DOI:
10.1039/C4SC03866E
(Edge Article)
Chem. Sci., 2015,
6, 2427-2433
Phenalenyl-fused porphyrins with different ground states†
Received
13th December 2014
, Accepted 3rd February 2015
First published on 4th February 2015
Abstract
Materials based on biradicals/biradicaloids have potential applications for organic electronics, photonics and spintronics. In this work, we demonstrated that hybridization of porphyrin and polycyclic aromatic hydrocarbon could lead to a new type of stable biradicals/biradicaloids with tunable ground state and physical property. Mono- and bis-phenalenyl fused porphyrins 1 and 2 were synthesized via an intramolecular Friedel–Crafts alkylation-followed-by oxidative dehydrogenation strategy. Our detailed experimental and theoretical studies revealed that 1 has a closed-shell structure with a small biradical character (y = 0.06 by DFT calculation) in the ground state, while 2 exists as a persistent triplet biradical at room temperature under inert atmosphere. Compound 1 underwent hydrogen abstraction from solvent during the crystal growing process while compound 2 was easily oxidized in air to give two dioxo-porphyrin isomers 11a/11b, which can be correlated to their unique biradical character and spin distribution. The physical properties of 1 and 2, their dihydro/tetrahydro-precursors 7/10, and the dioxo-compounds 11a/11b were investigated and compared.
Introduction
Stable π-conjugated biradicaloids have recently attracted intensive research interest due to their unique electronic, optical and magnetic properties and potential applications in organic electronics, non-linear optics, spintronics, and energy storage devices.1 Typical examples include bis(phenalenyls),2 zethrenes,3 indenofluorenes,4 extended p-quinodimethanes and vilogen,5 quinoidal oligothiophenes and thienoacenes,6 and zig-zag edged graphene molecules.7 In addition, a doubly linked corrole dimer8 and a meso-diketo-hexaphyrin9 were also reported by Osuka et al. to be singlet biradicaloids in the ground state. Among the various designs, phenalenyl monoradical10 as the smallest open-shell polycyclic aromatic hydrocarbon (PAH) showing remarkable thermodynamic stability has been used for the design of stable biradicaloids such as bis(phenalenyls)2 and zethrenes.3 Our particular interest here is to develop a new type of hybrid biradicals/biradicaloids by fusion of one or two phenalenyl units onto an aromatic porphyrin skeleton (Fig. 1). Although various PAH-fused porphyrins have been reported,11 none of them showed open-shell biradical character. The mono-phenalenyl fused porphyrin molecule can be drawn at least in two resonance structures, one closed-shell form containing a non-aromatic porphyrin, and an open-shell biradical form possessing an aromatic porphyrin unit (Fig. 1). Thus, it may be a singlet biradicaloid in the ground state. The bis-phenalenyl fused porphyrin however can only be drawn in an open-shell biradical form (Fig. 1). This difference raises the curiosity about their ground state, chemical reactivity and physical properties. Like all other biradicaloids, kinetic blocking of the high spin density sites is necessary for obtaining stable/persistent materials, thus the bulky mesityl-blocked and mono- and bis-phenalenyl fused Ni-porphyrins 1 and 2 (Fig. 1) were synthesized and investigated in this work.
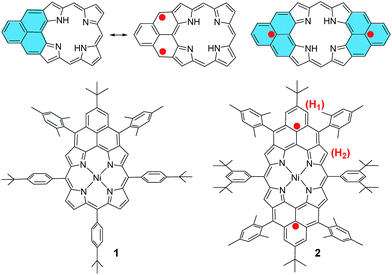 |
| | Fig. 1 Structures of mono- and bis-phenalenyl fused porphyrin biradicaloids and their derivatives 1 and 2. | |
Results and discussion
Synthesis
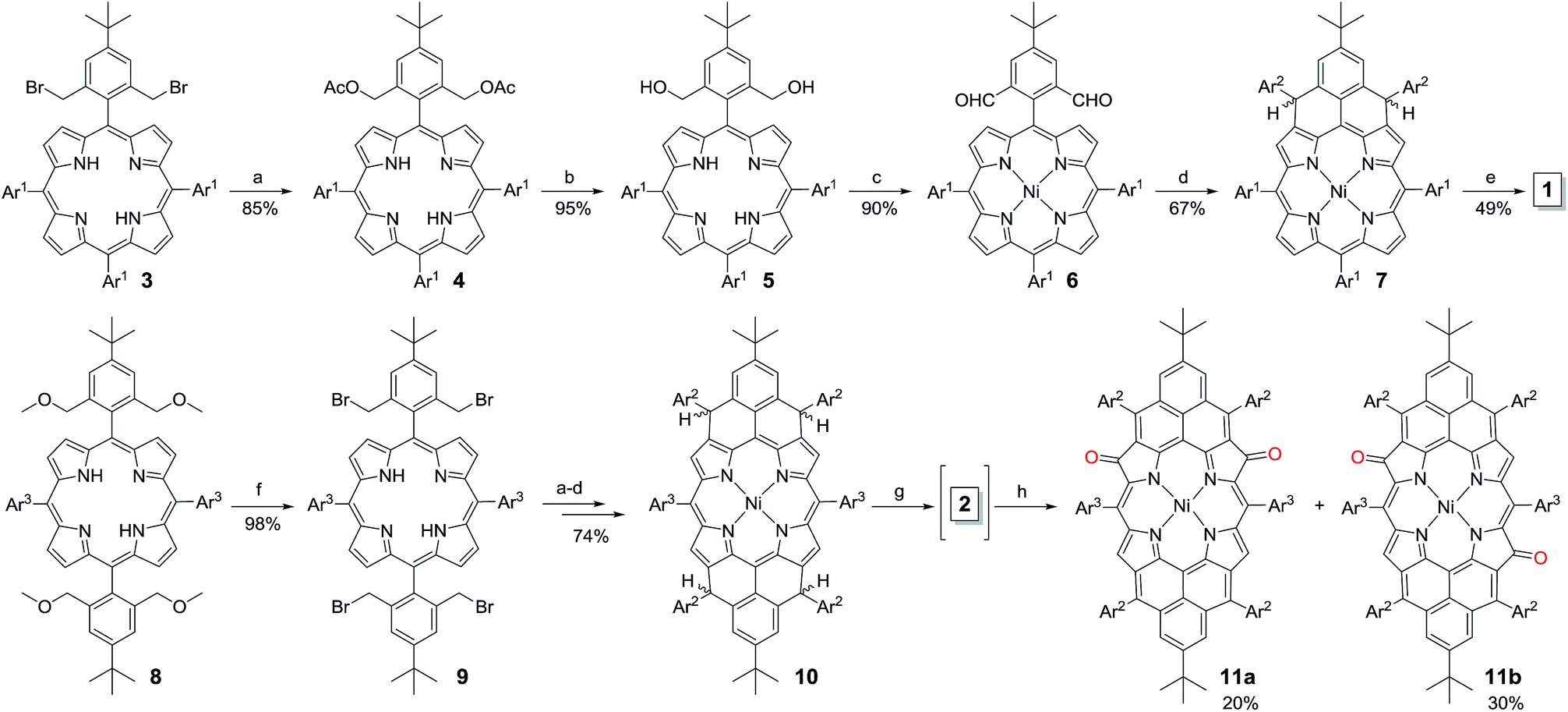
The fusion of one or two phenalenyl units onto the porphyrin core was successfully achieved by an intramolecular Friedel–Crafts alkylation-followed-by-oxidative dehydrogenation strategy (Scheme 1). For the synthesis of 1, the 2,6-bis(bromomethyl)-4-tert-butylphenyl substituted porphyrin 3 (ref. 12) was transformed into the Ni-porphyrin dialdehyde 6 in high yield via an esterification–hydrolysis–Swern oxidation–metallation sequence. Compound 6 was treated with mesitylmagnesium bromide to give the intermediate diol, which was subjected to a Friedel–Crafts alkylation reaction promoted by BF3·OEt2 to afford the dihydro-precursor 7 in 67% yield. Compound 1 was then obtained as a red solid in 49% yield by oxidative dehydrogenation of 7 with N-iodosuccinimide (NIS). It is worthy to note that the obtained Ni-porphyrin 1 showed largely enhanced stability compared to its zinc- and free base porphyrin analogues, which are very sensitive in air and difficult to separate. A similar strategy was used for the synthesis of 2. Treatment of the tetra(methylether)-substituted porphyrin 8 (ref. 13) with HBr in AcOH gave the tetra(bromomethyl)-substituted porphyrin 9, which underwent a similar esterification–hydrolysis–Swern oxidation–metallation–nucleophilic addition–Friedel–Crafts alkylation sequence to afford the tetrahydro-porphyrin precursor 10 in an overall 72% yield. Oxidative dehydrogenation of 10 with p-chloranil in dry dichloromethane (DCM) gave an air sensitive species (compound 2, vide infra) which could not be isolated in pure form. Upon exposure to air for 3 h, the reactive species was transformed into two dioxo-porphyrin isomers 11a (purple solid) and 11b (red solid), which can be separated by routine column chromatography in 20% and 30% yield, respectively.
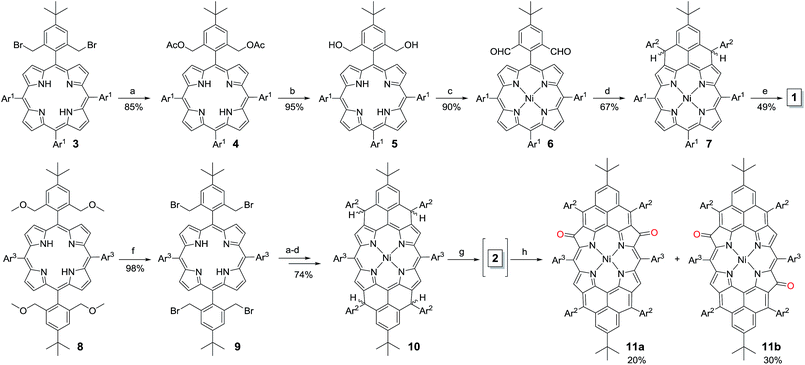 |
| | Scheme 1 Reagents and conditions: (a) KOAc, CH3CN, THF, reflux, 1 day; (b) LiOH·2H2O, dioxane, H2O, reflux, 2 days; (c) (i) oxalyl chloride, DMSO, DCM, Et3N; (ii) Ni(acac)2, toluene, reflux, 24 h; (d) (i) mesitylmagnesium bromide, THF, rt, 24 h; (ii) excess BF3·Et2O, DCM, 10 min; (e) NIS, DCM, 49%; (f) (i) HBr, AcOH, DCM, rt, (ii) NaHCO3 (aq.); (g) p-chloranil, DCM; (h) air. Ar1: 4-tert-butylphenyl, Ar2: mesityl, Ar3: 3,5-di-tert-butylphenyl. | |
Ground states of 1 and 2
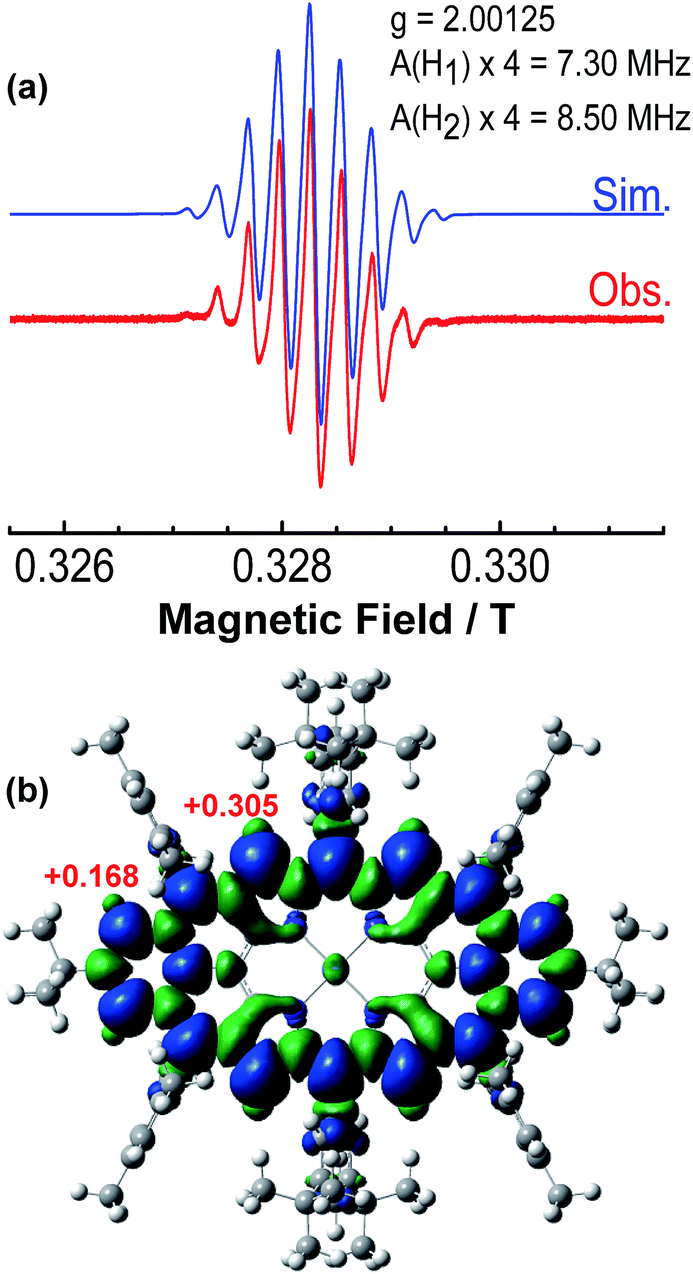
The intermediate species was identified to be the desired compound 2 as a triplet biradical. After addition of p-chloranil under argon, the colour of the solution changed from light-green to dark-brown in 10 minutes at room temperature (RT). High-resolution APCI mass spectroscopic measurement of the solution gave a peak at m/z = 1522.8235 ([M]+; calcd for 2 C108H112N4Ni: 1522.8240), indicating the successful removal of four hydrogens. The absorption spectrum of the solution showed a broad red-shifted absorption extending beyond 1200 nm, which is consistent with the calculated electronic transitions by UB3LYP method (Fig. S1 in ESI†). ESR measurement of the solution clearly exhibited a well-resolved spectrum (ge = 2.00125) (Fig. 2a), with a relative ESR intensity of 94% to the DPPH standard under the same concentration (10 and DPPH) and the same measurement conditions. Broken symmetry density functional theory (DFT) (UB3LYP/6-31G*) calculations suggested that compound 2 has an open-shell triplet ground state (〈s2〉 = 2.1194) with singlet-triplet energy gap (ΔES–T) of +6.98 kcal mol−1 based on Yamaguchi equation. This is reasonable since the structure of 2 cannot be drawn as a closed-shell resonance form, like many other reported triplet biradicals.14 On the basis of the molecular orbital (MO) characteristics, the triplet biradical 2 shows non-disjoint nature of the non-bonding molecular orbitals to avoid the Coulomb repulsion by filing the MO with two electrons, which prefer to the triplet electronic structure. The spin density map suggests that it is a π-electron based biradical species since the spin density on Ni atom is negligible (Fig. 2b). Simulation of the ESR spectrum was conducted by taking consideration of the spin-nucleus coupling with the four protons on the two fused benzene rings (H1 in Fig. 1) and the four β-protons on the pyrrole rings (H2 in Fig. 1), both possessing high spin densities (ρ(H1) = +0.168, ρ(H2) = +0.305, Fig. 2b). The simulated ESR spectrum (A(H1) = 7.30 MHz, A(H2) = 8.50 MHz) of the triplet biradical 2 was in good agreement with the experimentally observed spectrum (Fig. 2a). The VT ESR measurements on the frozen solution (173–113 K) revealed broadened ESR spectra with a hyperfine structure (Fig. S2 in ESI†), which can be well reproduced by similar spin Hamiltonian parameters (A(H1) = 8 MHz, A(H2) = 9 MHz, at 153 K). At the same time, the ESR intensity (I) increased with decreasing temperature (T), with I being approximately proportional to 1/T. The solvent was removed under nitrogen and the solid sample was also submitted to the temperature dependent ESR measurements (298–113 K) (Fig. S2 in ESI†). Similar to the frozen solution, broadened ESR spectra were observed and the ESR intensity showed a very good linear relationship to the 1/T. All these observations together with the DFT calculations supported a triplet biradical character of 2. However, The forbidden half-field Δms = ±2 transition ESR spectrum was not observed due to the large delocalization of the spin, which was also observed in other delocalized biradical systems.3,8,9 It is worth noting that there was no obvious change in spectral shape upon standing at RT in argon for 7 h except for a slight decrease in intensity, indicating good persistence of the triplet biradical under inert atmosphere, which is remarkable for a triplet biradical and can be explained by the efficient spin delocalization along the whole π-conjugated system. However, the pure biradical of 2 could not be isolated even after we tried different ways. The calculated large spin density at the β-pyrrolic carbon atoms also suggested the high reactivity of these sites. In fact, the biradical 2 can be easily oxidized by oxygen when stirring in air and mainly gave two stable dioxo-porphyrins 11a and 11b (Scheme 1).
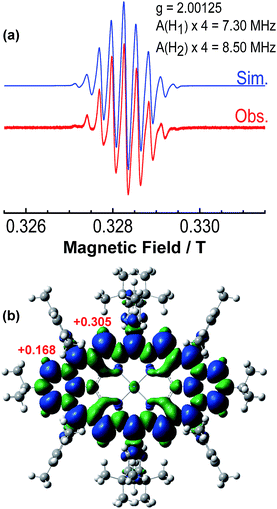 |
| | Fig. 2 (a) ESR spectrum of the in situ generated 2 recorded at 298 K and the simulated spectrum. (b) Calculated spin density distribution of the triplet biradical 2 (UB3LYP/6-31G*). Blue and green surfaces represent positive and negative spin densities, respectively. | |
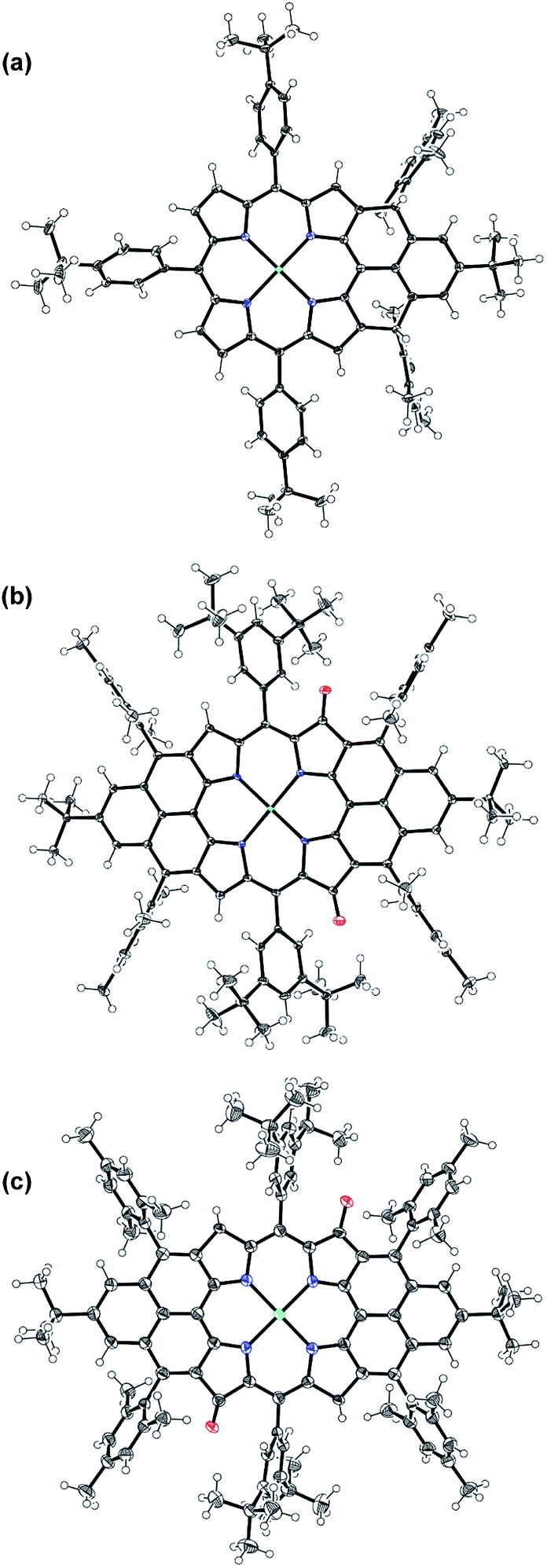
Compound 1 was identified as a closed-shell structure in the ground state based on the observation of sharp NMR spectrum even at elevated temperatures (Fig. S3 in ESI†) and it is also supported by DFT calculations (1(CS) ∼ 1(SB) > 1(TB); biradical character y = 0.06; 〈s2〉 = 0.0003; ΔES–T = −4.28 kcal mol−1 based on UB3LYP/6-31G* calculation). Attempted single crystal growth by slow diffusion of CH3CN into a solution of 1 in toluene however resulted in the dihydrogenated product 1-H2 which was confirmed by the crystallographic analysis (Fig. 3a) and high resolution APCI mass (m/z = 1155.5776 ([M+]; calcd for C80H81N4Ni: 1155.5809)).‡ The hydrogenation selectively took place at the two reactive sites with high spin densities in the phenalenyl unit (Fig. 1). Crystal growth in anhydrous toluene/CH3CN, DCM/CH3CN, and benzene/CH3CN all gave the dihydro-compound 1-H2, indicating that CH3CN likely is the hydrogen source. Compound 1 decomposed in protonic solvents such as methanol and ethanol. The structures of 11a and 11b were also identified by X-ray crystallographic analysis (Fig. 3b and c), implying that they are two isomers which differ only in the positions of the two oxygen atoms.‡ Both complexes have a flat central π-conjugated framework. 11b is centro-symmetric with Ni sitting on the inversion center but 11a is not. They are also the only two possible closed-shell structures that can be drawn for the oxidized products of 2 when the oxidation takes places at two of the four β-pyrrolic positions. Compounds 11a and 11b also showed different NMR spectra, which can be well assigned to their isomeric structures (ESI†).
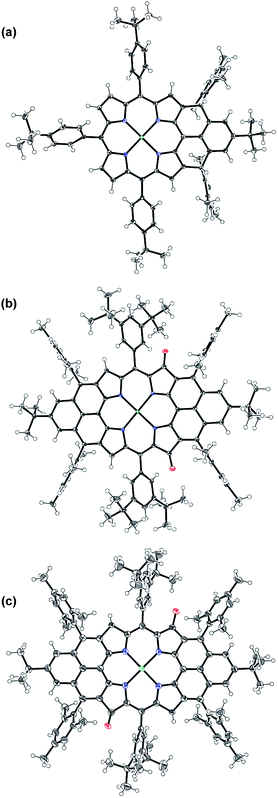 |
| | Fig. 3 X-ray crystallographic structures of 1-H2 (a), 11a (b) and 11b (c). Solvent molecules are omitted for clarity; ellipsoids are set to 50% probability. | |
Optical and electrochemical properties
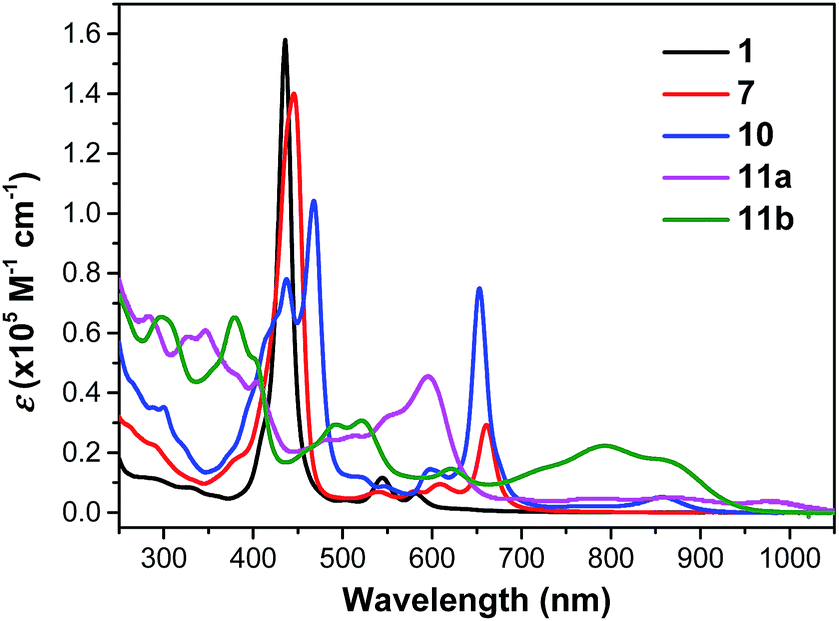
Compound 1 shows one intense Soret band at 436 nm and two weak Q bands at 545 and 582 nm in DCM, which are blue-shifted compared with the dihydro-precursor 7 (Fig. 4 and Table 1). Such a difference can be explained by partial destruction of aromaticity of the porphyrin ring after fusion of a phenalenyl unit. The 1H NMR spectrum of compound 1 however mainly showed an aromatic character possibly due to the contribution of the diradical form and a zwitterionic resonance form to the ground state (Fig. S4 in ESI†).15 The two dioxo-porphyrin isomers 11a and 11b exhibit very different absorption spectra in the UV-vis-NIR region (Fig. 4). The cis-isomer 11a displays a red-shifted absorption spectrum compared with the trans-isomer 11b presumably due to its asymmetric push–pull character. The observed band shape and intensity are well in agreement with the time-dependent DFT calculations for these two isomers (Fig. S5 and S6 in ESI†). Owing to their extended π-conjugation and intramolecular donor–acceptor interactions, both 11a and 11b have a smaller optical energy gap (1.17 eV for 11a and 1.27 eV for 11b) compared with compound 10 (1.37 eV).
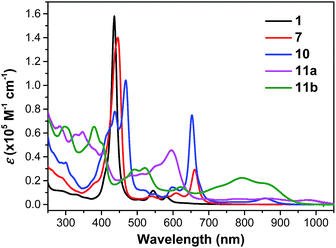 |
| | Fig. 4 UV-vis-NIR absorption spectra of 1, 7, 10, 11a and 11b in DCM. | |
Table 1 Photophysical and electrochemical data of the porphyrin derivatives 1, 7, 10, 11a and 11ba
|
|
λ
max (nm) |
ε
max (M−1 cm−1) |
E
1/2ox (V) |
E
1/2red (V) |
HOMO (eV) |
LUMO (eV) |
E
ECg (eV) |
E
Optg (eV) |
τ (ps) |
σ
(2) (GM) |
|
ε
max: molar extinction coefficient at the absorption maximum. E1/2ox and E1/2red are half-wave potentials of the oxidative and reductive waves, respectively, with potentials versus Fc/Fc+ couple. HOMO and LUMO energy levels were calculated according to equations: HOMO = −(4.8 + Eonsetox) eV and LUMO = −(4.8 + Eonsetred) eV, where Eonsetox and Eonsetred are the onset potentials of the first oxidative and reductive redox wave, respectively. EECg: electrochemical energy gap derived from LUMO–HOMO. EOptg: optical energy gap derived from lowest energy absorption onset in the absorption spectra. τ: excited lifetime based on the TA measurements. σ(2): TPA cross section.
|
|
1
|
436 |
158![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
0.50 |
|
−5.27 |
−3.18 |
2.09 |
1.96 |
300 |
— |
| 544 |
11600 |
0.93 |
−1.71 |
| 582 |
600 |
1.11 |
−2.16 |
|
|
|
1.30 |
|
| |
|
7
|
|
|
0.26 |
|
−4.98 |
−3.64 |
1.34 |
1.66 |
|
370 (1300 nm) |
| 446 |
139800 |
0.47 |
−0.95 |
1.1 (τ1) |
| 662 |
29000 |
0.65 |
−2.18 |
4.2 (τ2) |
|
|
|
1.08 |
|
|
| |
|
10
|
436 |
77500 |
0.02 |
|
−4.73 |
−3.62 |
1.11 |
1.37 |
|
|
| 467 |
103800 |
0.17 |
−0.90 |
4 (τ1) |
780 (1300 nm) |
| 652 |
74400 |
0.57 |
−1.29 |
32 (τ2) |
250 (1700 nm) |
| 858 |
5200 |
0.79 |
−1.72 |
|
|
|
|
|
0.93 |
|
|
|
| |
|
11a
|
346 |
61000 |
0.31 |
−1.13 |
−5.00 |
−3.74 |
1.26 |
1.17 |
2.4 (τ1), |
1000 (1200 nm) |
| 595 |
45700 |
0.95 |
−1.54 |
11.3 (τ2) |
| 997 |
3500 |
|
|
|
| |
|
11b
|
380 |
46000 |
|
|
−5.02 |
−3.66 |
1.36 |
1.27 |
8.9 |
980 (1600 nm) |
| 490 |
29200 |
0.31 |
−1.19 |
| 523 |
30600 |
0.99 |
−1.52 |
| 793 |
22400 |
1.21 |
|
| 862 |
17400 |
|
|
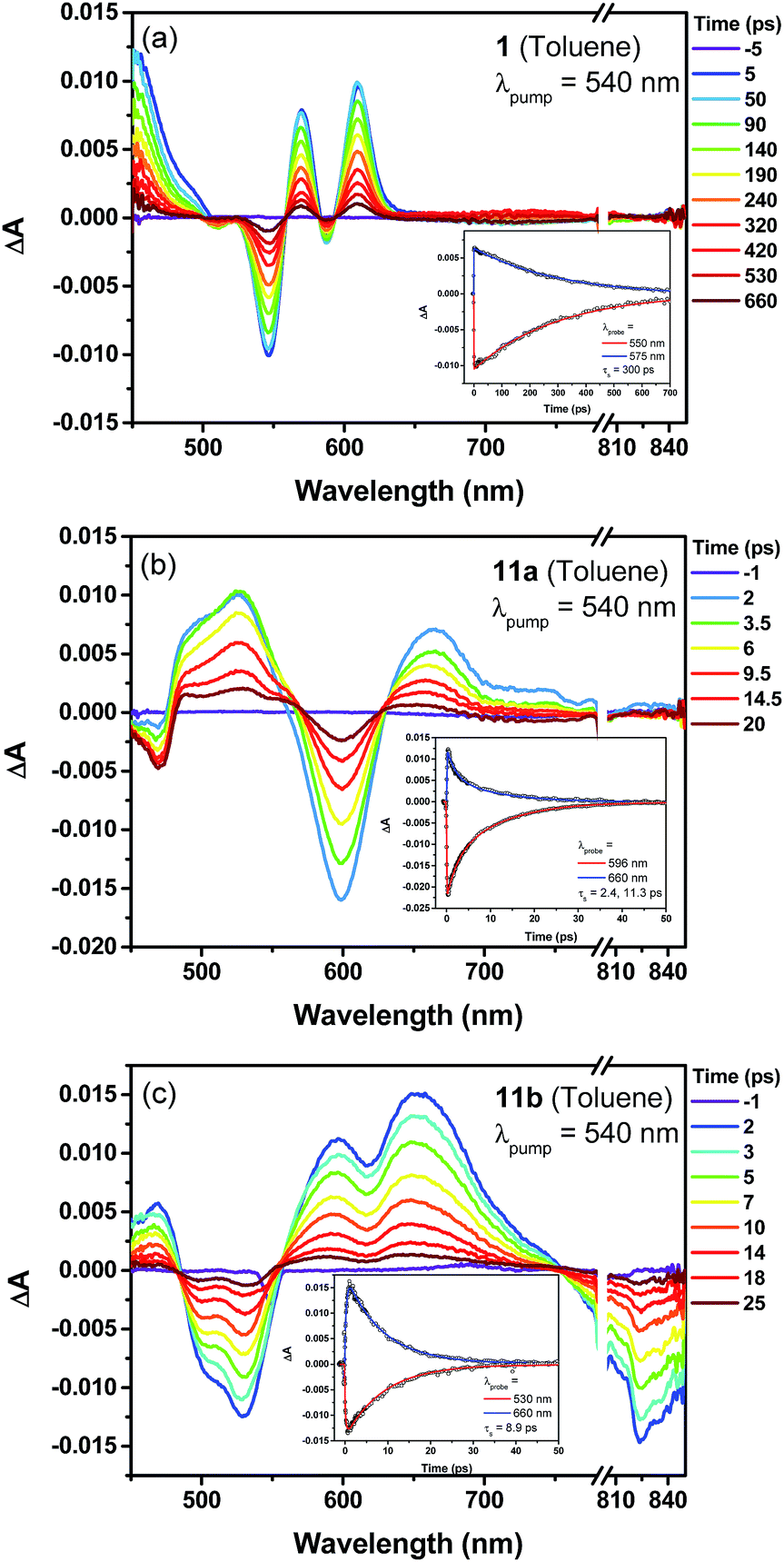
The excited-state dynamics of compound 1, the dihydro- (7) and tetrahydro- (10) precursors, and the dioxo-porphyrins 11a/11b were probed by femto-second transient absorption (TA) measurements (Fig. 5 and Fig. S10 in ESI†). The TA spectrum of 1 exhibited a ground-state bleaching signal around 545 nm together with two excited-state absorption bands at 570 and 610 nm, which is distinct from the dihydro-compound 7 (Fig. S10 in ESI†). At the same time, much longer singlet excited state lifetimes was determined for 1 (τ = 300 ps) than 7 (τ = 14.2 ps). This is out of our expectation since chromophore with small and moderate biradical character was theoretically predicted to show shorter singlet excited lifetime and larger two-photon absorption (TPA) cross sections.16 Such an unusual trend could be ascribed to the relatively larger energy gap of 1 compared with 7. The two dioxo-porphyrin isomers 11a and 11b exhibit very different TA spectra (Fig. 5) and both show shorter singlet excited state lifetime (τ = 11.3 ps for 11a and 8.9 ps for 11b) compared with the tetrahydro-porphyrin 10 (τ = 32 ps).
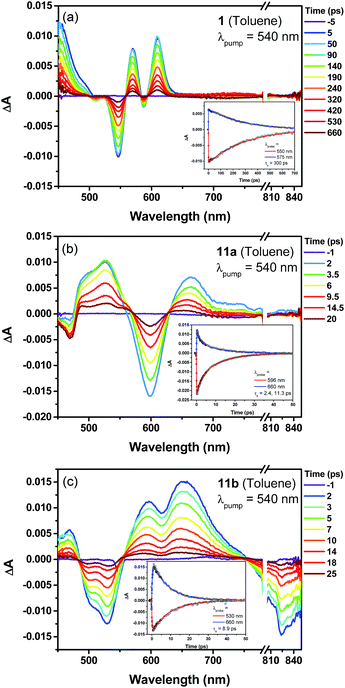 |
| | Fig. 5 Femtosecond transient absorption spectra and decay profiles (inset) of 1 (a), 11a (b) and 11b (c) in toluene measured at room temperature (296 K). For all, the excitation wavelength is 540 nm. | |
Due to the extended π-conjugation, compounds 11a and 11b also showed good third order non-linear susceptibility with large TPA cross sections in the near infrared region, with σ(2) = 1000 GM when excited at 1200 nm for 11a and σ(2) = 980 GM when excited at 1600 nm for 11b, both are larger than the tetrahydro-precursor 10 (σ(2) = 780 GM at 1300 nm and σ(2) = 250 GM at 1700 nm) (Fig. S11 and S12 in ESI†).§.
All the porphyrin compounds (1, 7, 10, 11a, 11b) showed multiple oxidation and reduction waves in the cyclic voltammetry and differential pulse voltammetry (Table 1 and Fig. S13 and S14 in ESI†) and the measured HOMO/LUMO energy levels and energy gaps are consistent with the DFT calculations and optical data (Fig. S7–S9, and Table S5 in ESI†).
Conclusion
In summary, phenalenyl-fused porphyrins 1 and 2 were synthesized by an intramolecular Friedel–Crafts alkylation-followed-by-oxidative dehydrogenation protocol. The mono-phenalenyl fused porphyrinoid 1 turned out to have a closed-shell ground state but the contribution of the biradical form makes it easy to be hydrogenated during the crystal growing process. The bis-phenalenyl fused porphyrinoid 2 cannot be drawn in a closed-shell structure and thus is a triplet biradical. It is persistent in inert atmosphere but can be easily oxidized into two dioxo-porphyrins in air. The observed physical properties and chemical reactivity can be well correlated to their biradical character and spin density distribution. Our research provided a good example of how to develop stable/persistent hybrid biradicaloids.
Acknowledgements
J. W. acknowledges the financial support from the MOE Tier 3 Programme, Tier 2 grant (MOE2014-T2-1-080) and A*STAR JCO grant (1431AFG100). The work at Yonsei University was supported by WCU (World Class University) programs (R32-2010-10217-0) and an AFSOR/APARD grant (no. FA2386-09-1-4092). We thank Dr Tan Geok Kheng and Dr Bruno Donnadieu for the crystallographic analysis.
Notes and references
-
(a) Y. Morita, K. Suzuki, S. Sato and T. Takui, Nat. Chem., 2011, 3, 197 CrossRef CAS PubMed;
(b) Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857 RSC;
(c) J. Casado, R. P. Ortiz and J. T. López Navarrete, Chem. Soc. Rev., 2012, 41, 5672 RSC;
(d) A. Shimizu, Y. Hirao, T. Kubo, M. Nakano, E. Botek and B. Champagne, AIP Conf. Proc., 2012, 1504, 399 CrossRef CAS PubMed;
(e) Z. Sun, Z. Zeng and J. Wu, Chem.–Asian J., 2013, 8, 2894 CrossRef CAS PubMed;
(f) M. Abe, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed;
(g) Z. Sun, Z. Zeng and J. Wu, Acc. Chem. Res., 2014, 47, 2582 CrossRef CAS PubMed.
-
(a) K. Ohashi, T. Kubo, T. Masui, K. Yamamoto, K. Nakasuji, T. Takui, Y. Kai and I. Murata, J. Am. Chem. Soc., 1998, 120, 2018 CrossRef CAS;
(b) T. Kubo, M. Sakamoto, M. Akabane, Y. Fujiwara, K. Yamamoto, M. Akita, K. Inoue, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2004, 43, 6474 CrossRef CAS PubMed;
(c) T. Kubo, A. Shimizu, M. Sakamoto, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Morita and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 6564 CrossRef CAS PubMed;
(d) A. Shimizu, M. Uruichi, K. Yakushi, H. Matsuzaki, H. Okamoto, M. Nakano, Y. Hirao, K. Matsumoto, H. Kurata and T. Kubo, Angew. Chem., Int. Ed., 2009, 48, 5482 CrossRef CAS PubMed;
(e) A. Shimizu, T. Kubo, M. Uruichi, K. Yakushi, M. Nakano, D. Shiomi, K. Sato, T. Takui, Y. Hirao, K. Matsumoto, H. Kurata, Y. Morita and K. Nakasuji, J. Am. Chem. Soc., 2010, 132, 14421 CrossRef CAS PubMed;
(f) A. Shimizu, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, M. Uruichi and K. Yakushi, Chem. Commun., 2012, 48, 5629 RSC.
-
(a) R. Umeda, D. Hibi, K. Miki and Y. Tobe, Org. Lett., 2009, 11, 4104 CrossRef CAS PubMed;
(b) T. C. Wu, C. H. Chem, D. Hibi, A. Shimizu, Y. Tobe and Y. T. Wu, Angew. Chem., Int. Ed., 2010, 49, 7059 CrossRef CAS PubMed;
(c) Z. Sum, K.-W. Huang and J. Wu, Org. Lett., 2010, 12, 4690 CrossRef PubMed;
(d) Z. Sun, K.-W. Huang and J. Wu, J. Am. Chem. Soc., 2011, 133, 11896 CrossRef CAS PubMed;
(e) Y. Li, K.-W. Heng, B. S. Lee, N. Aratani, J. L. Zafra, N. Bao, R. Lee, Y. M. Sung, Z. Sun, K.-W. Huang, R. D. Webster, J. T. López Navarrete, D. Kim, A. Osuka, J. Casado, J. Ding and J. Wu, J. Am. Chem. Soc., 2012, 134, 14913 CrossRef CAS PubMed;
(f) W. Zeng, M. Ishida, S. Lee, Y. Sung, Z. Zeng, Y. Ni, C. Chi, D.-H. Kim and J. Wu, Chem.–Eur. J., 2013, 19, 16814 CrossRef CAS PubMed;
(g) Z. Sun, S. Lee, K. Park, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, S. Das, Y. Li, C. Chi, R. Li, K. Huang, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2013, 135, 18229 CrossRef CAS PubMed;
(h) Z. Sun and J. Wu, J. Org. Chem., 2013, 78, 9032 CrossRef CAS PubMed;
(i) L. Shan, Z.-X. Liang, X.-M. Xu, Q. Tang and Q. Miao, Chem. Sci., 2013, 4, 3294 RSC;
(j) J. L. Zafra, R. C. González Cano, M. C. R. Delgado, Z. Sun, Y. Li, J. T. López Navarrete, J. Wu and J. Casado, J. Chem. Phys., 2014, 140, 054706 CrossRef PubMed;
(k) Y. Li, K.-W. Huang, Z. Sun, R. D. Webster, Z. Zeng, W. Zeng, C. Chi, K. Furukawa and J. Wu, Chem. Sci., 2014, 5, 1908 RSC;
(l) S. Das, S. Lee, M. Son, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, Z. Sun, W. Zeng, R.-W. Li, K.-W. Huang, J. Ding, D. Kim and J. Wu, Chem.–Eur. J., 2014, 20, 11410 CrossRef CAS PubMed;
(m) Z. Sun, B. Zheng, P. Hu, K.-W. Huang and J. Wu, ChemPlusChem, 2014, 79, 1549 CrossRef CAS.
-
(a) D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 1127 CrossRef CAS PubMed;
(b) A. Shimizu and Y. Tobe, Angew. Chem., Int. Ed., 2011, 50, 6906 CrossRef CAS PubMed;
(c) A. G. Fix, D. T. Chase and M. M. Haley, Top. Curr. Chem., 2012, 69, 890 Search PubMed;
(d) A. G. Fix, P. E. Deal, C. L. Vonnegut, B. D. Rose, L. N. Zakharov and M. M. Haley, Org. Lett., 2013, 15, 1362 CrossRef CAS PubMed;
(e) B. D. Rose, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Org. Lett., 2013, 14, 2426 CrossRef PubMed;
(f) A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076 CrossRef CAS PubMed;
(g) H. Miyoshi, S. Nobusue, A. Shimizu, I. Hisaki, M. Miyatab and Y. Tobe, Chem. Sci., 2014, 5, 163 RSC;
(h) B. S. Young, D. T. Chase, J. L. Marshall, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Chem. Sci., 2014, 5, 1008 RSC;
(i) D. Luo, S. Lee, B. Zheng, Z. Sun, W. Zeng, K.-W. Huang, K. Furukawa, D. Kim, R. D. Webster and J. Wu, Chem. Sci., 2014, 5, 4944 RSC.
-
(a) X. Zhu, H. Tsuji, H. Nakabayashi, S. Ohkoshi and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 16342 CrossRef CAS PubMed;
(b) Z. Zeng, Y. M. Sung, N. Bao, D. Tan, R. Lee, J. L. Zafra, B. S. Lee, M. Ishida, J. Ding, J. T. López Navarrete, Y. Li, W. Zeng, D. Kim, K.-W. Huang, R. D. Webster, J. Casado and J. Wu, J. Am. Chem. Soc., 2012, 134, 14513 CrossRef CAS PubMed;
(c) Z. Zeng, M. Ishida, J. L. Zafra, X. Zhu, Y. M. Sung, N. Bao, R. D. Webster, B. S. Lee, R.-W. Li, W. Zeng, Y. Li, C. Chi, J. T. López Navarrete, J. Ding, J. Casado, D. Kim and J. Wu, J. Am. Chem. Soc., 2013, 135, 6363 CrossRef CAS PubMed;
(d) Z. Zeng, S. Lee, J. L. Zafra, M. Ishida, X. Zhu, Z. Sun, Y. Ni, R. D. Webster, R.-W. Li, J. T. López Navarrete, C. Chi, J. Ding, J. Casado, D. Kim and J. Wu, Angew. Chem., Int. Ed., 2013, 52, 8561 CrossRef CAS PubMed;
(e) Z. Zeng, S. Lee, J. L. Zafra, M. Ishida, N. Bao, R. D. Webster, J. T. López Navarrete, J. Ding, J. Casado, D.-H. Kim and J. Wu, Chem. Sci., 2014, 5, 3072 RSC;
(f) W. W. Porter III, T. P. Vaid and A. L. Rheingold, J. Am. Chem. Soc., 2005, 127, 16559 CrossRef PubMed.
-
(a) V. Hernández, S. Calvo Losada, J. Casado, H. Higuchi and J. T. López Navarrete, J. Phys. Chem. A, 2000, 104, 661 CrossRef;
(b) J. Casado, T. M. Pappenfus, K. R. Mann, E. Ortí, P. M. Viruela, B. Milián, V. Hernández and J. T. López Navarrete, ChemPhysChem, 2004, 5, 529 CrossRef CAS PubMed;
(c) T. Takahashi, K. I. Matsuoka, K. Takimiya, T. Otsubo and Y. Aso, J. Am. Chem. Soc., 2005, 127, 8928 CrossRef CAS PubMed;
(d) R. P. Ortiz, J. Casado, V. Hernandez, J. T. López Navarrete, P. M. Viruela, E. Orti, K. Takimiya and T. Otsubo, Angew. Chem., Int. Ed., 2007, 46, 9057 CrossRef CAS PubMed;
(e) R. P. Ortiz, J. Casado, S. R. González, V. Hernández, J. T. López Navarrete, P. M. Viruela, E. Ortí, K. Takamiya and T. Otsubo, Chem.–Eur. J., 2010, 16, 470 CrossRef PubMed;
(f) E. V. Canesi, D. Fazzi, L. Colella, C. Bertarelli and C. Castiglioni, J. Am. Chem. Soc., 2012, 134, 19070 CrossRef CAS PubMed;
(g) G. E. Rudebusch, A. G. Fix, H. A. Henthorn, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Chem. Sci., 2014, 5, 3627 RSC;
(h) X. Shi, P. M. Burrezo, S. Lee, W. Zhang, B. Zheng, G. Dai, J. Chang, J. T. López Navarrete, K.-W. Huang, D. Kim, J. Casado and C. Chi, Chem. Sci., 2014, 5, 4490 RSC.
-
(a) A. Konishi, Y. Hirao, M. Nakano, A. Shimizu, E. Botek, B. Champagne, D. Shiomi, K. Sato, T. Takui, K. Matsumoto, H. Kurata and T. Kubo, J. Am. Chem. Soc., 2010, 132, 11021 CrossRef CAS PubMed;
(b) A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, R. Kishi, Y. Shigeta, M. Nakano, K. Tokunaga, K. Kamada and T. Kubo, J. Am. Chem. Soc., 2013, 135, 1430 CrossRef CAS PubMed.
- S. Hiroko, K. Furukawa, H. Shinokubo and A. Osuka, J. Am. Chem. Soc., 2006, 128, 12380 CrossRef PubMed.
-
(a) T. Koide, K. Furukawa, H. Shinokubo, J.-Y. Shin, K. S. Kim, D. Kim and A. Osuka, J. Am. Chem. Soc., 2010, 132, 7246 CrossRef CAS PubMed;
(b) M. Ishida, J.-Y. Shin, J. M. Kim, B. S. Lee, M.-C. Yoon, T. Koide, J. Sessler, A. Osuka and D. Kim, J. Am. Chem. Soc., 2011, 133, 15333 Search PubMed.
-
(a) P. B. Sogo, M. Nakazaki and M. Calvin, J. Chem. Phys., 1957, 26, 1343 CrossRef CAS PubMed;
(b) K. Goto, T. Kubo, K. Yamamoto, K. Nakasuji, K. Sato, D. Shiomi, T. Takui, M. Kubota, T. Kobayashi, K. Yakusi and J.-Y. Ouyang, J. Am. Chem. Soc., 1999, 121, 1619 CrossRef CAS;
(c) X. Chi, M. E. Itkis, B. O. Patrick, T. M. Barclay, R. W. Reed, R. T. Oakley, A. W. Cordes and R. C. Haddon, J. Am. Chem. Soc., 1999, 121, 10395 CrossRef CAS;
(d) P. A. Koutentis, Y. Chen, Y. Cao, T. P. Best, M. E. Itkis, L. Beer, R. T. Oakley, A. W. Cordes, C. P. Brock and R. C. Haddon, J. Am. Chem. Soc., 2001, 123, 3864 CrossRef CAS PubMed;
(e) M. E. Itkis, X. Chi, A. W. Cordes and R. C. Haddon, Science, 2002, 296, 1443 CrossRef CAS PubMed;
(f) S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley and R. C. Haddon, Science, 2005, 308, 281 CrossRef PubMed;
(g)
Y. Morita and S. Nishida, in Stable Radicals, ed. R. G. Hicks, Wiley-VCH, 2010, pp. 81–145 Search PubMed.
-
(a) T. D. Lash, T. M. Werner, M. L. Thompson and J. M. Manley, J. Org. Chem., 2001, 66, 3152 CrossRef CAS PubMed;
(b) S. Richeter, C. Jeandon, N. Kyritsakas, R. Ruppert and H. J. Callot, J. Org. Chem., 2003, 68, 9200 CrossRef CAS PubMed;
(c) H. S. Gill, M. Marmjanz, J. Santamaría, I. Finger and M. Scott, Angew. Chem., Int. Ed., 2004, 43, 485 CrossRef CAS PubMed;
(d) O. Yamane, K. Sugiura, H. Miyasaka, K. Nakamura, T. Fujimoto, K. T. Kaneda, Y. Sakata and M. Yamashita, Chem. Lett., 2004, 33, 40 CrossRef CAS;
(e) K. Kurotobi, K. S. Kim, S. B. Noh, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2006, 45, 3944 CrossRef CAS PubMed;
(f) M. Tanaka, S. Hayashi, S. Eu, T. Umeyama, Y. Matano and H. Imahori, Chem. Commun., 2007, 2069 RSC;
(g) N. K. S. Davis, M. Pawlicki and H. L. Anderson, Org. Lett., 2008, 10, 3945 CrossRef CAS PubMed;
(h) S. Tokuji, Y. Takahashi, H. Shinmori, H. Shinokubo and A. Osuka, Chem. Commun., 2009, 1028 RSC;
(i) N. K. S. Davis, A. L. Thompson and H. L. Anderson, Org. Lett., 2010, 12, 2124 CrossRef CAS PubMed;
(j) V. V. Diev, K. Hanson, J. D. Zimmerman, S. R. Forrest and M. E. Thompson, Angew. Chem., Int. Ed., 2010, 49, 5523 CrossRef CAS PubMed;
(k) C. Jiao, K.-W. Huang, Z. Guan, Q.-H. Xu and J. Wu, Org. Lett., 2010, 12, 4046 CrossRef CAS PubMed;
(l) C. Jiao, K.-W. Huang and J. Wu, Org. Lett., 2011, 13, 632 CrossRef CAS PubMed;
(m) J. P. Lewtak and D. T. Gryko, Chem. Commun., 2012, 48, 10069 RSC;
(n) J. Luo, M. Xu, R. Li, K.-W. Huang, C. Jiang, Q. Qi, W. Zeng, J. Zhang, C. Chi, P. Wang and J. Wu, J. Am. Chem. Soc., 2014, 136, 265 CrossRef CAS PubMed.
- N. Jux, Org. Lett., 2000, 2, 2129 CrossRef CAS PubMed.
- Synthesis of an analogue of 8 in which the Ar3 is replaced by 4-tert-butylphenyl was reported in ref. 12 and compounds 8 was synthesized by a similar protocol. See details in ESI.†.
-
(a)
Non-Kekulé Molecules as Reactive Intermediates, in Reactive Intermediate Chemistry, ed. R. A. Moss, M. S. Platz and M. Jones Jr, Wiley, 2004 Search PubMed;
(b) P. J. Boratynski, M. Pink, S. Rajca and A. Rajca, Angew. Chem., Int. Ed., 2010, 49, 5459 CrossRef CAS PubMed;
(c) A. Rajca, A. Olankitwanit and S. Rajca, J. Am. Chem. Soc., 2011, 133, 4750 CrossRef CAS PubMed;
(d) K. Okada, T. Imakura, M. Oda, A. Kajiwara, M. Kamachi and M. Yamaguchi, J. Am. Chem. Soc., 1997, 119, 5740 CrossRef CAS.
- Theoretical calculations (DFT-GIAO) on the NICS and NMR chemical shift implied that compound 1 and its free base analog 1′, should have significant non-aromatic character. We also managed to obtain a small amount of free base 1′ by using similar synthetic approach, which however is very sensitive to silica gel/Al2O3 column and air. The 1H NMR spectrum of 1′ (containing some impurities) showed a non-aromatic character as predicted (resonances for the β-H of pyrrole rings appeared at 5.9–6.3 ppm). The increased aromaticity after incorporation of Ni2+ is interesting and could be due to enhanced contribution of the biradical resonance form and the zwitterionic resonance form showed in Fig. S4 in ESI.† Similar phenomenon was also observed in Lash's work: A. M. Young, A. L. Von Ruden and T. D. Lash, Org. Biomol. Chem., 2011, 9, 6293 CAS.
- M. Nakano, T. Minami, K. Yoneda, S. Muhammad, R. Kishi, Y. Shigeta, T. Kubo, L. Rougier, B. Champagne, K. Kamada and K. Ohta, J. Phys. Chem. Lett., 2011, 2, 1094 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data of all new compounds; general methods for all physical characterizations; DFT calculation details; additional spectroscopic and crystallographic data. CCDC 1019251, 1019261 and 1019262. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03866e |
‡ Crystallographic data for 1-H2: C85.50H87N5Ni, Mw = 1243.31; triclinic; space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; a = 15.0498(12) Å, b = 15.6708(13) Å, c = 17.4668(15) Å, α = 111.901(3)°, β = 107.076(3)°, γ = 102.416(3)°; V = 3400.2(5) Å3; Z = 2; ρcalcd = 1.214 Mg m−3; R1 = 0.0441 (I > 2σ(I)), wR2 = 0.1134 (all data). CCDC no. 1019262. Crystallographic data for 11a: C136H142N4NiO2, Mw = 1923.24; triclinic; space group P; a = 12.8361(14) Å, b = 15.5717(18) Å, c = 15.9281(17) Å, α = 100.921(4)°, β = 109.976(4)°, γ = 108.180(4)°; V = 2682.0(5) Å3; Z = 1; ρcalcd = 1.191 Mg m−3; R1 = 0.0506 (I > 2σ(I)), wR2 = 0.1184 (all data). CCDC no. 1019251. Crystallographic data for 11b: C136H142N4NiO2, Mw = 1923.24; triclinic; space group P; a = 12.8500(12) Å, b = 15.5881(15) Å, c = 15.9283(15) Å, α = 100.951(5)°, β = 110.033(5)°, γ = 108.118(5)°; V = 2687.2(5) Å3; Z = 1; ρcalcd = 1.188 Mg m−3; R1 = 0.0771 (I > 2σ(I)), wR2 = 0.2360 (all data). CCDC no. 1019261. Oxygen atom disorder was observed in 11a but the structure can be identified by 1H NMR and TD DFT calculations, and it is the only possible closed-shell dioxo-isomer besides 11b. ; a = 15.0498(12) Å, b = 15.6708(13) Å, c = 17.4668(15) Å, α = 111.901(3)°, β = 107.076(3)°, γ = 102.416(3)°; V = 3400.2(5) Å3; Z = 2; ρcalcd = 1.214 Mg m−3; R1 = 0.0441 (I > 2σ(I)), wR2 = 0.1134 (all data). CCDC no. 1019262. Crystallographic data for 11a: C136H142N4NiO2, Mw = 1923.24; triclinic; space group P; a = 12.8361(14) Å, b = 15.5717(18) Å, c = 15.9281(17) Å, α = 100.921(4)°, β = 109.976(4)°, γ = 108.180(4)°; V = 2682.0(5) Å3; Z = 1; ρcalcd = 1.191 Mg m−3; R1 = 0.0506 (I > 2σ(I)), wR2 = 0.1184 (all data). CCDC no. 1019251. Crystallographic data for 11b: C136H142N4NiO2, Mw = 1923.24; triclinic; space group P; a = 12.8500(12) Å, b = 15.5881(15) Å, c = 15.9283(15) Å, α = 100.951(5)°, β = 110.033(5)°, γ = 108.118(5)°; V = 2687.2(5) Å3; Z = 1; ρcalcd = 1.188 Mg m−3; R1 = 0.0771 (I > 2σ(I)), wR2 = 0.2360 (all data). CCDC no. 1019261. Oxygen atom disorder was observed in 11a but the structure can be identified by 1H NMR and TD DFT calculations, and it is the only possible closed-shell dioxo-isomer besides 11b. |
| § Our TPA set-up is 1200–2400 nm and we cannot measure at OPA wavelengths shorter than 600 nm. Therefore, the TPA data of compound 1 was not obtained. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a