
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Compact, hydrophilic, lanthanide-binding tags for paramagnetic NMR spectroscopy†
M. D.
Lee
a,
C.-T.
Loh
b,
J.
Shin
a,
S.
Chhabra
a,
M. L.
Dennis
a,
G.
Otting
b,
J. D.
Swarbrick
*a and
B.
Graham
*a
aMonash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia. E-mail: bim.graham@monash.edu; james.swarbrick@monash.edu
bResearch School of Chemistry, Australian National University, Canberra, ACT 0200, Australia
First published on 25th February 2015
Abstract
The design, synthesis and evaluation of four novel lanthanide-binding tags for paramagnetic NMR spectroscopy are reported. Each tag is based on the ((2S,2′S,2′′S,2′′′S)-1,1′,1′′,1′′′-(1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrayl)tetrakis(propan-2-ol)) scaffold, featuring small chiral alcohol coordinating pendants to minimise the size and hydrophobic character of each tag. The tags feature different linkers of variable length for conjugation to protein via a single cysteine residue. Each tag's ability to induce pseudocontact shifts (PCS) was assessed on a ubiquitin A28C mutant. Two enantiomeric tags of particular note, C7 and C8, produced significantly larger Δχ-tensors compared to a previously developed tag, C1, attributed to the extremely short linker utilised, limiting the mobility of the bound lanthanide ion. The C7 and C8 tags' capacity to induce PCSs was further demonstrated on GB1 Q32C and 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK) S112C/C80A mutants. Whilst factors such as the choice of lanthanide ion, pH and site of conjugation influence the size of the PCSs obtained, the tags represent a significant advance in the field.
Introduction
The site-specific incorporation of paramagnetic metal ions into proteins allows access to unique NMR parameters that can provide valuable structural insights into protein structure and dynamics.1–3 These include pseudocontact shifts (PCS), residual dipolar couplings (RDC) and paramagnetic relaxation enhancement (PRE). PCSs are particularly attractive structural restraints as they are simple to measure (as the difference in chemical shift between a diamagnetic and paramagnetic sample) and encompass both distance and orientation information of nuclei relative to the magnetic susceptibility anisotropy (Δχ) tensor. The PCS of any nuclear spin can be back-calculated from knowledge of the Δχ-tensor:where Δχax and Δχrh are the axial and rhombic components of the Δχ-tensor and r, θ and φ are the polar coordinates of the nuclei with respect to the principal axes of the Δχ-tensor. The r−3 distance dependence of PCSs allows them to be measured for nuclei up to 40 Å or more away from the metal ion.4 Thus, PCSs provide long-range structural information that can be utilised in the study of protein structure and conformation,5–9 protein–protein10–13 and protein-small molecule interactions,14–17 and even de novo protein structure determination.18,19

Paramagnetic lanthanide(III) ions, except Gd3+, can be used to induce PCSs in the NMR spectra of macromolecules. Their anisotropic magnetic susceptibilities are inherently large (yet different) and, combined with their similar structure and bonding, allow the substitution of one lanthanide ion for another as a convenient route to vary the magnetic properties of a sample. However, most proteins do not natively bind lanthanide ions, which has spurred recent interest in the design of synthetic lanthanide-binding tags (LBTs)3,20 or peptides21,22 capable of introducing lanthanide ions into proteins in a site-specific manner.
Lanthanide ions are “hard” Lewis acids that can adopt high coordination numbers, thus polydentate ligands featuring hard bases (such as O and N atoms) are ideal candidates to ensure tight lanthanide ion binding. Rigidity of the lanthanide relative to the protein frame is paramount to prevent the deleterious averaging effects of tag mobility on measured PCSs and RDCs, which tend towards zero with increasing motion. Engineering tagging sites to take advantage of additional coordination to acidic side-chains of proteins,23–25 conjugation to proteins through multiple sites of attachment26,27 or the use of steric bulk28 have been successful strategies to limit tag mobility. It is imperative that the attached LBT must also give rise to a single observable species in solution, as multiple species in slow exchange can lead to highly complex spectra that are of limited practical utility.29–31
LBTs that bind lanthanide ions extremely tightly, without the need for additional protein interactions, are particularly attractive. They allow the study of proteins in the presence of their own native metal ions and metal ion-bound cofactors (e.g. metalloproteins and kinases) and remove any problems associated with excess free lanthanide ions that can result in line broadening in the NMR spectra. LBTs based on DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) have proved to be useful, having induced significant PCSs and RDCs in several proteins.28,32,33 Although capable of binding lanthanides with dissociation constants of the order of 10−23 to 10−25 M,34 lanthanide complexes of DOTA display a dynamic behaviour in solution at ambient temperature. Inversion of the cyclen ring (defined by the NCCN torsion angle as either δδδδ or λλλλ) and rotation of the pendant arms (defined by the NCCO torsion angles as either Δ or Λ) result in a dynamic equilibrium between square anti-prismatic (SAP) and twisted square anti-prismatic (TSAP) coordination geometries.35 When bound to a protein, this can lead to the presence of multiple stereoisomers in slow exchange, each producing their own paramagnetic effects that greatly complicate analysis of the spectra. In order to limit these conformational exchange processes and simplify the spectra, successful DOTA-based LBT designs have incorporated chiral elements into the pendant arms or cyclen ring,28,33 or employed two-points of conjugation to the protein.32,36
Previously, we demonstrated that attachment of three sterically bulky (S)- or (R)-phenethylacetamide pendant arms to a 1,4,7,10-tetraazacyclododecane (cyclen) macrocyclic ring (C1–C4, Fig. 1) was sufficient to generate a single apparent stereoisomer and to limit tag flexibility, allowing the observation of measureable and sizable paramagnetic effects.28,37 However, for some proteins in our laboratory, such as 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (HPPK), this family of tags was found to present issues in terms of protein stability, as evidenced by an increased tendency to precipitate during and post conjugation. We suggest this to originate from the incompatibility of the large hydrophobic nature of this tag series with these proteins. It is also foreseeable that the tags' hydrophobic character could complicate the study and screening of weak ligand–protein interactions, as small hydrophobic compounds (e.g. from fragment libraries) can potentially associate transiently with the LBT, leading to a transferred PCS effect and a skewed, “meaningless” average ligand PCS.
 | ||
| Fig. 1 Existing and newly developed LBTs referred to in the text. C2 and C8 are the enantiomers of C1 and C7, respectively. | ||
With this in mind, we have now developed a new series of tags that are much more structurally compact and hydrophilic in nature (C5–C8, Fig. 1). These tags are based on ((2S,2′S,2′′S,2′′′S)-1,1′,1′′,1′′′-(1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrayl)tetrakis(propan-2-ol)) ((S)-THP), a cyclen derivative featuring four chiral (S)-2-hydroxypropyl pendants. Multiple (S)-THP-Ln3+ (where Ln = La, Ce, Nd, Eu, Yb or Lu) complexes have been reported to show 1H NMR spectra that display a single set of resonances,38–40 which suggested that an (S)-THP based LBT could also produce a single set of PCSs to nuclei of a bound protein. The (S)-THP-Yb3+ complex specifically has been shown to adopt a Λ(λλλλ) TSAP geometry in solution.39
Conjugation of single-point attachment LBTs to proteins requires less prior structural knowledge of the target, fewer mutations for their introduction and can still produce useful effective Δχ-tensors when tag movements are limited.41 Thus, our initial focus has been on the development of (S)-THP derivatives featuring a single thiol-conjugatable group, so as to produce tags applicable to the study of as wide a range of protein systems as possible. The first of these (C5) utilises the same pyridyl disulfide-activated linker as our earlier reported C1 and C2 tags. Given the absence of the sterically bulky pendants of the latter tags, which were postulated to be an important element in limiting tag flexibility,28 it was anticipated that this tag might prove too mobile for NMR applications. Therefore, analogues with shorter linker groups were also engineered. C6 features a bidentate chelating 2-carboxylpyridine moiety with a conjugatable methylmercaptan group attached to the 4-position of the pyridine ring, and can be viewed as a hybrid of (S)-THP and the various DPA-based LBTs reported by Otting and co-workers.23,42–44C7, and its enantiomer C8, feature possibly the smallest practicable modification to (S)-THP that allows for bioconjugation: a pyridyl disulfide group is attached directly to one of the four chiral 2-hydroxypropyl pendants and the resulting protein-conjugated tags feature only a disulfide bond between the chirally pure (S)/(R)-THP-Ln3+ chelate and protein.
We now report the synthesis of the new tags (C5–C8) and demonstrate their utility in paramagnetic NMR structural studies using human ubiquitin and GB1 as model proteins, as well as the antimicrobial target, HPPK.45 As detailed below, the C5 and C6 tags are found to perform comparably to C1 in terms of the magnitude of the Δχ-tensors observed on ubiquitin. More significantly, however, the C7 and C8 tags produce considerably larger paramagnetic effects, indicating that the short linker present within these tags translates to a more restricted lanthanide ion attachment to the protein.
Results and discussion
Tag synthesis
C5 was prepared in good overall yield by nucleophilic substitution between the previously reported compounds, (1S,4S,7S)-1,4,7-tris(2-hydroxypropyl)-1,4,7,10-tetraazacyclododecane46 (1) and 2-chloro-N-(2-(pyridin-2-yldisulfanyl)ethyl)acetamide.28 (2) (Scheme 1).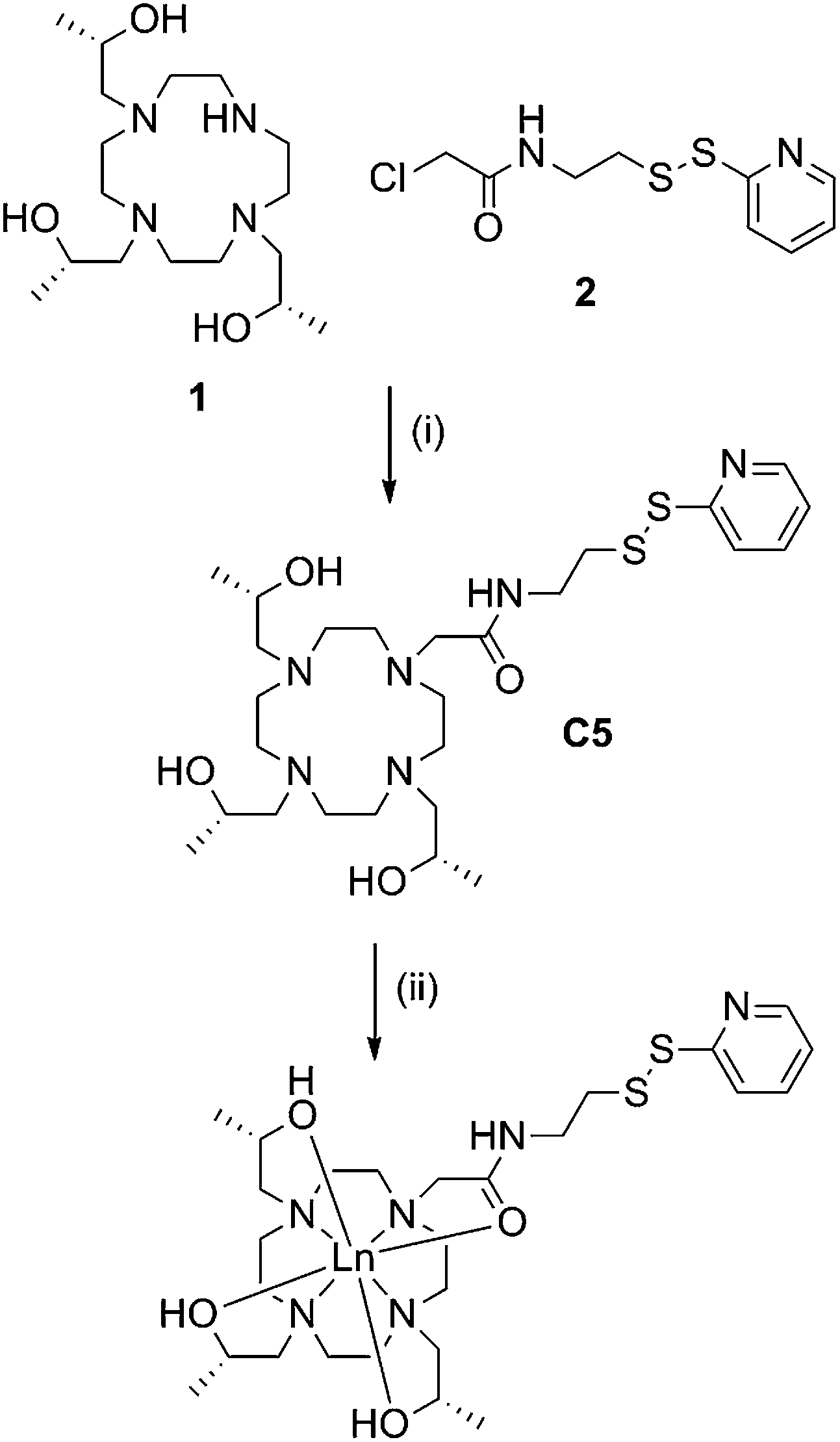 | ||
| Scheme 1 Synthesis of C5 and its lanthanide complexes. Reagents and conditions: (i) DIPEA, ACN, RT, 72 h, 61%; (ii) LnCl3, ACN, H2O, pH 7, reflux, overnight, quant. | ||
Synthesis of C6 (Scheme 2) required preparation of a novel carboxyl pyridine linker. Dimethyl 4-(hydroxymethyl)pyridine-2,6-dicarboxylate (3) was prepared following literature procedures23 and converted to the tert-butyl thioether 5via the mesylate derivative 4. Partial reduction with sodium borohydride and mesylation of the resulting hydroxyl group yielded 7, which was reacted with an excess of cyclen to form 8. Reaction with an excess of (S)-propylene oxide, followed by ester and tert-butyl deprotection yielded C6. We attempted to activate the thiol of C6 as a pyridyl disulfide, however the resulting product was unstable during purification, thus the free thiol was used for tagging (vide infra).
 | ||
| Scheme 2 Synthesis of C6 and its lanthanide complexes. Reagents and conditions: (i) MsCl, DIPEA, DCM, 0 °C, 30 min, quant; (ii) tBuSH, NaH, DMF, RT, 5 min, 47%; (iii) NaBH4, MeOH, DCM, RT, 2 h, 64%; (iv) MsCl, DIPEA, DCM, 0 °C, 30 min, 77%; (v) cyclen, CHCl3, RT, O/N, quant.; (vi) (S)-propylene oxide, MeOH, RT, 48 h, quant.; (vii) HCl (32%), reflux, 4 h, 85%; (viii) LnCl3, ACN, H2O, pH 7, reflux, overnight, quant. | ||
Metal complexes of C5 and C6 were prepared by heating the relevant tag with two equivalents of XCl3 salts (X = Y, Dy, Tb, Tm or Yb) at 80 °C in a water–acetonitrile mixture buffered at neutral pH overnight. Coordination of these tags was generally close to quantitative, with excess metal ions and uncomplexed tag removed via HPLC purification.
Due to the favourable properties of C7 (vide infra), its synthesis underwent several iterations in order to improve the yield (Scheme 3). Similarly to C5 and C6, the initial method involved synthesis of the tag, followed by metal ion complexation.
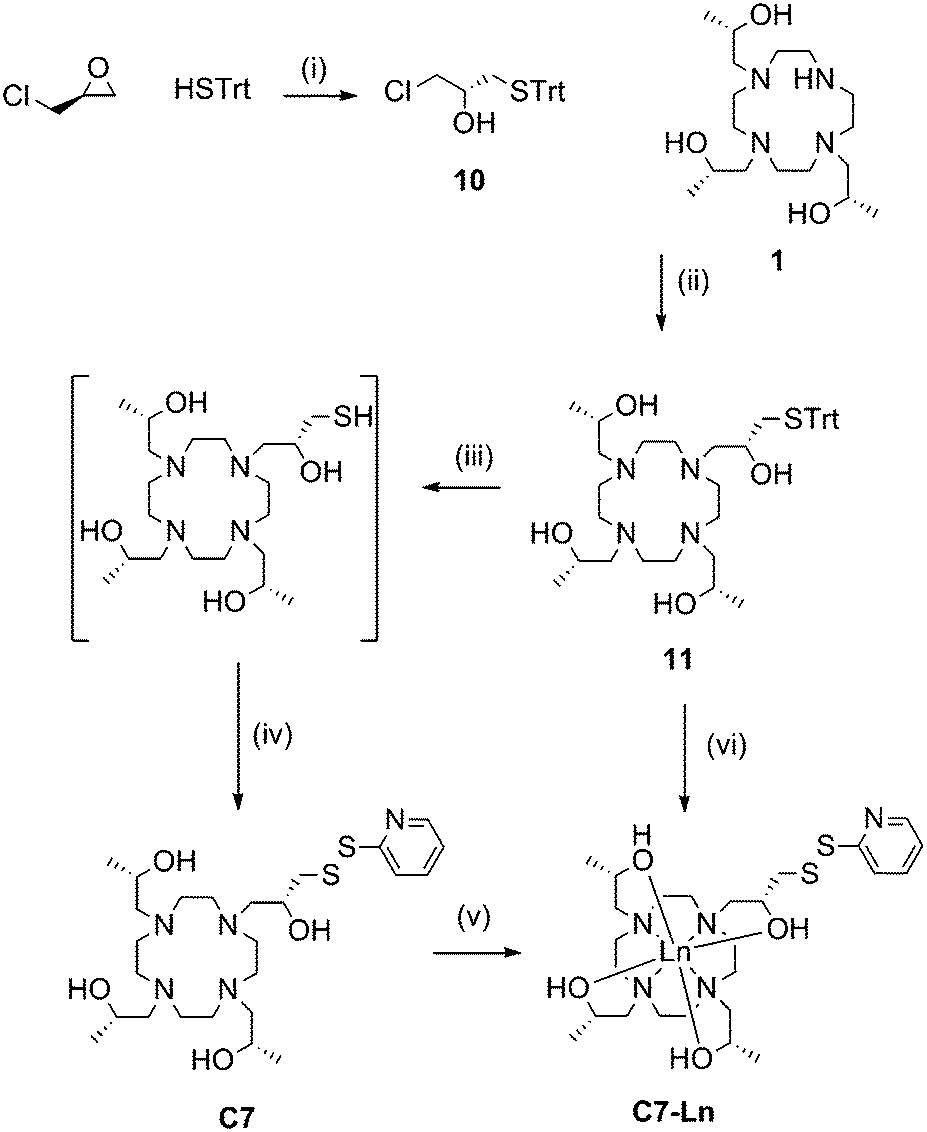 | ||
| Scheme 3 Synthesis of C7 and its lanthanide complexes. Reagents and conditions: (i) KF, MeOH, RT, 72 h, 95%; (ii) K2CO3, ACN, reflux, overnight, 60%; (iii) TFA, triethylsilane, DCM, RT, 1 h; (iv) 2,2′-dipyridyldisulfide, MeOH, RT, 15 min, 39% (from 11); (v) LnCl3, EtOH, DIPEA, reflux, overnight; (vi) LnCl3, MeOH, reflux, 4 h, 2,2′-dipyridyl disulfide, silver nitrate, RT, 2 h, 34%. | ||
Ring opening of (S)-epichlorohydrin with triphenylmethane thiol, in the presence of potassium fluoride, produced 10 in excellent yield (95%). 10 then underwent nucleophilic substitution by heating with 1 and potassium carbonate to form 11. Deprotection of the trityl group was carried out at room temperature with trifluoroacetic acid and triethylsilane. Subsequent thiol activation with 2,2′-dipyridyl disulfide and purification via HPLC produced C7 in 39% yield from 11.
Formation of C7-Ln3+ complexes was extremely slow in the presence of water and required heating in anhydrous ethanol. Complexation was still relatively slow compared to the other tags. Furthermore, if left for a prolonged period of time (e.g. greater than 48 h) noticeable amounts of disulfide rearrangement would occur, resulting in a chelate dimer and regeneration of 2,2′-dipyridyl disulfide. C7-Ln3+ complexes formed this way were thus generally purified from a mixture with uncomplexed C7, before a significant amount of disulfide rearrangement could occur, resulting in relatively poor yields.
Various attempts to optimise C7 complexation were made, including initial passage of C7 over anion exchange resin (to remove trifluoroacetic acid, present from prior HPLC purification) and addition of organic or inorganic bases to complexation reactions. However, we eventually found the most practical way of producing C7-Ln3+ complexes to be by forming metal complexes of 11, before trityl deprotection and thiol activation to the final product. Compound 11 was isolated as a neutral compound and readily formed 11-Ln3+ without side-product formation, by heating for several hours in methanol with two equivalents of the relevant metal chloride salt. 11-Ln3+ was then trityl deprotected with silver nitrate and thiol activated with 2,2′-dipyridyl disulfide, before reverse-phase HPLC purification to yield C7-Ln3+. This method allowed the formation of C7-Ln3+ complexes from 11 in “one pot” and required one less round of HPLC purification compared to the previous route, resulting in overall higher yields (34% from 11). The C8 tag and complexes followed the same procedures with the replacement of (S)-propylene oxide and (S)-epichlorohydrin with their (R)-enantiomers.
Fig. S1–S3† show the 1H NMR spectra of the Yb3+ complexes of C5–C7. Although greatly complicated by the pyridyl disulfide linker breaking the symmetry of the complex, the 1H NMR spectrum of C7-Yb3+ bears some resemblances to that of the (S)-THP-Yb3+ complex reported by Lelli et al.39 Comparing the most resolved signals, the peak at −28 ppm in (S)-THP-Yb3+ is split into four overlapping peaks of equal intensity in C7-Yb3+, while the peak at 52 ppm in (S)-THP-Yb3+ is split into three peaks in C7-Yb3+, one of which is twice the intensity of the other two. The 1H NMR spectra of the more structurally-varied C5-Yb3+ and C6-Yb3+ complexes show fewer similarities to (S)-THP-Yb3+. We did not attempt a complete assignment of the 1H NMR spectra of the C5–C7 complexes.
Testing of tags on a cysteine-bearing mutant of ubiquitin
A human ubiquitin A28C mutant was used as an initial model protein to assess the effects of the paramagnetic properties of each tag. Purified protein was first stirred with ten equivalents of DTT to reduce any oxidised cysteines. Excess DTT was removed by passage over a PD10 column equilibrated with 50 mM HEPES, pH 8.0. For the pyridyl disulfide-containing tags, C5, C7 and C8, five equivalents of the relevant lanthanide-complexed tag were added and the solutions stirred for 2 h at room temperature, before excess tag was removed by passage over a PD10 column. Tagging yields varied between 70% to quantitative, as determined by NMR analysis.In order to conjugate C6, reduced protein was first reacted with a ten-fold excess of DTNB for 1 h, before passage over a PD10 column followed by the addition of five equivalents of C6-Ln3+ complex. The reaction was allowed to stir for 2 h at room temperature, before removal of excess tag via a PD10 column. Tagging yields were generally quantitative.
15N-HSQC spectra of each lanthanide complex conjugated to ubiquitin A28C showed significant PCSs (Fig. 2 and S7–S9†). For each tag, the Y3+ complex produced minor chemical shift perturbations relative to the untagged protein, with larger shifts limited to residues in the vicinity of the tagging site. In each spectrum, only a single set of PCSs was observed. PCSs were measured as the difference in chemical shift of resonances between the paramagnetic (Dy3+, Tb3+, Tm3+ or Yb3+) and diamagnetic (Y3+) tagged samples. The Δχ-tensors were determined by fitting the measured PCSs (Tables S2 and S3†) to the first conformer of the NMR structure of ubiquitin (PDB ID 2MJB),47 both individually for each metal ion (Table 1) and simultaneously for each complex of a given tag with a common metal ion position (Table S4,†vide infra). Fig. 3 shows the correlations between measured and back-calculated PCSs for the individually derived Δχ-tensors, demonstrating their high quality, which is also reflected in the low Q-values.
 | ||
| Fig. 2 Overlays of 15N-HSQC spectra of C7 (top spectra) and C8 (bottom spectra) tagged ubiquitin A28C, loaded with either Y3+ (blue) or Tm3+ (green). The spectra were recorded at 25 °C and pH 8.0 at a 1H NMR frequency of 600 MHz. Selected PCSs are indicated with solid lines. | ||
| Tag | Ln3+ | # PCS | Δχax | Δχrh | Q | x | y | z | α | β | γ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a The axial and rhombic components of the Δχ-tensors are reported in units of 10−32 m3, and the Euler angles in degrees, using the zyz convention and unique tensor representation.53 Standard deviations (in brackets) were determined from random removal of 10% of the PCSs and recalculating the Δχ-tensor 1000 times, in some cases the z and y axes of the tensor were of similar magnitude and swapped in different fits, thus standard deviations were not determined. Quality factors (Q) were calculated as the root-mean-square deviation between the experimental and back-calculated PCSs divided by the root-mean-square of the experimental PCSs. b Metal ion coordinates (x, y, z) are reported relative to the NMR structure of ubiquitin (PDB ID 2MJB47). | |||||||||||
| C5 | Dy3+ | 39 | 8.2 | 5.3 | 0.04 | 2.908 | 2.285 | −15.138 | 141 | 88 | 71 |
| Tb3+ | 47 | 9.4 (0.5) | 2.2 (0.1) | 0.06 | 2.308 | −0.421 | −17.179 | 157 | 95 | 114 | |
| Tm3+ | 47 | −18.7 (1.7) | −6.9 (0.4) | 0.06 | 4.728 | −3.051 | −17.815 | 127 | 96 | 100 | |
| Yb3+ | 61 | −6.7 (0.4) | −2.1 (0.2) | 0.08 | 0.857 | −2.115 | −18.247 | 120 | 97 | 122 | |
| C6 | Dy3+ | 49 | −9.4 | −5.9 | 0.07 | 8.694 | 3.797 | −11.227 | 45 | 49 | 80 |
| Tb3+ | 47 | −14.6 (0.4) | −3.6 (0.1) | 0.04 | 7.019 | 2.304 | −13.622 | 44 | 68 | 97 | |
| Tm3+ | 51 | 11.5 (0.7) | 4.1 (0.4) | 0.10 | 7.018 | 3.097 | −12.773 | 41 | 71 | 130 | |
| Yb3+ | 51 | 2.0 (0.1) | 0.9 (0.0) | 0.08 | 9.178 | 2.136 | −12.736 | 42 | 60 | 103 | |
| C7 | Dy3+ | 35 | 26.6 (1.1) | 6.0 (0.5) | 0.03 | −0.734 | −3.238 | −13.305 | 71 | 29 | 32 |
| Tb3+ | 40 | 11.7 (0.3) | 1.7 (0.3) | 0.04 | −1.901 | −3.712 | −14.371 | 47 | 44 | 59 | |
| Tm3+ | 44 | −19.4 (0.7) | −7.8 (0.9) | 0.03 | −4.314 | −1.357 | −13.717 | 9 | 62 | 104 | |
| Yb3+ | 51 | 5.8 | 3.0 | 0.03 | −0.427 | −0.482 | −14.129 | 16 | 110 | 85 | |
| C8 | Dy3+ | 28 | 31.2 (0.7) | 7.1 (0.6) | 0.02 | 1.810 | −3.922 | −13.760 | 91 | 44 | 5 |
| Tb3+ | 37 | 14.3 (0.6) | 5.1 (0.7) | 0.05 | 2.003 | −1.928 | −13.959 | 73 | 38 | 19 | |
| Tm3+ | 43 | −16.3 | −10.2 | 0.04 | 0.862 | −3.716 | −15.075 | 67 | 26 | 46 | |
| Yb3+ | 46 | −4.2 (0.1) | −1.9 (0.2) | 0.04 | 1.679 | −3.184 | −14.267 | 140 | 22 | 169 | |
 | ||
| Fig. 3 Correlations between experimental and back-calculated PCSs for C5–C8 bound to ubiquitin A28C loaded with either Dy3+ (magenta), Tb3+ (blue), Tm3+ (green) or Yb3+ (red). Solid lines represent perfect correlation. | ||
Different pKa values for the deprotonation of a single alcohol pendant (or, possibly, bound water molecule) have been reported for different (S)-THP-Ln3+ complexes. These range from 8.4 for the lanthanum complex to 6.4 for lutetium, with a trend of decreasing pKa across the lanthanide series.39,48 Thus, at a given pH, different (S)-THP-Ln3+ complexes can exist across a range of equilibria between +3 and +2 charged states.
To investigate a potential change in the properties of different (S)-THP-Ln3+ tags with pH, we re-recorded the spectra of the C7-Ln3+-tagged ubiquitin samples at pH 6.5 (Fig. S10 and S11†). Most notably, the PCSs of the Dy3+-tagged sample were much smaller at pH 6.5 compared to pH 8 (slope 0.28, R2 0.55). The majority of the Tb3+ PCSs were also reduced at lower pH, though to a lesser degree than for Dy3+ (slope 0.47, R2 0.62). In contrast, the size of the PCSs observed in the Tm3+ (slope 0.95, R2 0.98) and Yb3+ (slope 0.94, R2 0.99) samples was not significantly affected by the change in pH. The pH-dependence of the PCSs induced by each C7-Ln3+ complex likely reflects protonation/deprotonation processes involving the pendants arms and/or aquo ligands, leading to changes in the average charge and coordination geometry of each complex, and thus potentially their interactions with the protein surface and resulting metal ion positions. At both pHs, each C7-Ln3+ complex produced a single PCS for each affected nucleus, indicating that any processes such as protonation/deprotonation (and their effect on coordination geometry) are fast on the NMR timescale, thus the spectra are straightforward to interpret. The Tm3+ and Yb3+ complexes are likely to prove of most practical use over a wider, biologically relevant pH range.
For the data recorded at pH 8, we determined Δχ-tensors for lanthanide ions both individually, allowing independent metal ion positions (Table 1), and simultaneously with a common metal ion position for complexes of a given tag (Table S4†). In some cases there were significant differences between the individually and simultaneously determined Δχ-tensors, with the Δχax component varying by up to 49% for the most extreme example of the C5-Dy3+ complex. Despite this, the Q-values of Δχ-tensors determined from either method were very respectable (0.02–0.12), demonstrating that the tensors from either approach are suitable for structural investigations. Individually derived Δχ-tensors produced Q-values that were universally lower than those of the simultaneously calculated tensors; however, the individually determined metal ion positions were up to 6.2 Å apart for different complexes of the same tag. This observation of different metal ion positions in individual Δχ-tensor fits has been noted previously24,49 and in those cases was attributed to the uncertainty in determining the metal ion position during the fitting procedure, which can also depend on the coverage and distribution of the PCSs over the tensor “space”. Thus, a common metal ion position that satisfactorily describes the PCSs of each metal ion is often used to increase stability of the metal ion coordinate and tensor components during the fitting. In this case, the apparent different sensitivities of each metal complex to pH (influencing their average charge, coordination geometry and possible interactions with the protein surface) could be seen as justification for the use of individually determined metal ion positions and Δχ-tensors. It is worth noting again that mobility of the metal ion, for instance due to flexibility of the tag linker, results in averaging of PCSs. By fitting a single tensor to these averaged values, we are describing an “effective Δχ-tensor”. The metal ion coordinate associated with this tensor should not be interpreted as a definitive point at which the metal ion is statically located.41 Unless specified otherwise, figures and values presented herein were derived using Δχ-tensors corresponding to individual metal ion positions.
Comparison of performance of the new tags with C1
With few exceptions, the lanthanide complexes of the new tags produced Δχ-tensors with Δχax components of similar or greater magnitude to those of the corresponding C1 tag conjugated to the same ubiquitin mutant.28 This is a particularly interesting and non-intuitive observation in the case of C5, as it suggests that any increase in mobility of the tag, due to the loss of the bulky phenyl amide pendants of C1, is compensated for by the altered coordination environment and ligand field associated with the alcohol pendants and/or changes in secondary interactions with the protein, allowing C5 to generate sizeable paramagnetic effects.In contrast to the case for C1, for which each lanthanide complex reliably produces PCSs of a predictable relative size and sign for a given nuclear spin (e.g. Tm3+ and Tb3+ PCSs are generally opposite in sign, with Tb3+ PCSs slightly larger in size), the relative order and size of PCSs induced by the new tags loaded with different lanthanide ions was quite variable (Fig. S12†). Correspondingly, the determined Euler angles of the Δχ-tensors from metal complexes of the same tag also varied to a larger extent than those observed for C1 (Fig. S13†), suggesting changes in coordination environment with each lanthanide ion, as alluded to above. The noted change in the orientation of the Δχ-tensor for each metal complex of the same tag is potentially a useful property, which can help resolve the redundant solutions that can be encountered in studies using PCSs (associated with the symmetry of the Δχ-tensor), without requiring multiple tagging sites or tags.50
Initially, only the (S)-enantiomer of each tag was synthesised and assessed. However, given the large Δχ-tensors and excellent fits observed for C7, its enantiomer C8 and the corresponding C8-Ln3+ complexes were also synthesised and conjugated to ubiquitin A28C. Despite the same coordination environment of the lanthanide ions in complexes of either tag enantiomer, different PCSs, Δχ-tensors and metal ion positions (Fig. 2, 3, S8 and S12; Tables 1, S3 and S4†) were observed, likely due to the differences in their interaction with the chiral protein surface, arising from the opposite stereochemistry of the pendant arms. On average, the Δχax components of each complex of the C7 and C8 tags were larger than those of the C1, C5 or C6 tags on ubiquitin A28C, suggesting that the very short linker is key to the tags' superior paramagnetic effects. A temperature titration of the C7-Tm3+ tagged sample showed no signs of additional cross-peaks due to conformational exchange (Fig. S14†), although at higher temperatures the observed PCSs were smaller, presumably due to increased tag mobility.
In order to investigate the rigidity of the C7 and C8 tags and their ability to induce partial alignment in the magnetic field, one bond 1H–15N RDCs (1DHN) of the Tm3+ complexes of C7 and C8 were measured relative to the Y3+-tagged protein. 1DHN RDCs up to 12.5 and 6.1 Hz were observed at 600 MHz for C7 and C8 respectively. Alignment tensors were determined by fitting the measured RDCs (Table S5†) to a structure of ubiquitin using single value decomposition within PALES51 (Table S6†). The measured and calculated 1DHN RDCs (Fig. 4A and B) are in good agreement and the principal axes of the alignment (Fig. 4C and D) and Δχ (Fig. 4E and F) tensors are very similar, demonstrating that the orientation of the tensors are relatively well defined for either enantiomer. The Δχ-tensor components derived from the alignment tensor parameters match very favourably with the PCS derived Δχ-tensor values for C7-Tm3+ (Tables 1 and S6; eqn S1†). However, for C8-Tm3+, the alignment tensor predicted Δχax and Δχrh are 62% and 54% of their respective PCS determined values, suggesting some degree of mobility is still present. It is not uncommon for alignment tensors to be smaller than Δχ-tensors.27,49,52 This is partly attributed to the greater sensitivity of RDCs to protein and tag movements than PCSs. The Q-factors of the alignment tensors are larger than those of the Δχ-tensors. Due to the chiral nature of the tags, C7 may be engaged in different secondary interactions with the protein, helping to limit its mobility to a greater degree than C8.
 | ||
| Fig. 4 Correlations between experimental and calculated 1DHN RDCs recorded at a 1H NMR frequency of 600 MHz for C7-Tm3+ (A) and C8-Tm3+ (B) tagged ubiquitin A28C. Solid lines represent perfect correlation. Orientations of the principal axes of the alignment (C C7, D C8) and Δχ (E C7, F C8) tensors. The points show where the principal axes of the tensors penetrate the sphere with the axes coloured as follows: z (blue), y (green), x (red). For the alignment tensors, 1000 replicates of SVD calculation using the structural noise Monte-Carlo method (‘-mcStruc’) within PALES are shown. For the Δχ-tensors, 1000 replicates with a random 10% of the PCS data removed each time are shown. The convention |z| > |y| > |x| is used to name the axes, resulting in swapping of the |z| and |y| axes in different fits when their magnitudes are similar. | ||
Further testing of C7 and C8 on a cysteine-bearing mutant of GB1 and HPPK
To demonstrate the general utility of the C7 and C8 tags, lanthanide complexes of both tags were conjugated to a GB1 Q32C mutant. Each sample produced a single set of PCSs from which Δχ-tensors were determined (Fig. S15 and S16; Tables 2, S7 and S8†). Differences between individual and simultaneously derived Δχ-tensors were apparent, though to a lesser extent than observed for ubiquitin. Both Tb3+ complexes, but particularly the C7 complex, resulted in only small PCSs and significantly smaller Δχ-tensors on GB1 compared to ubiquitin. Thus, the GB1 spectra, which were recorded at pH 6.5, seem consistent with the ubiquitin spectra recorded at pH 6.5, in that they suggest that the Tb3+ (and likely Dy3+) complexes are of less practical use at a lower pH. Conversely, both Tm3+ complexes resulted in sizable PCSs and Δχ-tensors. The Δχax components of C7-Tm3+ and C8-Tm3+ on GB1 Q32C are 73% and 91% of their respective values on ubiquitin A28C, demonstrating the influence of the tagging site and protein environment on the tags' performance.| Protein | Tag | Ln3+ | # PCS | Δχax | Δχrh | Q | x | y | z | α | β | γ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a See footnote a in Table 1. b Metal ion coordinates (x, y, z) for each tag are relative to the crystal structures of GB1 (PDB ID 1PGA)54 or HPPK (PDB ID 3QBC).45 | ||||||||||||
| GB1 | C7 | Tb3+ | 47 | 2.2 (1.1) | 1.0 (0.7) | 0.09 | 29.244 | 29.993 | 13.297 | 18 | 39 | 60 |
| Tm3+ | 37 | −14.2 (0.9) | −4.4 (0.5) | 0.06 | 31.745 | 29.577 | 12.618 | 145 | 56 | 80 | ||
| C8 | Tb3+ | 40 | 6.1 (0.4) | 3.1 (0.3) | 0.05 | 33.668 | 30.260 | 14.419 | 155 | 41 | 175 | |
| Tm3+ | 40 | −14.9 (0.4) | −6.4 (0.6) | 0.06 | 34.227 | 32.261 | 17.019 | 172 | 73 | 173 | ||
| HPPK | C7 | Tm3+ | 81 | 54.5 (0.5) | 12.5 (0.5) | 0.04 | 14.304 | 13.802 | 13.906 | 149 | 55 | 127 |
This variability was further observed in our investigations of the 20 kDa-sized protein HPPK (to be fully reported elsewhere). Tagging at different sites produced Δχ-tensors with varied Δχax components, up to 54.5 × 10−32 m3 for a HPPK S112C/C80A mutant tagged with C7-Tm3+ (Fig. S16 and S17; Tables 2 and S9†). Given that the PCS and RDC data for C7-Tm3+ tagged ubiquitin A28C had previously indicated that the chelate was relatively rigid on ubiquitin, such an increase in the Δχax component for HPPK S112C/C80A was highly unexpected. Spectra of both proteins were recorded at pH 8, thus different deprotonation/protonation equilibria based on solvent water alone are insufficient to explain such variance. However, different interactions with the protein surface could also affect the charged state of the tag. For this particular HPPK mutant, the cysteine for tagging was introduced on the β-sheet of a short β-hairpin, which features an aspartic acid (D107) on the adjacent β-sheet. The calculated metal ion position is above and between D107 and S112, which both point in the same direction in the HPPK crystal structure (Fig. S18†). The carboxyl group of D107 could conceivably be interacting with either the hydroxyl pendants of the tag or directly with the lanthanide ion to influence the charge of the chelate and its paramagnetic properties. In addition, HPPK samples tagged with C7 and C8 appeared more stable to precipitation than those tagged with C1 or C2, allowing the acquisition of multiple NMR experiments of each sample.
Experimental
Materials and methods
(1S,4S,7S)-1,4,7-Tris(2-hydroxypropyl)-1,4,7,10-tetraazacyclododecane,46 2-chloro-N-(2-(pyridin-2-yldisulfanyl)ethyl)acetamide28 and dimethyl 4-(hydroxymethyl)pyridine-2,6-dicarboxylate23 were prepared following literature procedures. The synthesis of C5 and C6 is described in the ESI.†Synthetic procedures
Formation of lanthanide complexes
Complexes of C5 and C6 were prepared by refluxing the ligands for 18 h with 2 equivalents of Y3+, Dy3+, Tb3+, Tm3+ or Yb3+-trichloride salts in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ACN:H2O solution adjusted to neutral pH, followed by purification by HPLC (0.1% TFA and a 0–80% ACN gradient on a C18 preparative column) to afford the complexes as off-white solids after lyophilisation. In the case of C6, TCEP was added prior to purification to prevent disulfide formation.
:1 ACN:H2O solution adjusted to neutral pH, followed by purification by HPLC (0.1% TFA and a 0–80% ACN gradient on a C18 preparative column) to afford the complexes as off-white solids after lyophilisation. In the case of C6, TCEP was added prior to purification to prevent disulfide formation.
Complexes of C7 and C8 were most readily prepared from 11. An example of the formation of the C7-Yb3+ complex follows. A solution of 11 (30 mg, 0.044 mmol) and YbCl3 (19 mg, 0.066 mmol) in MeOH (1.5 mL) was heated at 50 °C for 4 h, after which LCMS analysis indicated complete complexation. The solution was cooled to room temperature, then 2,2′-dipyridyldisulfide (29 mg, 0.13 mmol) and silver nitrate (37 mg, 0.22 mmol) added whilst vigorously stirring, forming a milky beige mixture, before formation of a beige precipitate that eventually turned grey. After 2 h, LCMS analysis indicated complete trityl deprotection and thiol activation, and the mixture was concentrated under reduced pressure. 0.1% TFA in H2O (5 mL) and DCM (5 mL) were added to the grey residue and the suspension transferred to a 15 mL centrifuge tube. The suspension was shaken vigorously and the precipitate sedimented and organic and aqueous phases separated, by centrifugation for 3 min at 2000 rcf. The aqueous phase was carefully removed and purified by reverse-phase HPLC (0.1% TFA and a 5–100% ACN gradient over 30 min on a C18 preparative column). Fractions containing pure product were lyophilised to yield the trifluoroacetate salt of C7-Yb3+ as an off-white solid. Yield: 17 mg (34%, assuming a tetratrifluoroacetate salt).
Working stock solutions of each metal complex were prepared at 20 mM in H2O and stored frozen at −20 °C when not in use.
1H NMR spectra and HRMS of the Yb3+ complexes of C5–C7, are shown in Fig. S1–S6.†
NMR sample preparation
Uniformly 15N-labelled human ubiquitin A28C was prepared as described.24 Prior to tagging the protein was first reduced by stirring with a 10-fold excess of DTT for 1 h at room temperature, before passage over a PD-10 column equilibrated with degassed buffer (50 mM HEPES, pH 8).For the C5 and C7/8 tags a 5-fold excess of the respective lanthanide complex was added to a solution of protein and stirred at room temperature for 2 h. Excess tag was removed by passage over a PD-10 column before the sample was concentrated using a Millipore ultrafilter (3 kDa) to a final protein concentration of approximately 100 μM.
In order to tag C6, the protein cysteines were first pre-activated by addition of 10 equivalents of 5,5′-dithiobis-2-nitrobenzoic acid (DTNB), producing a yellow coloured solution that was allowed to stir at room temperature for 1 h. Excess DTNB and TNB2− leaving group were removed by passage through a PD10 column, yielding a colourless solution. A 5-fold excess of the respective C6 complex was then added, forming a yellow solution that was stirred at room temperature for 2 h. Excess tag and TNB2− leaving group was removed by passage over a PD-10 column and samples were concentrated as above.
NMR spectroscopy
Spectra of differently tagged ubiquitin A28C in 90%/10% H2O/D2O, 50 mM HEPES, pH 8.0, were recorded at 25 °C on either Varian INOVA or Bruker Avance 600 MHz NMR spectrometers equipped with cryogenic probes. 1HN PCSs and 1DHN couplings were measured by recording 15N-fast-HSQC spectra with and without the 180° (1H) pulse during the 15N (t1) evolution time. 2D 15N-fast-HSQC were typically acquired with t1max (15N) = 51–62 ms and t2max (1H) = 142 ms.Calculation of Δχ and alignment tensors
Fitting of Δχ-tensors was carried out within the program Numbat.53 The tensors were fitted to the first conformer of the NMR structure of ubiquitin (PDB 2MJB47). Unambiguous PCS assignments were used to calculate an initial estimate of the Δχ-tensor, from which iterative cycles of further assignment and recalculation were made. The Δχ-tensors for GB1 and HPPK were determined in an analogous way, fitting to the crystal structures of GB1 (PDB 1PGA54) and HPPK (PDB 3QBC45).Backbone amide 1DHN RDCs were fitted to the first conformer of the NMR structure of ubiquitin (PDB 2MJB47) using single value decomposition via the “-bestFit” flag in PALES.51
Conclusions
We have presented the synthesis of three new LBT designs. Each tag is capable of binding lanthanide ions tightly and producing significant PCSs without need for the addition of free paramagnetic metal ions to protein samples. Each design features hydroxypropyl pendant arms, rendering the tags smaller and more hydrophilic than previously reported DOTA-style tags. The C5 tag can be readily synthesised and displayed comparable paramagnetic effects to C1, whose utility has been proven in several studies.55–57 The C6 tag also performed comparably with C1 on ubiquitin, however it features the longest synthesis of any of the tags and requires more protein handling via DTNB activation for conjugation. The C7/8 design features a particularly short linker, resulting in limited mobility relative to the protein surface, hence generating the largest paramagnetic effects on ubiquitin. The capability of C7 and C8 to produce paramagnetic effects on other proteins was further demonstrated on GB1 and HPPK. The performance of the tags varied with factors including the lanthanide used, pH and site of conjugation. Given their favourable properties, it is anticipated that C7 and C8 (particularly their Tm3+ complexes) will prove useful in the investigation of a wide range of biologically interesting proteins by paramagnetic NMR spectroscopy.Acknowledgements
An Australian Research Council Future Fellowship to BG (FT130100838) and Discovery Grants to GO and BG (DP120100561 and DP150100383) are gratefully acknowledged.Notes and references
- G. Otting, Annu. Rev. Biophys., 2010, 39, 387–405 CrossRef CAS PubMed.
- J. Koehler and J. Meiler, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 59, 360–389 CrossRef CAS PubMed.
- W.-M. Liu, M. Overhand and M. Ubbink, Coord. Chem. Rev., 2014, 273–274, 2–12 CrossRef CAS PubMed.
- M. Allegrozzi, I. Bertini, M. B. L. Janik, Y. M. Lee, G. Liu and C. Luchinat, J. Am. Chem. Soc., 2000, 122, 4154–4161 CrossRef CAS.
- L. Lee and B. D. Sykes, Biophys. J., 1980, 32, 193–210 CrossRef CAS.
- I. Baig, I. Bertini, C. Del Bianco, Y. K. Gupta, Y.-M. Lee, C. Luchinat and A. Quattrone, Biochemistry, 2004, 43, 5562–5573 CrossRef CAS PubMed.
- I. Bertini, Y. K. Gupta, C. Luchinat, G. Parigi, M. Peana, L. Sgheri and J. Yuan, J. Am. Chem. Soc., 2007, 129, 12786–12794 CrossRef CAS PubMed.
- S. Dasgupta, X. Hu, P. H. J. Keizers, W.-M. Liu, C. Luchinat, M. Nagulapalli, M. Overhand, G. Parigi, L. Sgheri and M. Ubbink, J. Biomol. NMR, 2011, 51, 253–263 CrossRef CAS PubMed.
- C. Luchinat, M. Nagulapalli, G. Parigi and L. Sgheri, J. Magn. Reson., 2012, 215, 85–93 CrossRef CAS PubMed.
- G. Pintacuda, A. Y. Park, M. A. Keniry, N. E. Dixon and G. Otting, J. Am. Chem. Soc., 2006, 128, 3696–3702 CrossRef CAS PubMed.
- T. Saio, M. Yokochi, H. Kumeta and F. Inagaki, J. Biomol. NMR, 2010, 46, 271–280 CrossRef CAS PubMed.
- P. H. J. Keizers, B. Mersinli, W. Reinle, J. Donauer, Y. Hiruma, F. Hannemann, M. Overhand, R. Bernhardt and M. Ubbink, Biochemistry, 2010, 49, 6846–6855 CrossRef CAS PubMed.
- Y. Hiruma, M. A. S. Hass, Y. Kikui, W. M. Liu, B. Olmez, S. P. Skinner, A. Blok, A. Kloosterman, H. Koteishi, F. Löhr, H. Schwalbe, M. Nojiri and M. Ubbink, J. Mol. Biol., 2013, 425, 4353–4365 CrossRef CAS PubMed.
- M. John, G. Pintacuda, A. Y. Park, N. E. Dixon and G. Otting, J. Am. Chem. Soc., 2006, 128, 12910–12916 CrossRef CAS PubMed.
- T. Zhuang, H. Lee, B. Imperiali and J. H. Prestegard, Protein Sci., 2008, 17, 1220–1231 CrossRef CAS PubMed.
- T. Saio, K. Ogura, K. Shimizu, M. Yokochi, T. R. Burke and F. Inagaki, J. Biomol. NMR, 2011, 51, 395–408 CrossRef CAS PubMed.
- J.-Y. Guan, P. H. J. Keizers, W.-M. Liu, F. Loehr, S. P. Skinner, E. A. Heeneman, H. Schwalbe, M. Ubbink and G. D. Siegal, J. Am. Chem. Soc., 2013, 135, 5859–5868 CrossRef CAS PubMed.
- C. Schmitz, R. Vernon, G. Otting, D. Baker and T. Huber, J. Mol. Biol., 2012, 416, 668–677 CrossRef CAS PubMed.
- H. Yagi, K. B. Pilla, A. Maleckis, B. Graham, T. Huber and G. Otting, Structure, 2013, 21, 883–890 CrossRef CAS PubMed.
- X.-C. Su and G. Otting, J. Biomol. NMR, 2010, 46, 101–112 CrossRef CAS PubMed.
- X.-C. Su, K. McAndrew, T. Huber and G. Otting, J. Am. Chem. Soc., 2008, 130, 1681–1687 CrossRef CAS PubMed.
- K. Barthelmes, A. M. Reynolds, E. Peisach, H. R. A. Jonker, N. J. DeNunzio, K. N. Allen, B. Imperiali and H. Schwalbe, J. Am. Chem. Soc., 2011, 133, 808–809 CrossRef CAS PubMed.
- X.-C. Su, B. Man, S. Beeren, H. Liang, S. Simonsen, C. Schmitz, T. Huber, B. A. Messerle and G. Otting, J. Am. Chem. Soc., 2008, 130, 10486–10487 CrossRef CAS PubMed.
- J. D. Swarbrick, P. Ung, S. Chhabra and B. Graham, Angew. Chem., Int. Ed., 2011, 50, 4403–4406 CrossRef CAS PubMed.
- H. Yagi, A. Maleckis and G. Otting, J. Biomol. NMR, 2013, 55, 157–166 CrossRef CAS PubMed.
- P. H. J. Keizers, J. F. Desreux, M. Overhand and M. Ubbink, J. Am. Chem. Soc., 2007, 129, 9292–9293 CrossRef CAS PubMed.
- T. Saio, K. Ogura, M. Yokochi, Y. Kobashigawa and F. Inagaki, J. Biomol. NMR, 2009, 44, 157–166 CrossRef CAS PubMed.
- B. Graham, C. T. Loh, J. D. Swarbrick, P. Ung, J. Shin, H. Yagi, X. Jia, S. Chhabra, N. Barlow, G. Pintacuda, T. Huber and G. Otting, Bioconjugate Chem., 2011, 22, 2118–2125 CrossRef CAS PubMed.
- T. Ikegami, L. Verdier, P. Sakhaii, S. Grimme, B. Pescatore, K. Saxena, K. M. Fiebig and C. Griesinger, J. Biomol. NMR, 2004, 29, 339–349 CrossRef CAS.
- G. Pintacuda, A. Moshref, A. Leonchiks, A. Sharipo and G. Otting, J. Biomol. NMR, 2004, 29, 351–361 CrossRef CAS.
- M. D. Vlasie, C. Comuzzi, A. M. C. H. van den Nieuwendijk, M. Prudêncio, M. Overhand and M. Ubbink, Chem. – Eur. J., 2007, 13, 1715–1723 CrossRef CAS PubMed.
- P. H. J. Keizers, A. Saragliadis, Y. Hiruma, M. Overhand and M. Ubbink, J. Am. Chem. Soc., 2008, 130, 14802–14812 CrossRef CAS PubMed.
- D. Häussinger, J. Huang and S. Grzesiek, J. Am. Chem. Soc., 2009, 131, 14761–14767 CrossRef PubMed.
- W. P. Cacheris, S. K. Nickle and A. D. Sherry, Inorg. Chem., 1987, 26, 958–960 CrossRef CAS.
- S. Aime, M. Botta, M. Fasano, M. P. M. Marques, C. F. G. C. Geraldes, D. Pubanz and E. Merbach, Inorg. Chem., 1997, 1669, 2059–2068 CrossRef PubMed.
- W.-M. Liu, S. P. Skinner, M. Timmer, A. Blok, M. A. S. Hass, D. V. Filippov, M. Overhand and M. Ubbink, Chem. – Eur. J., 2014, 20, 6256–6528 CrossRef CAS PubMed.
- C. T. Loh, K. Ozawa, K. L. Tuck, N. Barlow, T. Huber, G. Otting and B. Graham, Bioconjugate Chem., 2013, 24, 260–268 CrossRef CAS PubMed.
- K. A. Chin, J. R. Morrow, C. H. Lake and M. R. Churchill, Inorg. Chem., 1994, 33, 656–664 CrossRef CAS.
- M. Lelli, G. Pintacuda, A. Cuzzola and L. Di Bari, Chirality, 2005, 17, 201–211 CrossRef CAS PubMed.
- C.-H. Huang and J. R. Morrow, Inorg. Chem., 2009, 48, 7237–7243 CrossRef CAS PubMed.
- D. Shishmarev and G. Otting, J. Biomol. NMR, 2013, 56, 203–216 CrossRef CAS PubMed.
- B. Man, X.-C. Su, H. Liang, S. Simonsen, T. Huber, B. A. Messerle and G. Otting, Chem. – Eur. J., 2010, 16, 3827–3832 CrossRef CAS PubMed.
- X. Jia, A. Maleckis, T. Huber and G. Otting, Chem. – Eur. J., 2011, 17, 6830–6836 CrossRef CAS PubMed.
- Q.-F. Li, Y. Yang, A. Maleckis, G. Otting and X.-C. Su, Chem. Commun., 2012, 48, 2704–2706 RSC.
- S. Chhabra, O. Dolezal, B. M. Collins, J. Newman, J. S. Simpson, I. G. Macreadie, R. Fernley, T. S. Peat and J. D. Swarbrick, PLoS One, 2012, 7, e29444 CAS.
- J. Hammell, L. Buttarazzi, C.-H. Huang and J. R. Morrow, Inorg. Chem., 2011, 50, 4857–4867 CrossRef CAS PubMed.
- A. S. Maltsev, A. Grishaev, J. Roche, M. Zasloff and A. Bax, J. Am. Chem. Soc., 2014, 136, 3752–3755 CrossRef CAS PubMed.
- K. Chin and J. Morrow, Inorg. Chem., 1994, 33, 5036–5041 CrossRef CAS.
- F. Peters, M. Maestre-Martinez, A. Leonov, L. Kovačič, S. Becker, R. Boelens and C. Griesinger, J. Biomol. NMR, 2011, 51, 329–337 CrossRef CAS PubMed.
- Y. Kobashigawa, T. Saio, M. Ushio, M. Sekiguchi, M. Yokochi, K. Ogura and F. Inagaki, J. Biomol. NMR, 2012, 53, 53–63 CrossRef CAS PubMed.
- M. Zweckstetter, Nat. Protoc., 2008, 3, 679–690 CrossRef CAS PubMed.
- Y. Yang, J. Wang, Y. Pei and X. Su, Chem. Commun., 2015, 51, 2824–2827 RSC.
- C. Schmitz, M. J. Stanton-Cook, X.-C. Su, G. Otting and T. Huber, J. Biomol. NMR, 2008, 41, 179–189 CrossRef CAS PubMed.
- T. Gallagher, P. Alexander, P. Bryan and G. Gilliland, Biochemistry, 1994, 33, 4721–4729 CrossRef CAS.
- L. de la Cruz, T. H. D. Nguyen, K. Ozawa, J. Shin, B. Graham, T. Huber and G. Otting, J. Am. Chem. Soc., 2011, 133, 19205–19215 CrossRef CAS PubMed.
- H. Yagi, D. Banerjee, B. Graham, T. Huber, D. Goldfarb and G. Otting, J. Am. Chem. Soc., 2011, 133, 10418–10421 CrossRef CAS PubMed.
- K. Jaudzems, X. Jia, H. Yagi, D. Zhulenkovs, B. Graham, G. Otting and E. Liepinsh, J. Mol. Biol., 2012, 424, 42–53 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses of C5 and C6, 1H NMR spectra and HRMS of Yb3+ complexes of C5–C7, experimental details for expression, purification and tagging of GB1 Q32C and HPPK S112C/C80A, 15N-HSQC spectra of differently tagged ubiquitin A28C, GB1 Q32C and HPPK S112C/C80A, correlation between PCSs of C7 tagged ubiquitin A28C at pH 8 and pH 6.5, comparison of measured PCSs against residue number for C5–C8 tagged ubiquitin A28C, isosurface representations of Δχ-tensors of C1, C7 and C8 tagged ubiquitin A28C, simultaneously calculated Δχ-tensor properties for tagged ubiquitin A28C and GB1 Q32C, experimental RDCs and alignment tensor properties for C7-Tm3+ and C8-Tm3+ tagged ubiquitin A28C, experimental PCSs of tagged ubiquitin A28C, GB1 Q32C and HPPK S112C/C80A, 1D 1H and 13C NMR spectra of novel small molecule compounds, analytical HPLC traces of C5–C7. See DOI: 10.1039/c4sc03892d |
| This journal is © The Royal Society of Chemistry 2015 |