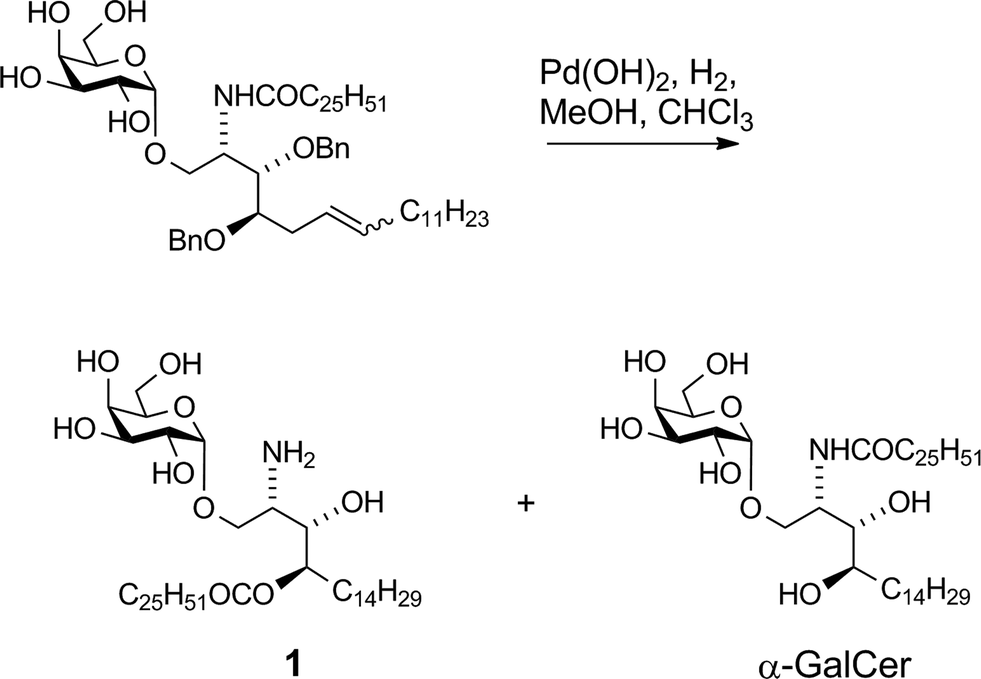
Our vaccine design stems from an unexpected finding during an earlier unpublished synthesis of α-GalCer. Hydrogenolytic deprotection of a late-stage intermediate,§ using Pd(OH)2/C in CHCl3/MeOH, led, in addition to the expected product α-GalCer, to significant quantities of a more polar product that was determined by 2D-NMR and mass spectral analysis to be amine 1 (Scheme 1). Although intramolecular N → O migrations of acyl groups are known in the literature they are usually promoted in strongly acidic media.25–27 We reasoned that, in the present case, HCl was produced from the solvent CHCl3 under the hydrogenolytic conditions, leading to the observed migration. A control experiment showed that, under similar reaction conditions but in the absence of a H2-atmosphere, α-GalCer did not isomerise to amine 1. Although the formation of HCl from CHCl3 by Pd-catalysed hydrogenolysis has been reported,28,29 its use (deliberate or otherwise) to isomerise amides to esters has not. Indeed, CHCl3 has been successfully used as a co-solvent in the final deprotection step (hydrogenolysis) of other syntheses of α-GalCer or analogues thereof, with no report of acyl-migration side-reactions.30–35
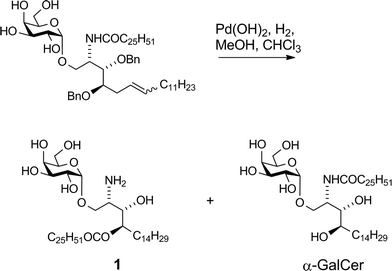 |
| | Scheme 1 Isolation of amine 1. | |
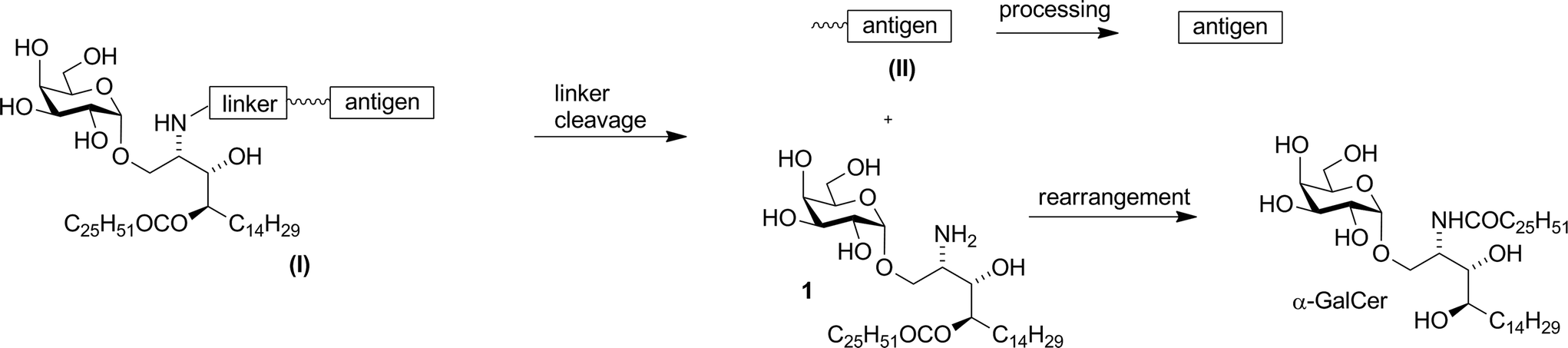
Compound 1 was shown to readily revert to α-GalCer under aqueous conditions,16 suggesting that capping of the amino group with enzymatically-cleavable linkers could generate an interesting new class of α-GalCer prodrugs, or “pro-adjuvants”. In particular, attachment of peptide antigens to 1 could lead to a new class of synthetic vaccines (structure (I), Fig. 2) that exploit the action of NKT cells in stimulating an antigen-specific immune response. Enzymatic cleavage of the self-immolative linker would give metabolites 1 and (II); further enzymatic processing of the peptide (II) would provide an epitope that can be presented on MHC class I, while O → N acyl transfer of 1 would produce α-GalCer, which is presented on CD1d.
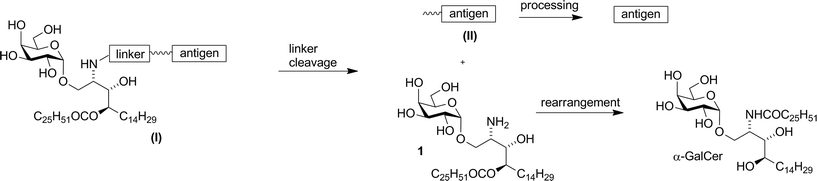 |
| | Fig. 2 Vaccine design and in vivo metabolism. | |
Synthesis
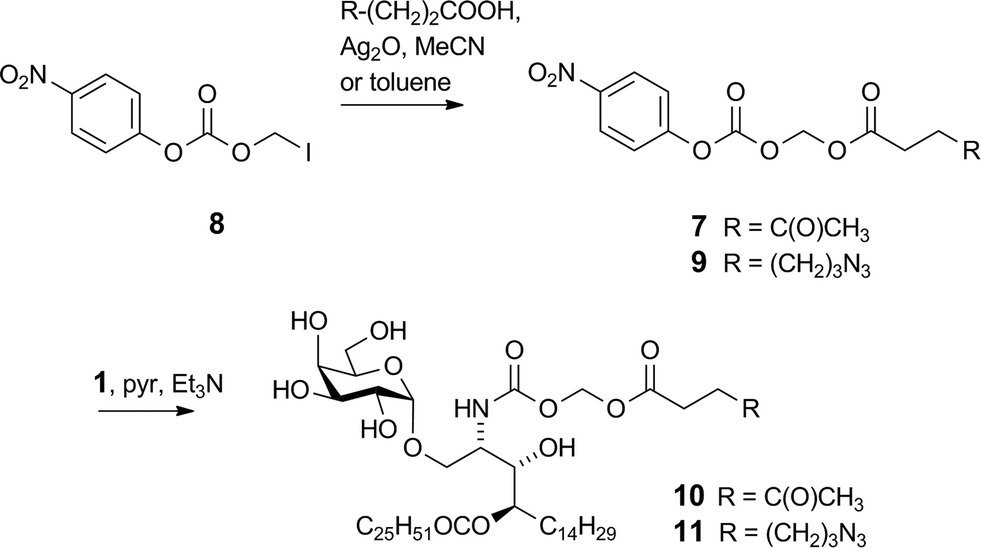
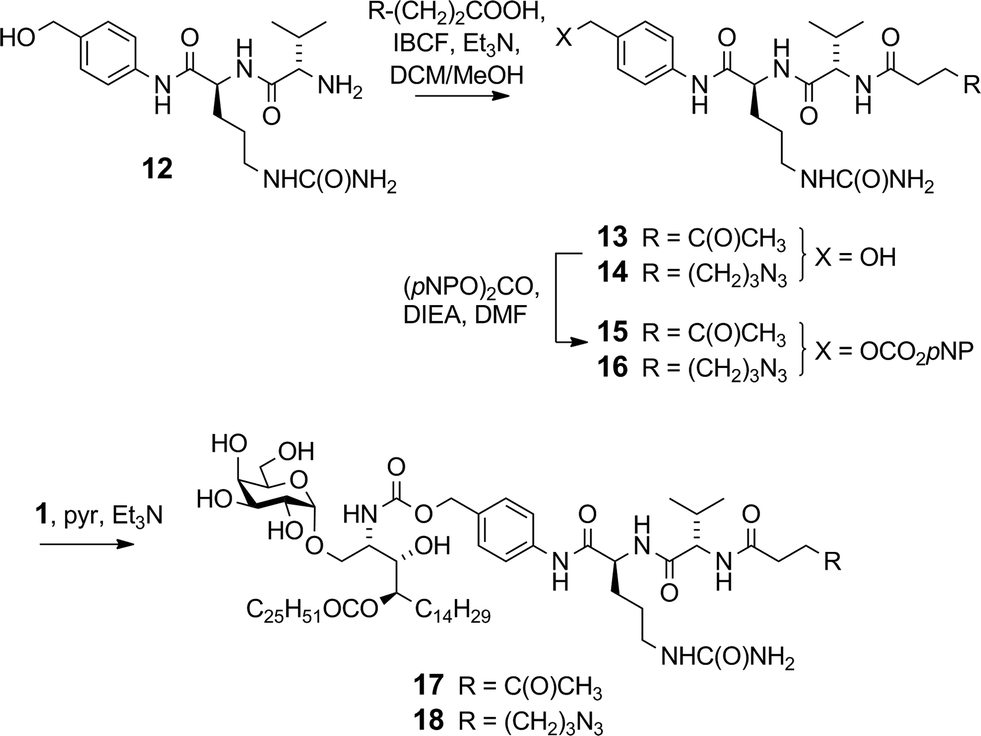
We previously reported16 the synthesis of conjugate 2 however we discuss here pertinent details of the linker synthesis and conjugation chemistry which were not previously disclosed, together with the synthesis of new targets 3–5. Conjugates were constructed from three components: 1; a linker with a reactive functional group for chemoselective ligation (either a ketone for oxime ligation, or an azide for CuAAC); and a synthetic peptide with a complementary N-terminal functional group. Compound 1 was efficiently obtained by the intentional acid-catalysed migration of the cerotic acid moiety from the phytosphingosine nitrogen to the 4′-OH group as reported previously.16 The synthesis of linkers for attachment to amine 1 is shown in Schemes 2 and 3. Acyloxymethyl carbonate linkers 7 and 9, containing keto and azido groups, respectively, were produced by alkylation of the appropriate acid with iodomethyl carbonate 8. The silver salt of the acid was formed in situ by the inclusion of silver(I) oxide (0.6 equiv.) in the reaction (Scheme 2). The inclusion of molecular sieves in both reactions improved product yields by preventing hydrolysis at the ester and/or carbonate groups. Carbonates 7 and 9 were then reacted with 1 in pyridine to afford ketone 10 (ref. 16) and azide 11, respectively. A small amount of α-GalCer was typically formed in these reactions, which could be removed by careful chromatography.
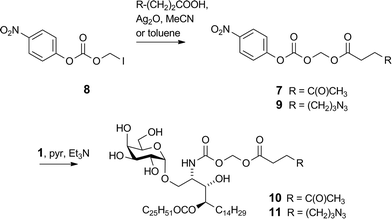 |
| | Scheme 2 Synthesis of acyloxymethyl carbamates 10 and 11. | |
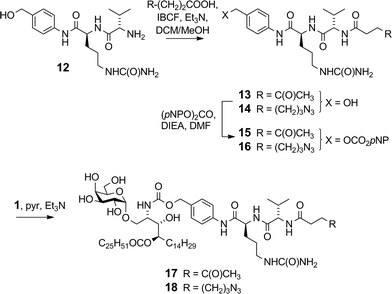 |
| | Scheme 3 Synthesis of VC–PAB carbamates 17 and 18. | |
The VC–PAB linkers were produced by acylation of known amine 12 (ref. 20) with levulinic acid or 6-azidohexanoic acid employing the mixed anhydride method (Scheme 3). The resulting alcohols 13 and 14 were converted to their p-nitrophenyl (pNP) carbonates 15 and 16, and subsequently reacted with 1 in pyridine with triethylamine to afford ketone 17 and azide 18.
Although widely used, aminooxy-terminated peptides can be difficult to obtain in pure form due to side reactions and purification difficulties, as reported in the literature36–38 and illustrated by our own difficulties encountered in trying to obtain sufficient quantities of peptide 19 from commercial sources. Instead, employing an Fmoc solid phase peptide synthesis strategy utilising an in-house manufactured aminomethyl polystyrene resin gave sufficient quantities of 19 without incident. Ketones 10 and 17 were reacted with aminooxy-terminated peptide 19 containing the OVA257 epitope SIINFEKL (Scheme 4). The disparate solubility properties of the glycolipid and peptide reaction partners necessitated a peculiar solvent cocktail at high dilution (2 mM in ketone). Preliminary experiments gave very poor conversion, even with catalysis by aniline (pH 4.7, 20 mM).39 Quenching of the aminooxy group with solvent-derived low-Mw aldehydes/ketones was a problem at high dilution, which could be minimised by distilling solvents from 2,4-dinitrophenylhydrazine before use, however reactions were still unacceptably slow. Conversion of ketone 17 was 63% after 1 day at 25 °C, while in the case of 10, the major reaction pathway was hydrolysis of the levulinoyl ester, leading to the production of 1 and formaldehyde, with consequent quenching of the aminooxy group. Conversion of 17 improved to 92% when using 100 mM aniline, and by reducing the pH to ∼4.3 the ester cleavage in compound 10 was almost completely eliminated. A small excess of peptide (0.3 equiv.) was sufficient to achieve >95% conversion of the ketone after 2 days at 25 °C affording conjugates 2 (ref. 16) and 3 in >95% purity after preparative HPLC.
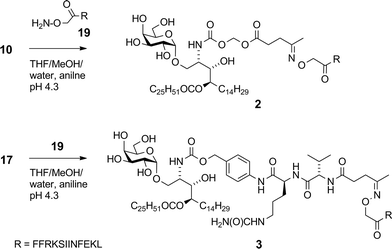 |
| | Scheme 4 Synthesis of conjugate vaccines 2 and 3. | |
Azides 11 and 18 were reacted with alkynyl-terminated peptide 20 in the presence of CuSO4, a reducing agent and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) ligand (Scheme 5).40 Again, the solubility properties of the reaction partners dictated an unusual solvent combination consisting of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3:1 DMSO–chloroform–methanol–water. While no reaction was observed using the standard ascorbate reducing agent, a small piece of copper foil in the reaction mixture generated sufficient Cu(I) for the reaction to proceed to completion after 24 h at 20 °C.41¶ In the absence of TBTA, under otherwise similar conditions, conversion was approximately 30% after three days. The proportion of DMSO was kept at no more than 30% due to its reported40 propensity to counteract the accelerating effect of TBTA at high concentration – a phenomenon we also observed. Using an initial azide concentration of ∼3 mM, at least 1.5 equiv. of peptide was required to achieve full conversion within a reasonable timeframe (24 h). Longer reaction times as necessitated by using less peptide, removing TBTA or increasing the DMSO concentration all resulted in formation of close-running impurities as observed by HPLC. Although we have not been able to fully characterise these by-products, LCMSMS data suggests the occurrence of side-reactions within the peptide chain – an interesting finding in this normally very tolerant click reaction, especially considering that this peptide does not contain any of the usual reactive suspects (e.g. Cys, Met, Trp) in its sequence. After preparative HPLC, conjugates 4 and 5 were obtained in 44% and 46% yields in high purity (>95%).
:3:3:1 DMSO–chloroform–methanol–water. While no reaction was observed using the standard ascorbate reducing agent, a small piece of copper foil in the reaction mixture generated sufficient Cu(I) for the reaction to proceed to completion after 24 h at 20 °C.41¶ In the absence of TBTA, under otherwise similar conditions, conversion was approximately 30% after three days. The proportion of DMSO was kept at no more than 30% due to its reported40 propensity to counteract the accelerating effect of TBTA at high concentration – a phenomenon we also observed. Using an initial azide concentration of ∼3 mM, at least 1.5 equiv. of peptide was required to achieve full conversion within a reasonable timeframe (24 h). Longer reaction times as necessitated by using less peptide, removing TBTA or increasing the DMSO concentration all resulted in formation of close-running impurities as observed by HPLC. Although we have not been able to fully characterise these by-products, LCMSMS data suggests the occurrence of side-reactions within the peptide chain – an interesting finding in this normally very tolerant click reaction, especially considering that this peptide does not contain any of the usual reactive suspects (e.g. Cys, Met, Trp) in its sequence. After preparative HPLC, conjugates 4 and 5 were obtained in 44% and 46% yields in high purity (>95%).
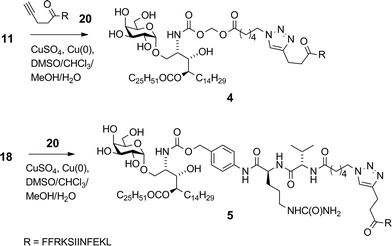 |
| | Scheme 5 Synthesis of conjugate vaccines 4 and 5. | |
To gauge the stability of the linkers in an aqueous medium, we developed an HPLC-MSMS method to detect and quantify low-level formation of α-GalCer which would be expected after any background hydrolysis leading to linker collapse. We have already shown in previous work that once the phytosphingosine amino group is revealed formation of α-GalCer is facile in an aqueous system.16 Aqueous samples at pH 3, 5, 7 and 9 were incubated at 20 °C and monitored periodically for α-GalCer over eight days. Compounds 2 and 4 generated low levels of α-GalCer at physiological and alkaline pH (Table 1, ESI†). No formation of α-GalCer was detected (detection limit = 0.05%) over this time for conjugates 3 and 5 at all pH values indicating greater stability of the VC–PAB carbamate-containing conjugates.
Biological analysis
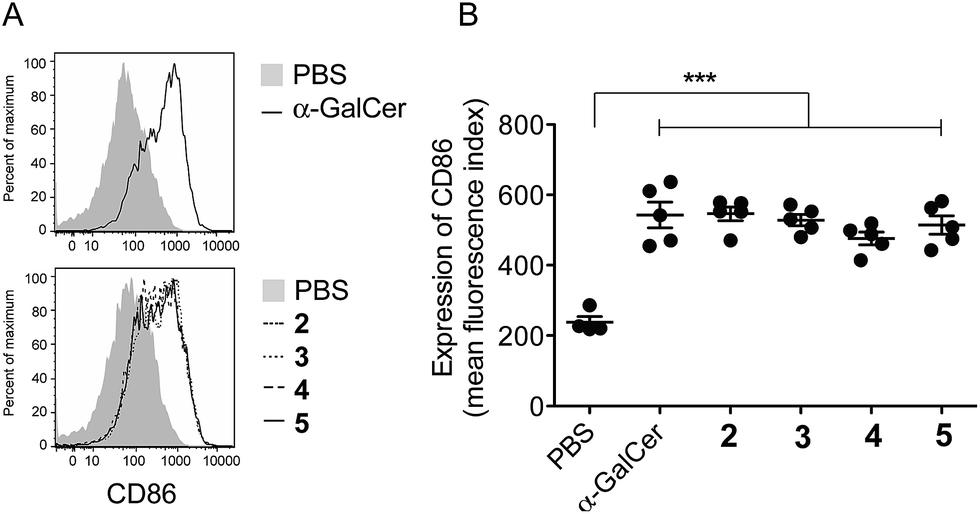
To ensure that the different enzymatic cleavage sites enabled release of a CD1d-binding NKT cell agonist, we tested vaccines 2–5 for their induction of NKT cell activity. By comparing these four compounds, we were able to independently assess the impact of the enzymatic cleavage group (esterase-labile in 2 and 4, versus protease-labile in 3 and 5) and the conjugation chemistry employed (oxime ligation in 2 and 3, versus CuAAC in 4 and 5). The ability of the vaccines to activate NKT cells in mice in vivo was determined indirectly by assessing NKT cell-mediated activation of DCs in the spleen by flow cytometry.11 Using up-regulation of the activation marker CD86 as a readout, all four vaccines induced marked DC activation, resulting in similar levels of CD86 expression to that seen after injection of free α-GalCer (Fig. 3). These results suggest that in vivo cleavage of the linkers in each case releases α-GalCer after elimination and rearrangement. While we cannot rule out some intrinsic agonist activity of the intact conjugates, or of cleaved intermediates, it is pertinent that the N-acetyl derivative of 1, which is unable to collapse to α-GalCer, does not activate NKT cells.16
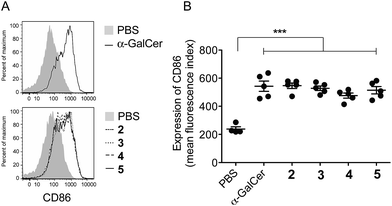 |
| | Fig. 3 Assessment of NKT cell activity in response to i.v. administration of the indicated compounds into C57BL/6 mice, as determined by assessing up-regulation of CD86 as a marker of APC activation in the spleen by flow cytometry. Graphs represent the mean ± SEM of fluorescence intensity for CD86 on DCs (CD11c+ cells) in groups of three animals. Data are representative of two separate experiments. *p < 0.05. | |
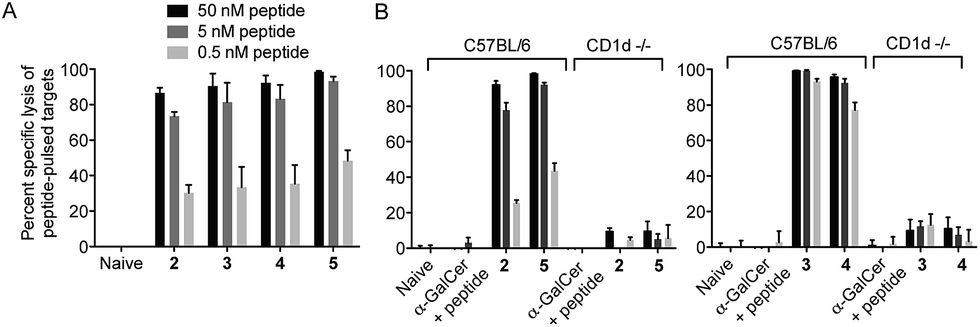
We next tested vaccines 2–5 in an in vivo assay of cytotoxic activity against peptide-loaded target cells. Each of the four vaccines induced significantly increased antigen-specific T cell cytotoxicity compared to the unvaccinated control, with no notable differences in levels of cytolytic activity between the different conjugates (Fig. 4A). These results indicate that all of the vaccines were able to efficiently release the MHC class I-binding peptide epitope and induce a peptide-specific CTL response, and the design of linker had little impact on biological activity. The importance of conjugation is further highlighted by comparing these results to those obtained from treatment with the admixed unconjugated vaccine components (i.e. α-GalCer and OVA257 peptide) at equivalent molar concentrations. Compared to each of the vaccines injection with the admixture resulted in no detectable lysis of target cells (Fig. 4B). Additional evidence that CTL activity was mediated through the NKT cell pathway is provided by the observed loss of activity of the vaccines in CD1d-deficient mice (Fig. 4B).
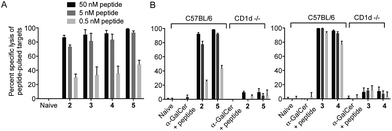 |
| | Fig. 4 Conjugated peptide vaccines stimulate peptide-specific cytotoxic immune responses irrespective of linker design. Assessment of the cytotoxic response was assessed seven days after administration of the indicated compounds compared to administration of unconjugated components (α-GalCer and OVA257 peptide). Cytolytic activity was assessed on injected populations of fluorescent syngeneic splenocytes that had been loaded with different doses of the targeted OVA257 peptide. An additional population of fluorescent splenocytes without peptide served as internal negative control. Lysis was assessed by flow cytometry of blood, and is expressed as percent reduction in peptide loaded cells relative to unloaded control. Data from five mice per group are shown with mean percentage of specific lysis ± SEM indicated. (A): Assessment of 2–5; there were no significant differences in activity between treatment groups. (B): Assessment in wild-type versus CD1d-deficient hosts. Mean percent lysis observed after injection of vaccines in wild-type hosts was significantly enhanced over injection of admixed components, regardless of peptide dose on targets (p < 0.001). | |
To assess the impact of conjugation of the glycolipid on antigen presentation, equivalent molar doses of the free peptide epitope OVA257 and N-terminally capped peptide 21 (see ESI† for structure and synthesis) were cultured with splenocytes from OT-I mice, which express a transgenic TCR for the OVA257 epitope but harbour no NKT cells. Analysis of T cell proliferation in this assay, where there is no adjuvant activity, provides a sensitive measure of presentation of the minimal peptide epitope. Peptide 21 contains the Phe–Phe–Arg–Lys cleavage sequence used in all four vaccines and is therefore required to be processed before MHC class I presentation, however it lacks any glycolipid moiety.24 This requirement for additional processing caused a reduction in peptide presentation relative to cultures containing free OVA257 (Fig. 5A), although it is not clear where this was occurring, and extracellular processing cannot be ruled out. Interestingly, when conjugates 2–5 were examined in this assay, presentation was reduced even further. Reduced presentation of the T cell epitope was also seen when conjugate 5 was cultured with dendritic cells, in this case using flow cytometry to assess the appearance of H-2Kb/OVA257 complexes on the cell surface (Fig. 5B). Interestingly, in dendritic cell cultures containing free OVA257 the levels of expression of H-2Kb/OVA257 complexes decreased over time, whereas levels of these complexes increased in cultures containing conjugate 5 over the same period (Fig. 5C). These data suggest that the extra processing steps enforced by modification of the minimal peptide epitope have a significant negative impact on the immediate presentation of peptide on the cell surface, but nonetheless H-2Kb/OVA257 complexes do ultimately appear on the cell surface with time. These delayed kinetics, which could reflect increased durability of the modified peptide from extracellular degradation, or continued presentation from intracellular pools, may be of more relevance in the in vivo environment, and may contribute to the strong CD8+ T cell responses seen in the presence of NKT cell-mediated adjuvancy. However, further investigation is required to fully elucidate the in vivo significance of these in vitro findings.
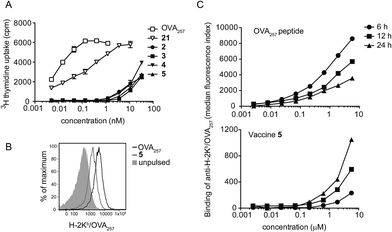 |
| | Fig. 5 Conjugation alters peptide presentation on MHC molecules in vitro. (A): Graded doses of OVA257 peptide, peptide 21 or conjugates 2, 3, 4 or 5 were cultured with splenocytes from OT-I mice, and then 72 h later, proliferation was assessed by uptake of 3H thymidine over the last 8 h of culture. (B) and (C): Graded doses of OVA257 peptide or conjugate 5 were cultured with DC2114 dendritic cells and then expression of H-2Kb/OVA257 complexes determined on the cell surface by flow cytometry 6, 12 and 24 h later. Representative flow cytometry histograms showing binding of anti-H-2Kb/OVA257 24 h after pulsing with 5.7 μM OVA257 peptide, or 5.7 μM conjugate 5, are shown in (B), and graphs showing binding at the indicated times after culture with graded doses are shown in (C). | |
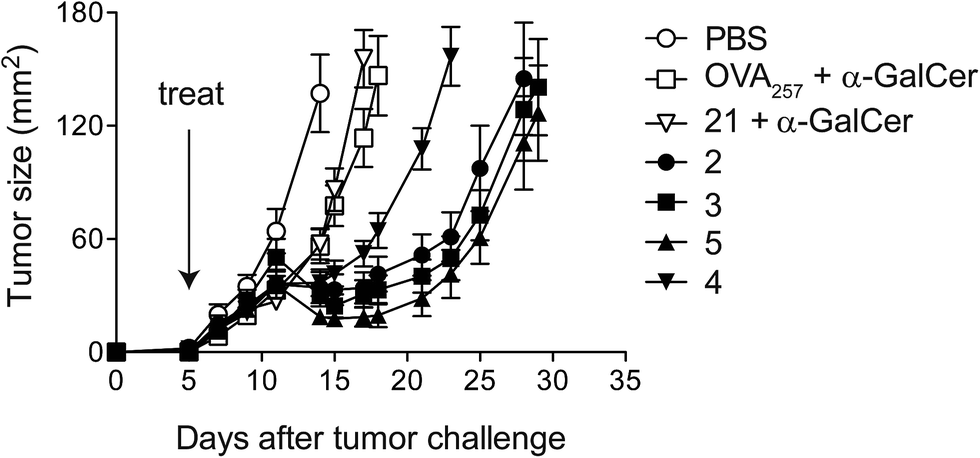
Given that potent cytotoxic activity was induced by all four vaccines, we tested them all as therapeutic agents against the aggressive mouse melanoma, B16.OVA.42 This transplantable tumour cell line has been modified to express ovalbumin as a model of a tumour-specific antigen. To explore the impact of conjugation, injection of admixed unconjugated components (OVA257 peptide and α-GalCer) was used as a control. Additionally, α-GalCer was admixed with the N-terminally modified peptide, 21, to investigate whether any observed differences between vaccines 2–5 and the unconjugated components were simply due to peptide modification43 rather than conjugation to the glycolipid. Once the tumours were engrafted and palpable, a single administration of each of the vaccines (500 pmol, ca. 1.5 μg) resulted in marked anti-tumour activity that was significantly superior to that seen in either group treated with admixed components (Fig. 6). Importantly, although a number of elegant studies have been undertaken with similarly well-defined lipopeptide vaccines, we are unaware of any that report such pronounced single-agent activity in a therapeutic tumour model.9,44–48 Interestingly, despite vaccine 4 inducing similar levels of cytolytic activity to the other vaccines (see Fig. 4), in repeated experiments this conjugate induced less anti-tumour activity. Therefore, it would appear that vaccines with the combination of acyloxymethyl carbamate and triazole groups in the linker induce a different quality of T cell response, which requires further investigation. While vaccines 2, 3 and 5 showed similar anti-tumour activity, the convenient chemistry used to make 5, together with the superior stability of the VC–PAB carbamate linkage it contains (Table 1, SI†), led us to select this design for further analysis.
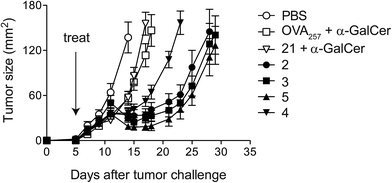 |
| | Fig. 6 Conjugated peptide vaccines stimulate potent anti-tumour activity. A single dose (500 pmol) of each of the conjugate vaccines was administered intravenously five days after challenge with B16.OVA melanoma cells (n = 8 per group). Control groups received admixed peptide and α-GalCer, or the N-terminally modified peptide 21 and α-GalCer. Mean tumour sizes per group ± SEM over time are shown. Data are representative of two separate experiments. | |
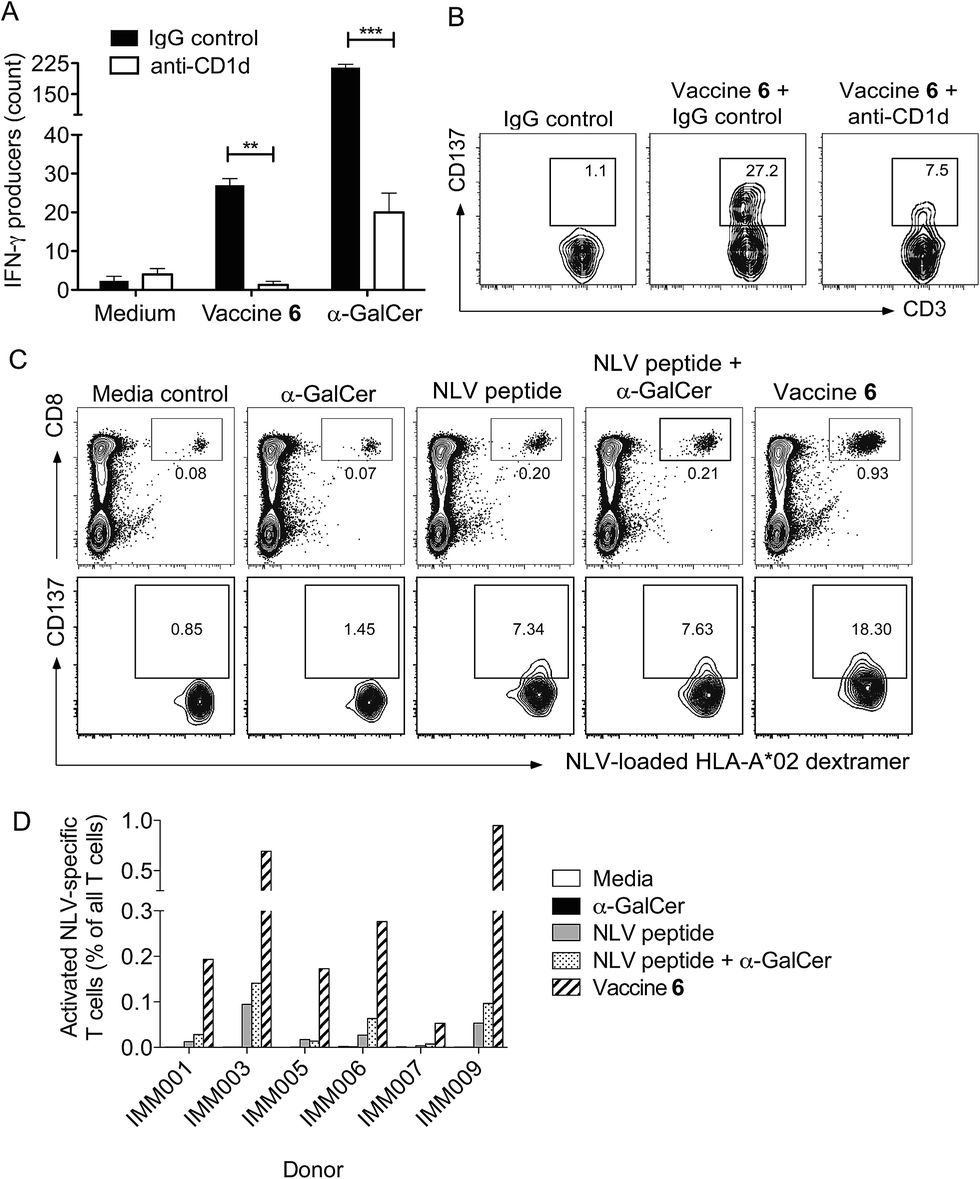
We next investigated whether conjugation would also result in increased T cell activity in assays of human cells. Conjugate vaccine 6, (Fig. 1) incorporating peptide sequence NLVPMVATV (NLV), an HLA-A*02-restricted epitope from cytomegalovirus (CMV) pp65 protein, was synthesised using the experimental conditions developed for vaccine 5. This peptide was chosen because it is possible to detect NLV-specific CD8+ T-cells in peripheral blood mononuclear cells (PBMC) of HLA-A2+ CMV-seropositive donors by flow cytometry using peptide-loaded MHC Class I dextramers.49 In addition, it is notable that CMV proteins are currently being considered as useful tumour-associated antigens for immunotherapy. They are reported to be present in glioblastoma (GBM) while absent from the normal brain tissue, and patient-derived CMV pp65-specific T cells are capable of recognizing and killing autologous GBM tumour cells.50 In initial experiments, the capacity of vaccine 6 to stimulate human NKT cells was assessed in an HLA-A2 negative donor. By simply incubating 6 with PBMC in the presence or absence of a blocking antibody for CD1d, we observed that the conjugate stimulated NKT cells to produce interferon-γ (IFN-γ) in a CD1d-dependent manner, as determined by ELISpot assay (Fig. 7A). Interestingly, significantly fewer IFN-γ-producing NKT cells were seen relative to cultures supplemented with free α-GalCer, which may reflect the extra processing steps required to release the NKT cell agonist from the conjugate. As additional evidence of NKT cell activation in the cultures, analysis by flow cytometry showed that the NKT cells had upregulated CD137, a marker of T cell activation, and this was also blocked by inclusion of anti-CD1d (Fig. 7B). Importantly, after seven days of culture in blood from HLA-A2+ CMV seropositive donors the proportion of NLV peptide-specific CD8+ T-cells was enhanced following treatment with conjugate 6 as compared to peptide alone or admixed peptide and α-GalCer (Fig. 7C). This result was seen in PBMCs from several donors (Fig. 7D). This enhanced proliferation of human peptide-specific T cells shows that the effect of conjugation is not restricted to the mouse system, or indeed the choice of peptide, and that the NKT cell-dependent mechanism of action can be exploited in a human setting. The fact that the result was reproducible in several donors is an important finding given the lower abundance of NKT cells in humans compared to mice,51 and supports the results of a recent study that showed NKT cell-mediated adjuvant activity in a high proportion of participants in a trial of a derivative of α-GalCer.52
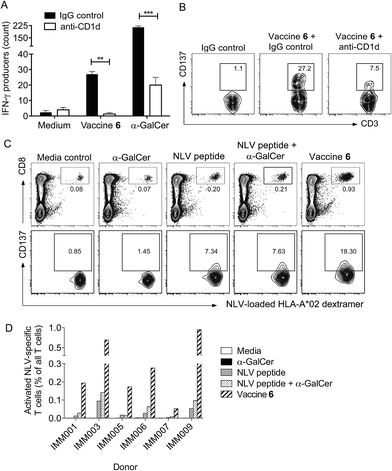 |
| | Fig. 7 Conjugate vaccine 6 activates NKT cells in a CD1d-dependent manner, and enhances expansion of NLV peptide-specific CD8+ T cells in human PBMCs in vitro. (A): PBMCs from an HLA-A*02 negative donor were cultured for 18 hours in the presence of vaccine 6 (500 μg mL−1) and anti-CD1d monoclonal antibody (20 μg mL−1) or IgG isotype control on an ELISpot plate coated with IFN-γ capture antibody. Experiments were conducted in triplicate, and numbers represent mean number of IFN-γ ELISpots per well. **p < 0.01, ***p < 0.001. (B): Typical flow cytometry plots showing expression of CD137 on NKT cells after 18 hours. Numbers represent the percentage of cells activated. (C): PBMCs from an HLA-A*02 positive CMV seropositive donor were cultured in the presence of vaccine 6 (500 μg mL−1), NLV peptide or α-GalCer or both, at equimolar concentrations. The frequency of NLV-specific CD8+ T cells was determined by flow cytometry after seven days. Typical flow cytometry plots are presented showing gated NLV-specific cells (top panels), with numbers representing NLV-specific CD8+ T cell frequency as a percentage of all live T cells. Lower panels show expression of CD137 on these cells. (D): Summary of flow cytometry data from several donors, showing activated NLV-specific cells (CD137+) as percentage of all T cells. | |
Conclusions
In summary we have developed and refined NKT cell-dependent lipopeptide vaccines that show potent therapeutic activity in an aggressive melanoma model. All of the conjugates tested showed superior biological activity to unconjugated admixed controls. While the choice of linker had only limited impact on vaccine potency, the increased stability of the VC–PAB carbamate at physiological pH and ease of synthesis of the triazole linkage are important attributes for future good manufacturing practice development. Importantly, in vitro data suggest the mechanism for the increased T cell activity works in the human setting. The application of this synthetic vaccine technology to other relevant antigens, and other disease conditions, is currently under further investigation.
Acknowledgements
We thank the personnel of the Biomedical Research Unit of the Malaghan Institute of Medical Research for animal husbandry. This research was supported by the New Zealand Ministry of Science and Innovation (C08X0808), Genesis Oncology Trust (GOT-1352-RPG), and New Zealand Health Research Council (Project 14-500).
Notes and references
- P. W. Kantoff, C. S. Higano, N. D. Shore, E. R. Berger, E. J. Small, D. F. Penson, C. H. Redfern, A. C. Ferrari, R. Dreicer, R. B. Sims, Y. Xu, M. W. Frohlich and P. F. Schellhammer, N. Engl. J. Med., 2010, 363, 411–422 CrossRef CAS PubMed.
- J. D. Wolchok, H. Kluger, M. K. Callahan, M. A. Postow, N. A. Rizvi, A. M. Lesokhin, N. H. Segal, C. E. Ariyan, R. A. Gordon, K. Reed, M. M. Burke, A. Caldwell, S. A. Kronenberg, B. U. Agunwamba, X. Zhang, I. Lowy, H. D. Inzunza, W. Feely, C. E. Horak, Q. Hong, A. J. Korman, J. M. Wigginton, A. Gupta and M. Sznol, N. Engl. J. Med., 2013, 369, 122–133 CrossRef PubMed.
- Y. Hailemichael, Z. Dai, N. Jaffarzad, Y. Ye, M. A. Medina, X. F. Huang, S. M. Dorta-Estremera, N. R. Greeley, G. Nitti, W. Peng, C. Liu, Y. Lou, Z. Wang, W. Ma, B. Rabinovich, K. S. Schluns, R. E. Davis, P. Hwu and W. W. Overwijk, Nat. Med., 2013, 19, 465–472 CrossRef CAS PubMed.
- S. A. Rosenberg, J. C. Yang and N. P. Restifo, Nat. Med., 2004, 10, 909–915 CrossRef CAS PubMed.
- S. G. Reed, M. T. Orr and C. B. Fox, Nat. Med., 2013, 19, 1597–1608 CrossRef CAS PubMed.
- J. T. van Dissel, S. M. Arend, C. Prins, P. Bang, P. N. Tingskov, K. Lingnau, J. Nouta, M. R. Klein, I. Rosenkrands, T. H. Ottenhoff, I. Kromann, T. M. Doherty and P. Andersen, Vaccine, 2010, 28, 3571–3581 CrossRef CAS PubMed.
- T. H. Ottenhoff, T. M. Doherty, J. T. van Dissel, P. Bang, K. Lingnau, I. Kromann and P. Andersen, Hum. Vaccines, 2010, 6, 1007–1015 CrossRef CAS.
- T. H. Wright, A. E. Brooks, A. J. Didsbury, J. D. MacIntosh, G. M. Williams, P. W. Harris, P. R. Dunbar and M. A. Brimble, Angew. Chem., Int. Ed., 2013, 52, 10616–10619 CrossRef CAS PubMed.
- H. Cai, M. S. Chen, Z. Y. Sun, Y. F. Zhao, H. Kunz and Y. M. Li, Angew. Chem., Int. Ed., 2013, 52, 6106–6110 CrossRef CAS PubMed.
- S. Fujii, K. Shimizu, C. Smith, L. Bonifaz and R. M. Steinman, J. Exp. Med., 2003, 198, 267–279 CrossRef CAS PubMed.
- I. F. Hermans, J. D. Silk, U. Gileadi, M. Salio, B. Mathew, G. Ritter, R. Schmidt, A. L. Harris, L. Old and V. Cerundolo, J. Immunol., 2003, 171, 5140–5147 CrossRef CAS.
- D. I. Godfrey and M. Kronenberg, J. Clin. Invest., 2004, 114, 1379–1388 CrossRef CAS.
- A. Bendelac, P. B. Savage and L. Teyton, Annu. Rev. Immunol., 2007, 25, 297–336 CrossRef CAS PubMed.
- M. Morita, K. Motoki, K. Akimoto, T. Natori, T. Sakai, E. Sawa, K. Yamaji, Y. Koezuka, E. Kobayashi and H. Fukushima, J. Med. Chem., 1995, 38, 2176–2187 CrossRef CAS.
- L. Kain, B. Webb, B. L. Anderson, S. Deng, M. Holt, A. Constanzo, M. Zhao, K. Self, A. Teyton, C. Everett, M. Kronenberg, D. M. Zajonc, A. Bendelac, P. Savage and L. Teyton, Immunity, 2014, 41, 543–554 CrossRef CAS PubMed.
- R. J. Anderson, C. W. Tang, N. J. Daniels, B. J. Compton, C. M. Hayman, K. A. Johnston, D. A. Knight, O. Gasser, H. C. Poyntz, P. M. Ferguson, D. S. Larsen, F. Ronchese, G. F. Painter and I. F. Hermans, Nat. Chem. Biol., 2014, 10, 943–949 CrossRef CAS PubMed.
- M. Cavallari, P. Stallforth, A. Kalinichenko, D. C. Rathwell, T. M. Gronewold, A. Adibekian, L. Mori, R. Landmann, P. H. Seeberger and G. De Libero, Nat. Chem. Biol., 2014, 10, 950–956 CrossRef CAS PubMed.
- J. Alexander, R. Cargill, S. R. Michelson and H. Schwam, J. Med. Chem., 1988, 31, 318–322 CrossRef CAS.
- A. Younes, A. K. Gopal, S. E. Smith, S. M. Ansell, J. D. Rosenblatt, K. J. Savage, R. Ramchandren, N. L. Bartlett, B. D. Cheson, S. de Vos, A. Forero-Torres, C. H. Moskowitz, J. M. Connors, A. Engert, E. K. Larsen, D. A. Kennedy, E. L. Sievers and R. Chen, J. Clin. Oncol., 2012, 30, 2183–2189 CrossRef CAS PubMed.
- G. M. Dubowchik, R. A. Firestone, L. Padilla, D. Willner, S. J. Hofstead, K. Mosure, J. O. Knipe, S. J. Lasch and P. A. Trail, Bioconjugate Chem., 2002, 13, 855–869 CrossRef CAS PubMed.
- G. M. Dubowchik and R. A. Firestone, Bioorg. Med. Chem. Lett., 1998, 8, 3341–3346 CrossRef CAS.
- S. Guha, M. Rajani and H. Padh, Indian J. Biochem. Biophys., 2007, 44, 443–449 CAS.
- W. Zeng, K. J. Horrocks, G. Robevska, C. Y. Wong, K. Azzopardi, M. Tauschek, R. M. Robins-Browne and D. C. Jackson, J. Biol. Chem., 2011, 286, 12944–12951 CrossRef CAS PubMed.
- J. B. Flechtner, K. P. Cohane, S. Mehta, P. Slusarewicz, A. K. Leonard, B. H. Barber, D. L. Levey and S. Andjelic, J. Immunol., 2006, 177, 1017–1027 CrossRef CAS.
- H. Baadsgaard and W. D. Treadwell, Helv. Chim. Acta, 1955, 38, 1669–1679 CrossRef CAS PubMed.
- G. Drefahl and H.-H. Hörhold, Chem. Ber., 1961, 94, 1641–1656 CrossRef CAS PubMed.
- S. K. Johansen, H. T. Kornø and I. Lundt, Synthesis, 1999, 171, 177 Search PubMed.
- J. A. Secrist and M. W. Logue, J. Org. Chem., 1972, 37, 335–336 CrossRef CAS.
- W. W. Turner, R. N. Booher, S. E. Smits and A. Pohland, J. Med. Chem., 1977, 20, 1065–1068 CrossRef CAS.
- T. Tashiro, N. Hongo, R. Nakagawa, K. Seino, H. Watarai, Y. Ishii, M. Taniguchi and K. Mori, Bioorg. Med. Chem., 2008, 16, 8896–8906 CrossRef CAS PubMed.
- J. J. Park, J. H. Lee, S. C. Ghosh, G. Bricard, M. M. Venkataswamy, S. A. Porcelli and S. K. Chung, Bioorg. Med. Chem. Lett., 2008, 18, 3906–3909 CrossRef CAS PubMed.
- J. M. H. Cheng, S. H. Chee, D. A. Knight, H. Acha-Orbea, I. F. Hermans, M. S. M. Timmer and B. L. Stocker, Carbohydr. Res., 2011, 346, 914–926 CrossRef CAS PubMed.
- P. Matto, E. Modica, L. Franchini, F. Facciotti, L. Mori, G. de Libero, G. Lombardi, S. Fallarini, L. Panza, F. Compostella and F. Ronchetti, J. Org. Chem., 2007, 72, 7757–7760 CrossRef CAS PubMed.
- K. Murata, T. Toba, K. Nakanishi, B. Takahashi, T. Yamamura, S. Miyake and H. Annoura, J. Org. Chem., 2005, 70, 2398–2401 CrossRef CAS PubMed.
- S.-Y. Luo, S. S. Kulkarni, C.-H. Chou, W.-M. Liao and S.-C. Hung, J. Org. Chem., 2006, 71, 1226–1229 CrossRef CAS PubMed.
- I. P. Decostaire, D. Lelièvre, H. Zhang and A. F. Delmas, Tetrahedron Lett., 2006, 47, 7057–7060 CrossRef CAS PubMed.
- C. Bure, D. Lelievre and A. Delmas, Rapid Commun. Mass Spectrom., 2000, 14, 2158–2164 CrossRef CAS.
- L. E. Canne, A. R. Ferre- D'Amare, S. K. Burley and S. B. H. Kent, J. Am. Chem. Soc., 1995, 117, 2998–3007 CrossRef CAS.
- A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581–7584 CrossRef CAS PubMed.
- S. I. Presolski, V. Hong, S. H. Cho and M. G. Finn, J. Am. Chem. Soc., 2010, 132, 14570–14576 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- D. M. Brown, T. L. Fisher, C. Wei, J. G. Frelinger and E. M. Lord, Immunology, 2001, 102, 486–497 CrossRef CAS.
- S. Zwaveling, S. C. Ferreira Mota, J. Nouta, M. Johnson, G. B. Lipford, R. Offringa, S. H. van der Burg and C. J. Melief, J. Immunol., 2002, 169, 350–358 CrossRef CAS.
- V. Lakshminarayanan, P. Thompson, M. A. Wolfert, T. Buskas, J. M. Bradley, L. B. Pathangey, C. S. Madsen, P. A. Cohen, S. J. Gendler and G. J. Boons, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 261–266 CrossRef CAS PubMed.
- B. L. Wilkinson, S. Day, L. R. Malins, V. Apostolopoulos and R. J. Payne, Angew. Chem., Int. Ed., 2011, 50, 1635–1639 CrossRef CAS PubMed.
- H. Cai, Z. Y. Sun, M. S. Chen, Y. F. Zhao, H. Kunz and Y. M. Li, Angew. Chem., Int. Ed., 2014, 53, 1699–1703 CrossRef CAS PubMed.
- S. Ingale, M. A. Wolfert, J. Gaekwad, T. Buskas and G. J. Boons, Nat. Chem. Biol., 2007, 3, 663–667 CrossRef CAS PubMed.
- G. G. Zom, S. Khan, C. M. Britten, V. Sommandas, M. G. Camps, N. M. Loof, C. F. Budden, N. J. Meeuwenoord, D. V. Filippov, G. A. van der Marel, H. S. Overkleeft, C. J. Melief and F. Ossendorp, Cancer Immunol. Res., 2014, 2, 756–764 CrossRef CAS PubMed.
- J. W. Gratama, J. W. van Esser, C. H. Lamers, C. Tournay, B. Lowenberg, R. L. Bolhuis and J. J. Cornelissen, Blood, 2001, 98, 1358–1364 CrossRef CAS PubMed.
- S. K. Nair, J. H. Sampson and D. A. Mitchell, OncoImmunology, 2014, 3, e29289 CrossRef PubMed.
- R. Weinkove, C. R. Brooks, J. M. Carter, I. F. Hermans and F. Ronchese, Haematologica, 2013, 98, 376–384 CrossRef CAS PubMed.
- J. N. Tefit, S. Crabe, B. Orlandini, H. Nell, A. Bendelac, S. Deng, P. B. Savage, L. Teyton and V. Serra, Vaccine, 2014, 32, 6138–6145 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc03599b |
| ‡ These authors contributed equally to this work. |
| § Synthesised via Gervay-Hague glycosylation methodology. |
| ¶ We have observed in subsequent experiments that the reaction proceeds similarly well in the absence of CuSO4, indicating that Cu(0) alone can act as a source of Cu(I).41 |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Benjamin J.
Compton‡
a,
Benjamin J.
Compton‡