
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInherently chiral electrodes: the tool for chiral voltammetry
Serena
Arnaboldi
*a,
Tiziana
Benincori
b,
Roberto
Cirilli
c,
Włodzimierz
Kutner
de,
Mirko
Magni
a,
Patrizia Romana
Mussini
*a,
Krzysztof
Noworyta
d and
Francesco
Sannicolò
*a
aUniversità degli Studi di Milano, Dipartimento di Chimica and C.I.Ma.I.NA, via Golgi 19, 20133 Milano, Italy. E-mail: serena.arnaboldi@unimi.it; patrizia.mussini@unimi.it; francesco.sannicolo@unimi.it; Tel: +39 0250314211
bDipartimento di Scienza e Alta Tecnologia, Università degli Studi dell'Insubria, via Valleggio 11, 22100 Como, Italy
cDipartimento del Farmaco, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Roma, Italy
dInstitute of Physical Chemistry, Polish Academy of Sciences (IPC PAS), Kasprzaka 44/52, 01-224 Warsaw, Poland
eFaculty of Mathematics and Natural Sciences, School of Sciences, Cardinal Stefan Wyszynski University in Warsaw, Woycickiego 1/3, 01-938 Warsaw, Poland
First published on 13th January 2015
Abstract
2,2′-Bis[2-(5,2′-bithienyl)]-3,3′-bithianaphthene oligomers are a model case of electroactive films endowed with “inherent chirality”, originating from a stereogenic element coinciding with the whole electroactive backbone, thus resulting in impressive manifestations. This study highlights their applicative potentialities as low-cost and easy-to-prepare artificial enantiopure electrode surfaces, which display an unprecedented ability to pronouncedly separate voltammetry peaks of enantiomers of quite different chiral probes of applicative interest, concurrently with linear dynamic ranges for peak currents, affording enantiomer excess determination. Thus inherently chiral enantiopure electrodes can indeed be regarded as a key to chiral voltammetry.
Introduction
Development of “intelligent” electrodes capable of discriminating enantiomers, in particular those of compounds of biological and pharmaceutical importance, remains as one of the major challenges in electroanalysis.Enantiomeric molecules, being mirror-image structures, have identical physico-chemical scalar properties (properties unchanged by reflection, like m.p., nD, λmax, etc.), but opposite pseudo-scalar ones (properties inverted by reflection, like αD, CD ellipticity, enantiodiscrimination capacity, etc.). When interacting with a racemic probe, chiral molecules are able to recognize the enantiomers through diastereomeric interactions. Classic examples are the selective dispersion and absorption of the two opposite circularly polarized components of plane-polarized light effected by chiral materials, and the chiral stationary phases in HPLC, which control the elution times of two enantiomers. Similarly, chiral electrodes are required for enantioselective electroanalysis.
The only efficient examples of chiral electroanalytical enantiodiscrimination already in use are provided by biosensors, composite systems based on chiral biological materials, chiefly enzymes, as recognition layers, coupled with prevailing amperometric or potentiometric signal transduction. In this context, the development of chiral electrode surfaces and sensors based on artificial enzymes or prepared using synthetic materials is very attractive. In the last few years, chiral platinum surfaces,1–5 SAM-modified electrodes,6,7 surface-modified nanoporous opal film electrodes,8 metals doped with chiral organic molecules,9 films imprinted with enantiopure templates,10–13 or including a chiral crown ether derivative14, a sensitivity-enhanced field-effect chiral sensor hinging on a thiophene-based monomer,15 and mesoporous chiral metal surfaces16 were described.
However, even the most successful attempts at chiral discrimination almost invariably resulted in just the detection of a difference in current intensity between the signals of the two antipodes of a model probe, without differentiation of their redox potentials. Moreover, the chiral enantioselective layer was in many instances specifically tailored for a given probe molecule. Finally, many procedures of surface modification appeared to be very sophisticated and/or the active films very fragile. In this context, most desirable is the development of a low-cost and easy-to-prepare artificial chiral electrode material, stable and capable of discrimination of a wide variety of antipode pairs. Above all, however, discrimination should result in a well pronounced differentiation in the antipode redox potentials, with the corresponding currents exhibiting a linear dynamic range, so that the two antipodes could be recognized and the enantiomeric excess estimated.
Chiral conducting polymers and oligomers represent an attractive pool where a solution to the above issues could be looked for. However, the approaches exploited so far,17 namely, the attachment of chiral pendants to common achiral monomers, or the adoption of chiral templating conditions, have led to poor or unstable chiral manifestations, possibly on account of the “external” nature of the chirality source.
Recently, we devised an innovative approach to chiral conducting thiophene oligomers, based on the concept of “inherent chirality”.18–21 In these oligomers, the stereogenic element is a tailored torsion intrinsic to the whole electroactive backbone. This torsion arises from insertion of atropisomeric biheteroaromatic scaffolds, rather than stereocenters localized in attached pendants. Therefore, in these oligomers, the stereogenic element responsible for chirality is also responsible for electroactivity, since it constitutes the conjugated backbone. Moreover, chemical or electrochemical oligomerization of the 2,2′-bis[2-(5,2′-bithienyl)]3,3′-bithianaphthene (BT2-T4) parent monomer mostly yields cyclic oligomers (BT2-T4)n, predominantly dimers and trimers. They constitute rings of fully conjugated thiophene units, idealizing conducting polymers without ends (Scheme 1).
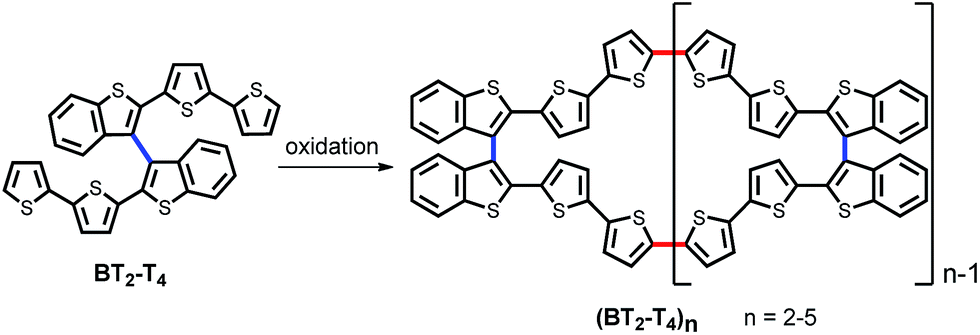 | ||
| Scheme 1 Planar presentation of monomer BT2-T4 and cyclic oligomers (BT2-T4)n. | ||
This outstanding feature combination yields exceptional chirality manifestations: among others, circularly polarized luminescence, and, above all, the capability, as an electrode material, to discriminate the two enantiomers of a commercial ferrocene-based chiral probe in terms of redox potential. That is, two neatly separated reversible voltammetric peaks were reproducibly recorded.18,19 However, several issues still remain to be addressed, in particular those involving the assessment of this material’s aptitude to perform as a chiral electrode material. This assessment includes verification of the enantiorecognition ability of the same material towards different chiral redox probes, possibly of pharmaceutical interest. Moreover, it involves verification of the possibility to work in solvents other than the ionic liquid (IL) applied in our initial tests, thus opening up a large field of experiments governed by the solubility of the analytes. Then, analytical parameters, including detectability, sensitivity, and the existence of a linear dynamic concentration range, should be assessed in order to meet the requirements for the determination of chiral analytes, including quantification of their enantiomeric excess.
Accordingly, we herein provide preliminary, but precisely affirmative, answers to each of the above issues. Thus, we show that this new concept should be regarded as an extraordinarily valuable asset for chiral electroanalysis.
Result and discussions
(S)- and (R)-(BT2-T4)n films, deposited either by potentiodynamic electrooxidation of enantiopure BT2-T4 monomers in 1-butyl-3-methylimidazolium hexafluoro-phosphate (BMIM)PF6 as mixtures of several cyclic oligomer products,18,19,21 or drop-cast from solutions of single cyclic oligomers,19,21 revealed spectacular CV peak potential separation between the (S)- and (R)-antipodes of N,N-dimethyl-1-ferrocenylethylamine. The separation is as high as ∼100 mV if the probe enantiomers are tested individually, and it is even higher for the racemate. The electrode processes are electrochemically and chemically reversible and their first oxidation potential is either slightly lower than18 or close to19 that of the (BT2-T4)n oxidation. These very attractive results were obtained using modified screen printed electrodes (in particular Au-SPEs) in a drop of IL, resulting in the optimal electrodeposition medium, consistent with former literature evidences.22–24 Moreover, not only was the anodic peak potential separation of the enantiomeric probes impressive, but the separation was quite specular on enantiomeric electrodes and reproducible in consecutive experiments performed with the same electrode, after regenerating it with a few CV cycles in probe-free IL. As a possible explanation of such splitting of the antipode peak potentials, it was suggested that as the process takes place inside the chiral film (since it is not significantly charged at the working potentials), the interactions of the enantiopure probes with the surrounding enantiopure environment should be of a diastereomeric nature, and therefore significantly affect the energetics of the electron-transfer process in both thermodynamic and kinetic terms.18 Importantly, the enantiopure films studied so far were not imprinted or templated with the chiral probe employed. In fact, these probes were selected only on account of being commercially available and having a clear reversible signal in a suitable potential range. Another interesting feature concerns the dependence of the separation of the peaks of the enantiomeric probes upon the enantiomeric ratio. In fact, peak separation was much higher when testing the racemate probe compared to the enantiopure antipodes. A reasonable interpretation resorts to antipodal interaction phenomena, first recognized by Wynberg and Feringa a long time ago:25–28 the energetics of a binary system, constituted by a single enantiopure probe in the environment of the enantiopure chiral film, can be significantly different from that of a ternary system formed by a pair of probe enantiomers in the environment of the same enantiopure chiral film.The major issue still to be addressed was whether the enantioselectivity of the same chiral films would be comparable towards other chiral probes of different bulkiness and, desirably, of practical importance. To this aim, we herein tested the films using two different chiral probes of pharmaceutical interest, namely 3,4-dihydroxy-phenylalanine (DOPA), a precursor of catecholamine neurotransmitters, and ofloxacin, a very popular antibiotic. Since the IL medium was appropriate for neither one of them, we turned to conventional solvents. Moreover, we used conventional glassy carbon (GC) electrodes because acetonitrile (MeCN) is not compatible with the insulating paints of commercial SPEs. These conditions afforded further validation of the “inherent chirality” approach to electroanalytical enantioselectivity.
Thus, the enantioselectivity tests were performed for 4.0 mM solutions of L-DOPA and D-DOPA in acidic aqueous solution (50 mM HCl), working on (S)- and (R)-(BT2-T4)n films, deposited by potentiodynamic electrooxidation on commercial GC electrodes from 0.5 mM solutions of the starting monomers in MeCN + 0.1 M tetrabutylammonium hexafluororphosphate (TBA)PF6 (Fig. 1).
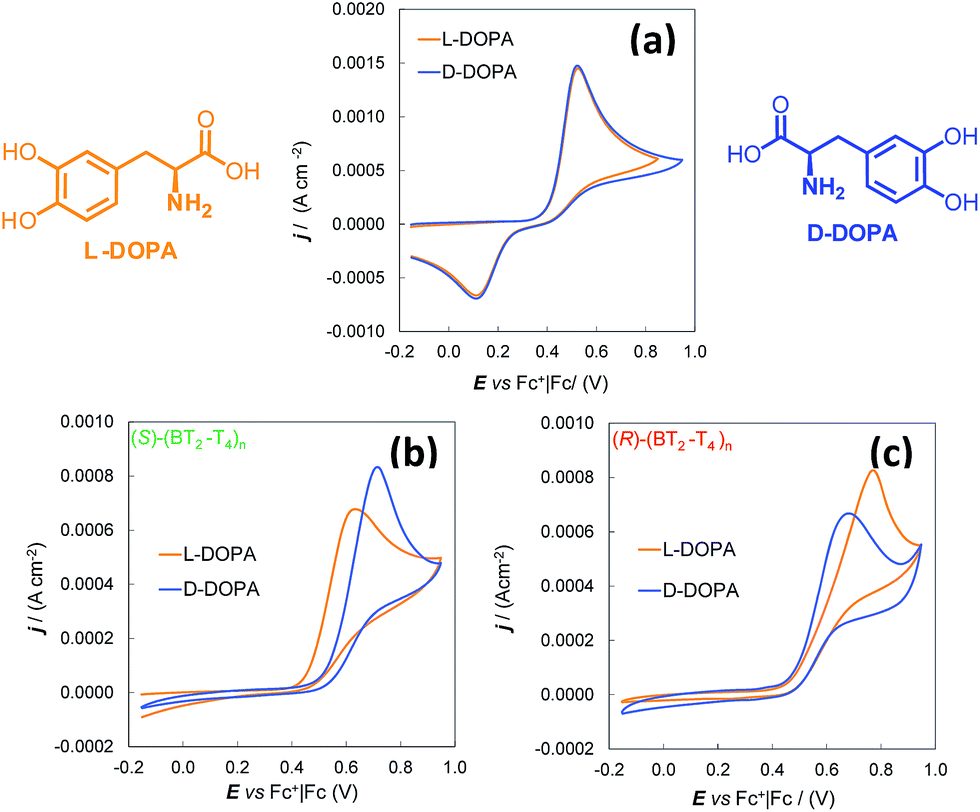 | ||
| Fig. 1 Cyclic voltammetry characteristics of DOPA enantiomers (4 mM in H2O + 50 mM HCl). (a) Bare GC electrode. (b) and (c) GC electrode coated with (S)-(BT2-T4)n and (R)-(BT2-T4)n film, respectively. | ||
Enantioselectivity tests were also performed on the ofloxacin enantiomers, i.e., for the 3.5 mM dextro (R)-enantiomer and for the 3.5 to 8 mM more available levo (S)-enantiomer. In this case, the same MeCN + 0.1 M (TBA)PF6 electrolyte solution was used for both the deposition of the chiral (BT2-T4)n oligomer films on the GC supports by potentiodynamic electrooxidation and the subsequent enantiodiscrimination tests. Under these experimental conditions, the ofloxacin first anodic peak was superimposed on the onset of the (BT2-T4)n film oxidation, implying the need to work at high probe concentrations or to apply background subtraction to enhance the signal-to-background ratio. However, a neat difference of about 80 mV was observed between the oxidation peak of a given enantiomer (in our case levo-ofloxacin, available at higher concentrations) on the specular (S) and (R) surfaces (Fig. 2a and b, green and red thick lines). Symmetrically, the same neat potential difference was observed for the two levo- and dextro-enantiomers on the same (R) enantiomeric surface (Fig. 2a and b, thick and thin red lines) on which the dextro- enantiomer is the first sensed, the results clearly visible even at a concentration of 3.5 mM. The separation of the 3.5 mM levo- and dextro-enantiomers on the (S) surface can be observed too, by CV with background subtraction (Fig. 2c).
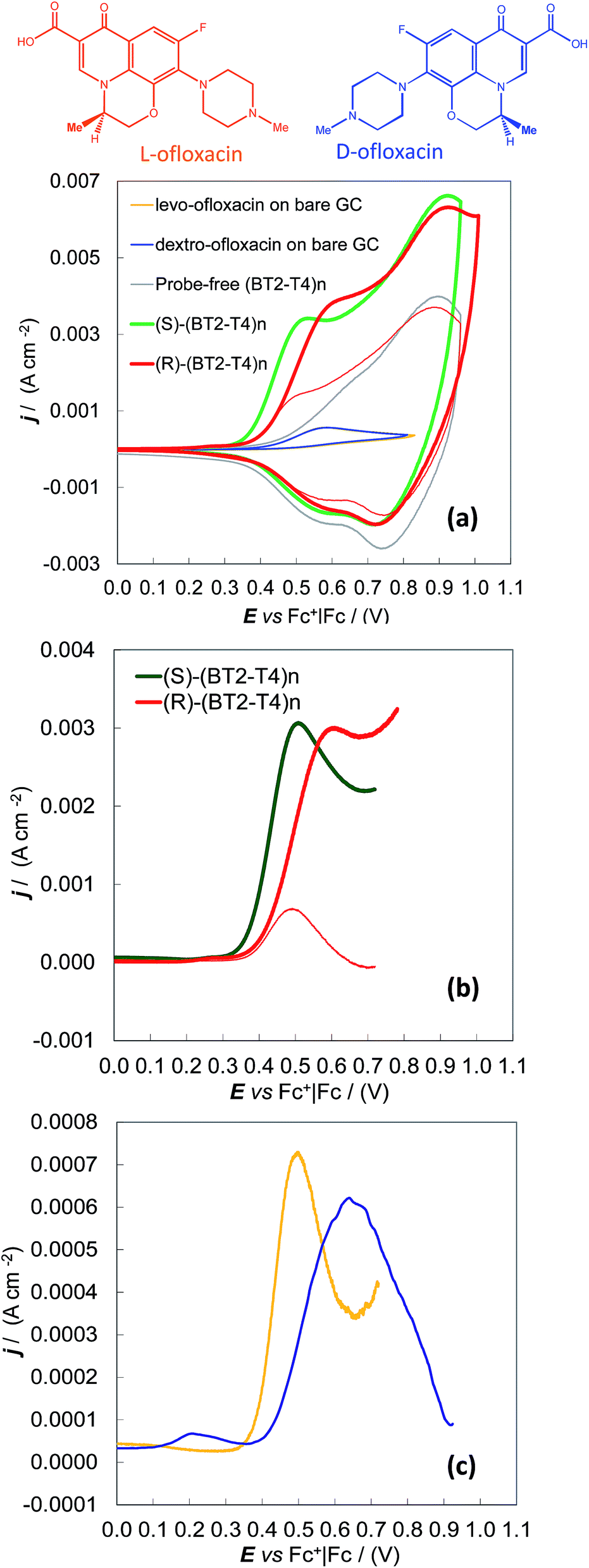 | ||
| Fig. 2 Discrimination of levo- and dextro-ofloxacin enantiomers on GC disk electrodes coated with (S)-(BT2-T4)n and (R)-(BT2-T4)n films, in MeCN + 0.1 M (TBA)PF6 medium. (a) CV patterns and (b) background-subtracted CV patterns recorded for 8 mM levo-ofloxacin on (S) and (R) electrodes (green and red thick lines), and for 3.5 mM dextro-ofloxacin on a (R) electrode (red thin line). (c) Background-subtracted CV patterns for 3.5 mM levo- and dextro-ofloxacin (yellow and blue line respectively) on a (S) electrode. For comparison's sake, plot (a) also includes the CV features of 3.5 mM ofloxacin enantiomers on the bare GC electrode (yellow and blue dotted lines) and the CV features of the probe-free (R)-(BT2-T4)n film electrodeposited on GC (grey line). | ||
Finally, some preliminary quantitative analysis tests were performed in search of a linear dynamic concentration range for the chiral probes on the enantiopure film coated electrodes and for exploiting these tests in estimation of the enantiomeric excess when analysing non-equimolar mixtures of enantiomers.
Concerning the first issue, CV patterns were recorded for levo-ofloxacin in MeCN + 0.1 M (TBA)PF6 on the (S)-(BT2-T4)n film coated GC electrode. Despite the levo-ofloxacin first anodic peak being located at the onset of the (S)-(BT2-T4)n film oxidation, the CV anodic peak current was proportional to the levo-ofloxacin concentration in the range 1.0 mM to 10 mM (Fig. 3), yielding a calibration plot which obeyed the linear regression equation of Ianodic/(A) = (1.7 ± 0.1) × 10−2clevo-ofloxacin/(mol dm−3) + (9 ± 9) × 10−6 with the correlation coefficient R2 = 0.996. The sensitivity and detectability were 0.017 ± 0.001 A M−1 and (6.2 ± 0.2) × 10−4 M, respectively, at a signal-to-noise ratio of 3. Thus, the studied chiral film coated electrodes are suitable for quantitative enantioselective analysis.
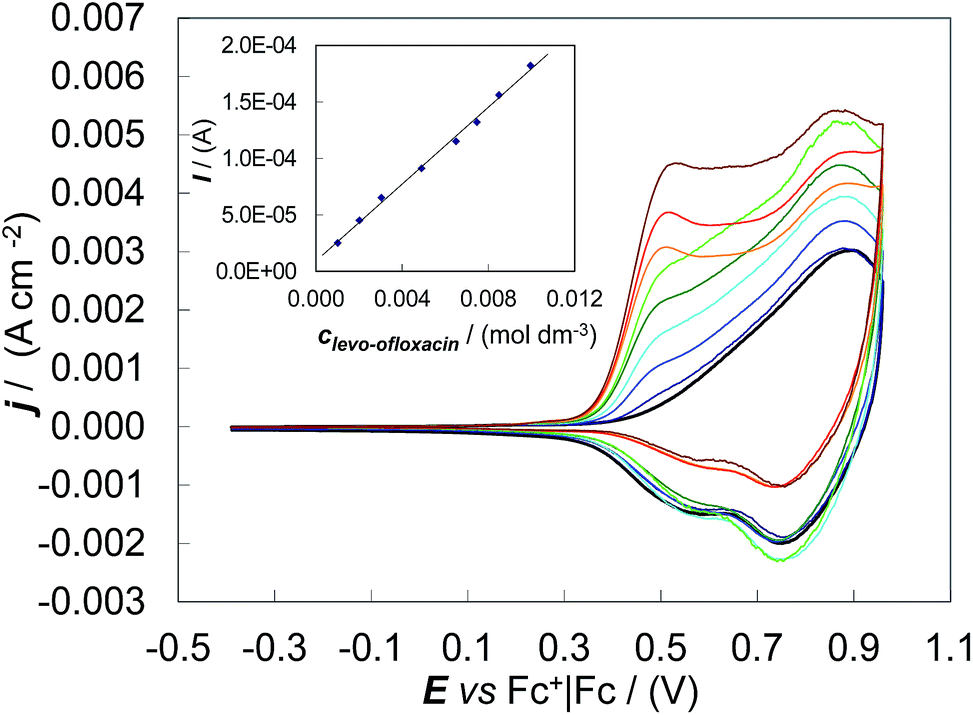 | ||
| Fig. 3 Quantitative CV analysis preliminary tests on (S)-(BT2-T4)n film electrodeposited on GC disk, in MeCN + 0.1 M (TBA)PF6. The inset shows the linear dynamic range for the levo-ofloxacin antibiotic. | ||
The possibility of enantiomeric excess evaluation has been achieved, for the first time in electroanalysis, with (S)- and (R)-N,N-dimethyl-1-ferrocenylethylamine probes on oligo-(S)-(BT2-T4)n films, electrodeposited on Au-SPEs in (BMIM)PF6 (Fig. 4). Two symmetrical series of (BMIM)PF6 solutions with increasing S/R or R/S ferrocenyl probe ratios were analysed by DPV, depositing ∼0.025 cm3 drops on the functionalized Au-SPEs. Each measurement sequence was performed with the use of the same film coated Au-SPE by regenerating the surface between subsequent measurements by a few CV cycles in monomer free IL. For both series, ratios of the DPV peak area for the enantiomers correlated well with the enantiomer concentration ratios, as confirmed by a nearly unit slope of the plot in Fig. 4c.
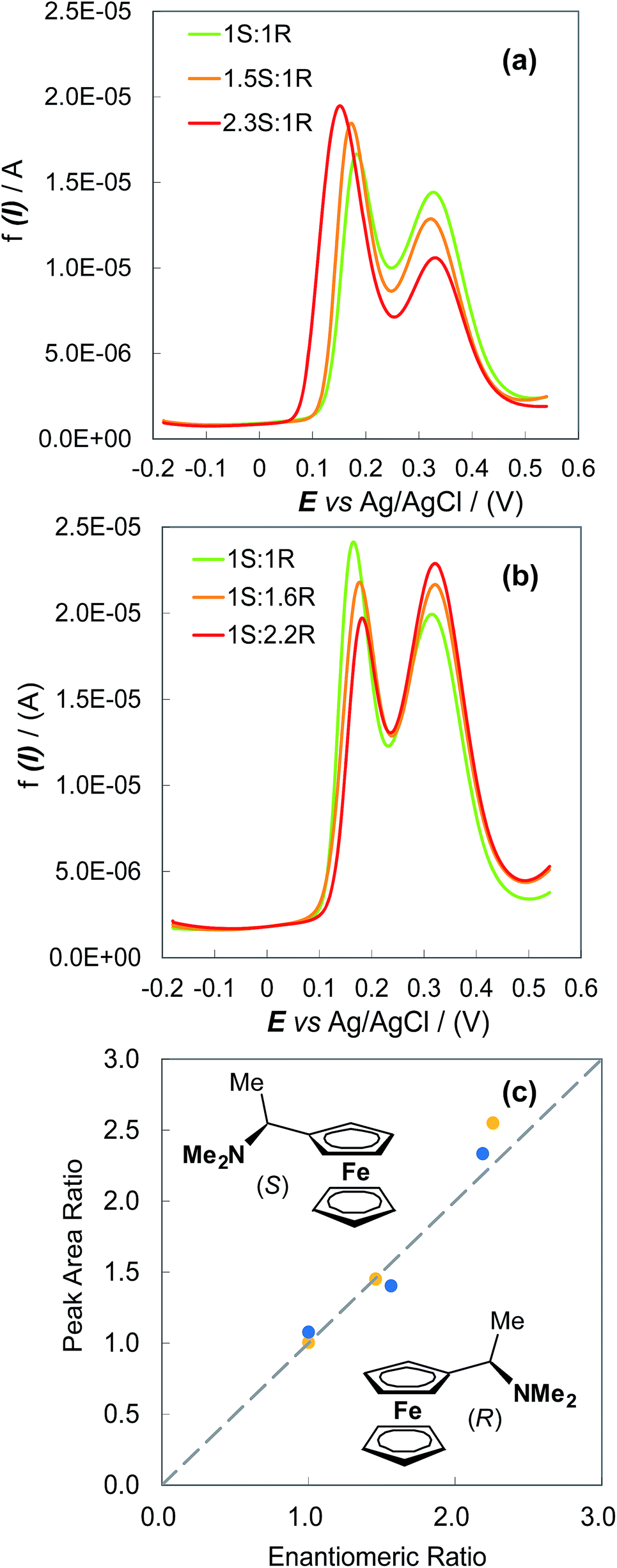 | ||
| Fig. 4 (a) and (b) DPV evaluation of the enantiomeric excess for a ferrocenyl chiral probe, on (S)-(BT2-T4)n film electrodeposited on Au-SPE electrode, in about 0.025 cm3 (BMIM)PF6. (c) DPV peak area ratios plotted against ferrocenyl enantiomer S/R (yellow dots) and R/S (blue dots) molar ratios. The initial concentration of each probe enantiomer was ∼0.026 mol dm−3. | ||
Experimental
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) experiments were performed using Autolab PGSTAT potentiostats (Eco-Chemie (Utrecht, The Netherlands)), controlled by a PC with GPES software from the same manufacturer. All measurements were performed in traditional solvents, acetonitrile (MeCN, Sigma-Aldrich, anhydrous, ≥99.8%) and water (H2O, Sigma-Aldrich, TraceSELECT®). Measurements were carried out using a three-electrode V-shaped minicell (with 3 cm3 of solution), with a glass-embedded glassy carbon disk (GC, Metrohm, S = 0.031 cm2) as the working electrode, a Pt disk as the counter electrode, and an aqueous saturated calomel electrode (SCE) as the reference electrode operating in a double bridge filled with the working medium to avoid water and KCl leakage into the working solutions. N2 purging before measurement was not necessary since all experiments were included in a potential range more positive than O2 reduction. The optimised preliminary polishing procedure for the GC disk electrode consisted of treatment with a diamond powder of 1 μm diameter (Aldrich) on a wet DP-Nap cloth (Struers).Electrodepositions of the conducting chiral (R)- and (S)-(BT2-T4)n oligomer sensing films were performed on the GC disk electrode with 0.5 mM enantiopure (R)- or (S)-BT2-T4 monomer solutions in acetonitrile with 0.1 M tetrabutylammonium hexafluorophosphate as the supporting electrolyte (Fluka, ≥98%). 36 consecutive oxidative potential cycles at 0.2 V s−1 around the first oxidation peaks of BT2-T4 were performed, followed by repeated potential cycling in a monomer-free solution, until the CV curves became stable.
Enantiodiscrimination tests with (R)-ofloxacin (Carbosynth) and (S)-ofloxacin (Sigma-Aldrich, ≥ 98.0%), and L- and D-DOPA ((S)- and (R)-3,4-dihydroxy-phenylalanine respectively, Sigma-Aldrich, ≥98%) were carried out with 1–10 mM ofloxacin solution in acetonitrile with (TBA)PF6 0.1 M, and with 4 mM DOPA aqueous solution with 0.05 M hydrochloric acid (Sigma-Aldrich, ≥37%, for trace analysis) as the supporting electrolyte.
For the preliminary enantiomeric excess experiments on screen printed electrodes, (BMIM)PF6 IL (Sigma-Aldrich, ≥98.5%), a medium granting very high reproducibility and regularity of oligomer deposition, was employed. Enantiopure conducting oligomer films (S)- and (R)-(BT2-T4)n were electrodeposited from a drop (0.02–0.03 cm3) of the respective 12 mM (S)- and (R)-BT2-T4 monomer solutions, dispensed on screen-printed gold working electrodes (Au-SPEs, Metrohm, 61208210) by 36 consecutive oxidative CV cycles at a 0.05 V s−1 scan rate, followed by several potential cycles in a monomer-free blank medium. The voltammetric characteristics of the (R)- and (S)-N,N-dimethyl-1-ferrocenylethylamine probes (Sigma-Aldrich, ≥ 97%) were then recorded, using the probe dissolved again in a drop of (BMIM)PF6. Solutions with (R)-![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(S)-ferrocenyl probe enantiomeric ratios of (i) 1:1; (ii) 1:1.5 and (iii) 1:2, and the specular ones, were employed. The subsequent DPV recordings at increasing (S)/(R) or (R)/(S) ratios were performed on the same electrodeposited film, regenerating it in probe-free conditions between subsequent readings by (a) washing off the probe solution drop with MeCN, then (b) carrying out a few redox cycles in probe-free ionic liquid, then (c) washing off the ionic liquid again with MeCN. In such conditions the film can undergo some modifications that can influence its response to the probe concentration in absolute terms but not in relative ones, as demonstrated by the good correlation in Fig. 4c.
:(S)-ferrocenyl probe enantiomeric ratios of (i) 1:1; (ii) 1:1.5 and (iii) 1:2, and the specular ones, were employed. The subsequent DPV recordings at increasing (S)/(R) or (R)/(S) ratios were performed on the same electrodeposited film, regenerating it in probe-free conditions between subsequent readings by (a) washing off the probe solution drop with MeCN, then (b) carrying out a few redox cycles in probe-free ionic liquid, then (c) washing off the ionic liquid again with MeCN. In such conditions the film can undergo some modifications that can influence its response to the probe concentration in absolute terms but not in relative ones, as demonstrated by the good correlation in Fig. 4c.
Conclusions
The present preliminary tests clearly highlight the impressive enantiodiscrimination aptitude of inherently chiral (BT2-T4)n films to pronouncedly separate voltammetry peaks of enantiomers of chiral probes of quite different chemical and electrochemical properties, and of quite different structural features. Moreover, such aptitude can be exploited, when necessary, in organic and aqueous media and on common electrode supports, besides the optimal combination of ionic liquid medium and screen printed electrodes. Finally, the peak currents recorded on the electrode, coated with the inherently chiral films, display a linear dynamic range, to be exploited, for instance, to quantify enantiomeric excesses on disposable SPEs, testing small drops of enantiomer solutions. It is also remarkable that, while usual chiral recognition methods are based on selectors of natural origin and therefore available as a single enantiomer, this approach offers availability of both selector enantiomers.Although each of the above issues will be the object of specific, more detailed studies in the near future, the present preliminary results confirm that the new inherently chiral enantiopure electrodes can indeed be regarded as a very promising key to chiral voltammetry.
Acknowledgements
This work was financially supported by Fondazione Cariplo (Materiali avanzati 2011-0417 “Inherently Chiral Multifunctional Conducting Polymers”). The authors are also grateful to Laura Santagostini for providing the DOPA enantiopure probes.Notes and references
- A. Ahmadi and G. Attard, Langmuir, 1999, 15, 2420 CrossRef CAS.
- G. A. Attard, A. Ahmadi, J. Feliu, A. Rodes, E. Herrero, S. Blais and G. Jerkiewicz, J. Phys. Chem. B, 1999, 103, 1381 CrossRef CAS.
- D. J. Watson and G. A. Attard, Electrochim. Acta, 2001, 46, 3157 CrossRef CAS.
- G. A. Attard, C. Harris, E. Herrero and J. Feliu, Faraday Discuss., 2002, 121, 253 RSC.
- O. A. Hazzazi, G. A. Attard and P. B. Wells, J. Mol. Catal. A: Chem., 2004, 216, 247 CrossRef CAS PubMed.
- Y. Inose, S. Moniwa, A. Aramata, A. Yamagishi and K. Naing, Chem. Commun., 1997, 11 Search PubMed.
- J. Zhou, L. Wang, Q. Chen, Y. Wang and Y. Fu, Surf. Interface Anal., 2012, 44, 170 CrossRef CAS.
- J. Cichelli and I. Zharov, J. Am. Chem. Soc., 2006, 128, 8130 CrossRef CAS PubMed.
- H. Yang, D. Chi, Q. Sun, W. Sun, H. Wang and J. Lu, Chem. Commun., 2014, 50, 8868 RSC.
- K. Ariga, G. J. Richards, S. Ishihara, H. Izawa and J. P. Hill, Sensors, 2010, 10, 6796 CrossRef CAS PubMed.
- Y. Zhou, B. Yu and K. Levon, Chem. Mater., 2003, 15, 2774 CrossRef CAS.
- S. Fireman-Shoresh, I. Turyan, D. Mandler, D. Avnir and S. Marx, Langmuir, 2005, 21, 7842 CrossRef CAS PubMed.
- R. B. Pernites, S. K. Venkata, B. D. B. Tiu, A. C. C. Yago and R. C. Advincula, Small, 2012, 8, 1669 CrossRef CAS PubMed.
- M.-H. Piao, J. Hwang, M.-S. Won, M. H. Hyun and Y.-B. Shim, Electroanalysis, 2008, 20, 1293 CrossRef CAS.
- L. Torsi, G. M. Farinola, F. Marinelli, M. C. Tanese, O. H. Omar, L. Valli, F. Babudri, F. Palmisano, P. G. Zambonin and F. Naso, Nat. Mater., 2008, 8, 1669 Search PubMed.
- C. Wattanakit, Y. B. Saint Come, V. Lapeyre, P. A. Bopp, M. Heim, S. Yadnum, S. Nokbin, C. Warakulwit, J. Limtrakul and A. Kuhn, Nat. Commun., 2014, 5, 4325 Search PubMed.
- L. A. P. Kane-Maguire and G. G. Wallace, Chem. Soc. Rev., 2010, 39, 2545 RSC.
- F. Sannicolò, S. Arnaboldi, T. Benincori, V. Bonometti, R. Cirilli, L. Dunsch, W. Kutner, G. Longhi, P. R. Mussini, M. Panigati, M. Pierini and S. Rizzo, Angew. Chem., Int. Ed., 2014, 53, 2623 CrossRef PubMed.
- F. Sannicolò, P. R. Mussini, T. Benincori, R. Cirilli, S. Abbate, S. Arnaboldi, S. Casolo, E. Castiglioni, G. Longhi, R. Martinazzo, M. Panigati, M. Pappini, E. Quartapelle Procopio and S. Rizzo, Chem.–Eur. J., 2014, 20, 15298 CrossRef PubMed.
- G. Longhi, S. Abbate, G. Mazzeo, E. Castiglioni, P. Mussini, T. Benincori, R. Martinazzo and F. Sannicolò, J. Phys. Chem. C, 2014, 118, 16019 CAS.
- F. M. E. Sannicolò, P. R. Mussini, S. Arnaboldi, E. Quartapelle Procopio, M. Panigati, R. Martinazzo, E. Selli, G. L. Chiarello, T. Benincori, G. Longhi, S. Rizzo, R. Cirilli and A. Penoni, Patent Application MI-2014-A-000948, 23/5/2014.
- M. Freemantle, An Introduction to Ionic Liquids, Royal Society of Chemistry, London, 2009 Search PubMed.
- P. Hapiot and C. Lagrost, Chem. Rev., 2008, 108, 2238 CrossRef CAS PubMed.
- F. Endres, D. MacFarlane and A. Abbott, Electrodeposition from Ionic Liquids, Wiley-VCH, Weinheim, 2008 Search PubMed.
- H. Wynberg and B. Feringa, Tetrahedron, 1976, 32, 2831 CrossRef CAS.
- C. Girard and H. B. Kagan, Angew. Chem., Int. Ed., 1998, 37, 2922 CrossRef.
- C. Puchot, E. Samuel, E. Dunach, S. Zhao, H. Agami and H. Kagan, J. Am. Chem. Soc., 1986, 108, 2353 CrossRef CAS PubMed.
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |