
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adaptive binding and selection of compressed 1,ω-diammonium-alkanes via molecular encapsulation in water†
Dan
Dumitrescu‡
,
Yves-Marie
Legrand
,
Eddy
Petit
,
Arie
van der Lee
and
Mihail
Barboiu
*
Adaptive Supramolecular Nanosystems Group, Institut Européen des Membranes, ENSCM/UMII/UMR-CNRS 5635, Pl. Eugène Bataillon, CC 047, 34095 Montpellier, Cedex 5, France. E-mail: mihail-dumitru.barboiu@univ-montp2.fr
First published on 12th January 2015
Abstract
Guest molecules confined inside hollow molecular assemblies and thus protected from their environment can show unexpected structural behavior or special reactivity compared to their behavior in a bulk, unprotected environment. A special case is the coiling behavior of variable-length alkane chains in rigid hydrogen-bonded molecular cages. It has been found before that coiling may occur in such circumstances, but no experimental evidence concerning the exact conformation of the chains has yet been presented. We reveal in this study the self-assembly of a molecular cage in water and the crystalline state from three distinct components in which linear 1,ω-diammonium-alkanes chains are confined with different degrees of compression. The exact coiling behavior is determined from atomic resolution X-ray diffraction showing crenel-like conformations in the compressed state. Chemical selection can be obtained from mixtures of alkane chains via the encapsulation of kinetically stable conformations observed during the encapsulation of pure components. Moreover, it was found that uncompressed and compressed chains can be competitively trapped inside the capsule. These findings may provide insight in areas to a better understanding of biological processes, such as the fatty acid metabolism.
Introduction
Encapsulation chemistry is a fascinating domain, as the properties of the guests are generally different inside host capsules compared with the bulk solution. Crossing the solution/capsule barrier, unexpected phenomena can be observed within a “compartmentalized” chemical space, opening the door for new emergent areas of chemistry and physics under confined conditions.1–6 Compartmentalization is also a basic feature of biological processes, as most of the physiological processes occur in cells and depend on selective exchanges of metabolites between the cell and its exterior.7One of the intriguing phenomena observed to date, concerns the compression of n-alkanes inside self-assembled H-bonded capsules in water. Attempts to encapsulate alkane molecules in confined spaces have generally furnished very interesting details on their constitutional flexibility, inducing synergetic dynamic shape changes of the capsule/host system.8–11 Internal order can in some cases be reinforced by synergetic supplementary strong interactions between guests and host capsules.11 Stabilization of large alkanes, adapting their conformation in smallest capsules, has been demonstrated to shed light evidence of constrained conformations and give answers on how alkane compression occurs.8 Dynamic selection of congruent alkyl-ester hosts can be adaptively obtained on guest encapsulation.9
We recently reported the exact behaviors of compressed alkanes at the molecular level constrained within crystalline molecular capsules. They were determined from atomic resolution X-ray diffraction and confirmed the previous molecular modeling as well as the spectroscopic NMR studies in aqueous solution.12 For this reason, we designed a crystalline superstructure “Pyrene box” from available commercial materials, containing the 1,3,5,8 pyrenetetrasulfonate anions, PTS4−, the guanidinium cations, G+ and 1,ω-diammonium-alkanes.
The confinement of the 1,10-diammonium-decane, 1, 1,11-diammonium-undecane, 2 and 1,12-diammonium-dodecane, 3 within the PTSG host-matrix has been readily realized in aqueous solutions of G+Cl−, PTS4−4Na+ and 1, 2 or 3 resulting in the formation of the inclusion single crystals of PTSG{1}, PTSG{2} and PTSG{3}, respectively. These preliminary results showed crenel-like conformations of the larger alkanes in the compressed states. H-bonding and van der Waals interactions between alkane guest and the resulted capsule host are strongly and synergistically contributing for the assembly of the alkane-capsule systems in water.12 At his point, two questions arise: (a) how important are the presence of molecular components and how does they contribute to the stabilization of the overall host–guest superstructure? (b) How selective is alkane encapsulation within competitive conditions?
In order to answer these questions, herein we expanded our initial study to different capsule systems and/or to competitive encapsulation screenings of mixtures of alkanes. To our surprise, we observed that adaptive encapsulation occur and both highly compressed and non-compressed chains are equally favored. This is not so expected, as it would imply that a hydrophobic pocket of a given finite volume would not be specific for a particular alkane guest. A brief survey of the literature data available for biological entities indicated a similar lack of selectivity occurring in the fatty acid metabolism. More specifically, medium chain acyl-CoA dehydrogenase shows a weak specificity towards C8 alkane chains, but can also competitively encapsulate C10, C12 and C14 alkane chains under certain conditions.13
Results and discussion
Strategy
Our previous experience in the confinement of unstable species inside a crystalline guanidinium–sulfonate, GS molecular flask, led us to consider a similar approach for compressing alkanes.5 Alkanes, however, are not a preferred building blocks in crystal engineering, due to their very high number of possible conformations generating disorder and their general lack of affinity.In practice, it is very difficult to co-crystalize a long alkane, especially in a precise way. Furthermore, from the chemical point of view, alkanes are not very compatible with GS networks. In order to generate the PTSG {1,ω-diammonium-alkanes} host–guest, we found it important to play on the hydrophobic/hydrophilic balance. Using water as solvent, the encapsulating components had to have both hydrophobic and hydrophilic behaviors; the 1,3,5,8-pyrene-tetrasulfonate PTS4− anion being a good candidate, ideal to make H-bonded ionic complexes with donor H-bonding cationic species.5 Guanidinium cations, G+ were chosen as suitable counter-ions and H-bond donors. The 1,ω-diammonium-alkanes have been used for their higher solubility in water compared with alkanes. Moreover the ammonium groups might be considered as tethering points by interacting with the sulfonate groups of PTS4− anions via H-bonding and ionic interactions (Fig. 1). A series of 1,ω-diammonium-alkanes of varying lengths have been chosen to screen the molecular chain behaviours inside a fairly robust PTSG cage, with fixed dimensions.
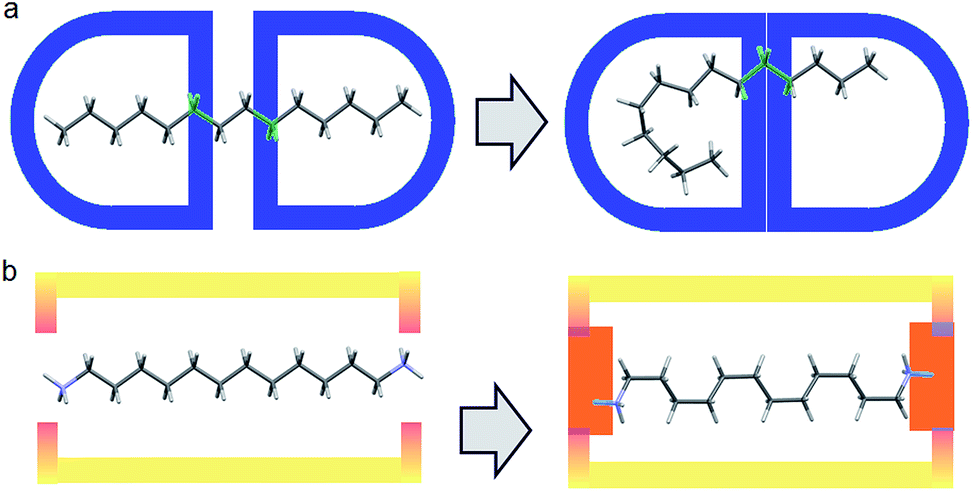 | ||
| Fig. 1 (a) Free motion of unfixed of alkane inside a capsule host; (b) Diammonium alkane encapsulation, fixed at the ends with fewer degrees of freedom via H-bonding and charge interactions. | ||
NMR studies in aqueous solution
The ion-pairing proxy interactions between 1,ω-diammoniumalkanes 1–3 and PTS4− anions can be detected in aqueous solution. The formation of PTS{1–3}2 and PTSG{1–3} complexes is confirmed by the upfield shifts of the protons of encapsulated 1–3 when compared with the spectra of 1–3 recorded in the absence of PTSG capsule (Fig. 2). In all cases the prevalent interaction observed is the ion-pairing between PTS4− and the 1,ω-diammonium-alkanes, even in the absence of G+, which results in a shielding of the methylene groups shifts in the middle of the chain by about 1 ppm (Fig. 2). The broad upfield protons signals, showing –C–H(alkane chains)/π(PTS) interactions are reminiscent of important dynamic behaviors of alkane chains within the ion-pairing system PTS{1}2 but not for PTS{2}2 and PTS{3}2. This conformational motion of dication 1 interacting with PTS4− anions, is largely reduced in the presence of G+ cations, as shown by the presence of sharp peaks (Fig. 2), reminiscent with the strong H-bonding/ion-pairing interactions in aqueous solution. This means that the presence of the G+ cation is required for the stabilization of the “Pyrene box”, in which the motion of alkane chains is restricted. The slight upfield shift decrease with increasing length of alkane chains of 2 to 3 and can be explained only if the alkane chains adopt coiled conformations within the PTSG capsule, spatially allowing the folding within the confined space, as previously observed.15 The signal corresponding to the methylene groups in the 1 and ω terminal positions of the alkane chain remains unchanged in the presence PTS4−. (Fig. 2). This is in good agreement with the crystal structures (see below), where the middle of the chain is shielded by the pyrene rings and the terminal atoms are close to the edge, almost out of the box. In general, higher concentrations of G+ resulted in a stronger shielding, with the exception of 1,10 diammonium-decane where a dynamic process is observed (Fig. 2a). The stoichiometry of the complex present in solution was determined by titration experiments. A molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 PTS4−:G+:1–3 is always enough to generate the confined systems in solution. Although not clearly evident from the NMR titration experiments, it should be noted that crystals could only be obtained in PTS4−:G+:1–3 ratios of 1:4:1, with the exception of PTSG{1}2.
:2:1 PTS4−:G+:1–3 is always enough to generate the confined systems in solution. Although not clearly evident from the NMR titration experiments, it should be noted that crystals could only be obtained in PTS4−:G+:1–3 ratios of 1:4:1, with the exception of PTSG{1}2.
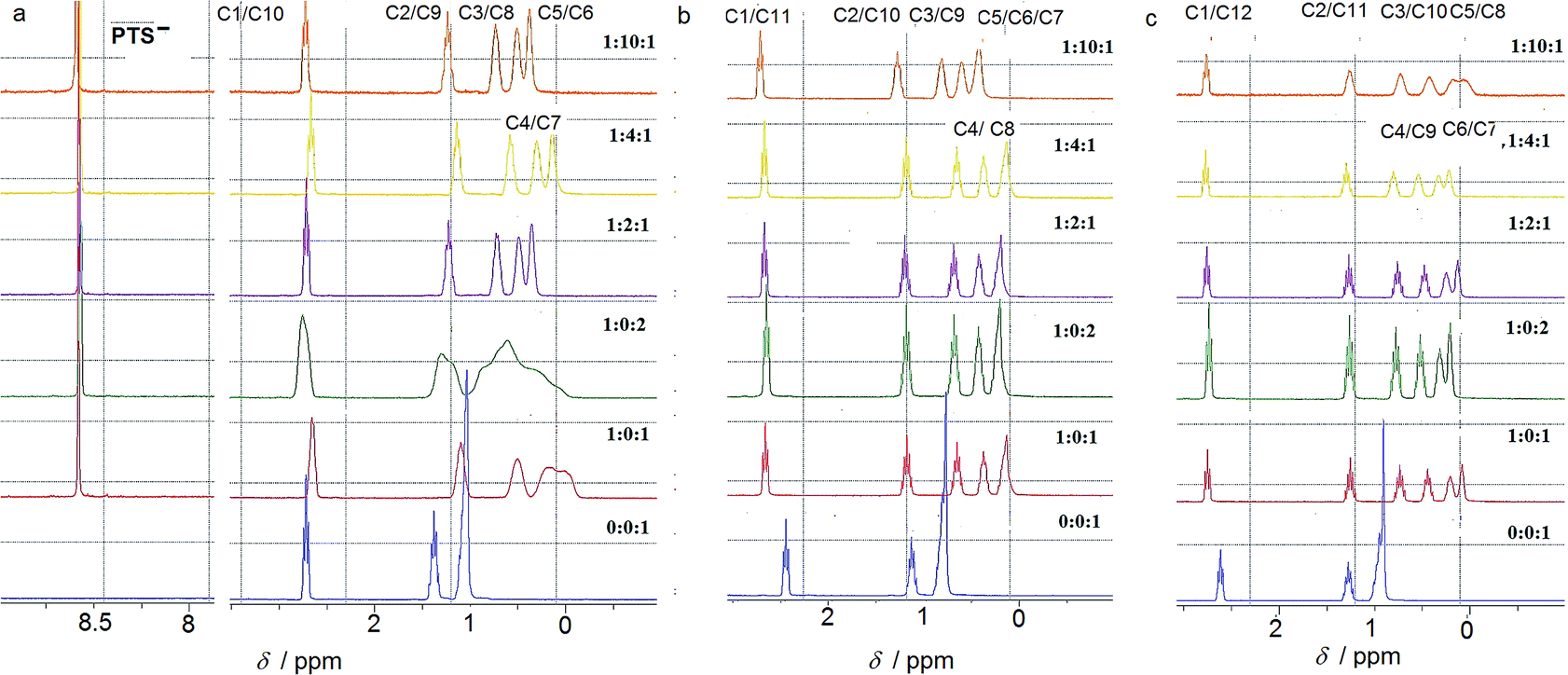 | ||
| Fig. 2
1H-NMR spectra at different molar ratios of PTS4−:G+:1,ω-diammonium alkane in D2O at 25 °C for (a) 1,10-diammonium-decane, 1, (b) 1,11-diammonium-undecane, 2 and (c) 1,12-diammonium-dodecane, 3 guest compounds. | ||
The compression of the alkane chains is confirmed by COSY/ROESY experiments (Fig. 3). In the case of PTSG{1}, the alkyl chain appears to have an elongated zig–zag conformation, with no correlation observed on a distance larger than 2 carbons (distinct NOE effects are found between the protons H1 to H3) (Fig. 3a). In the case of PTSG{2} (Fig. 3b) and PTSG{3} (Fig. 3c) spatial correlations are visible up to 4 carbon atoms away (distinct NOE effects are found between the protons H1 to H4). Furthermore, the DOSY spectroscopy (Fig. 4) showed that only one dimensionally constant capsule is present in solution for the all PTSG{1–3} systems (Table 1). The calculated diffusion coefficients get progressively smaller from PSTG{1} to PTSG{3}, which is consistent with a mass increase of the capsule (Table 1).
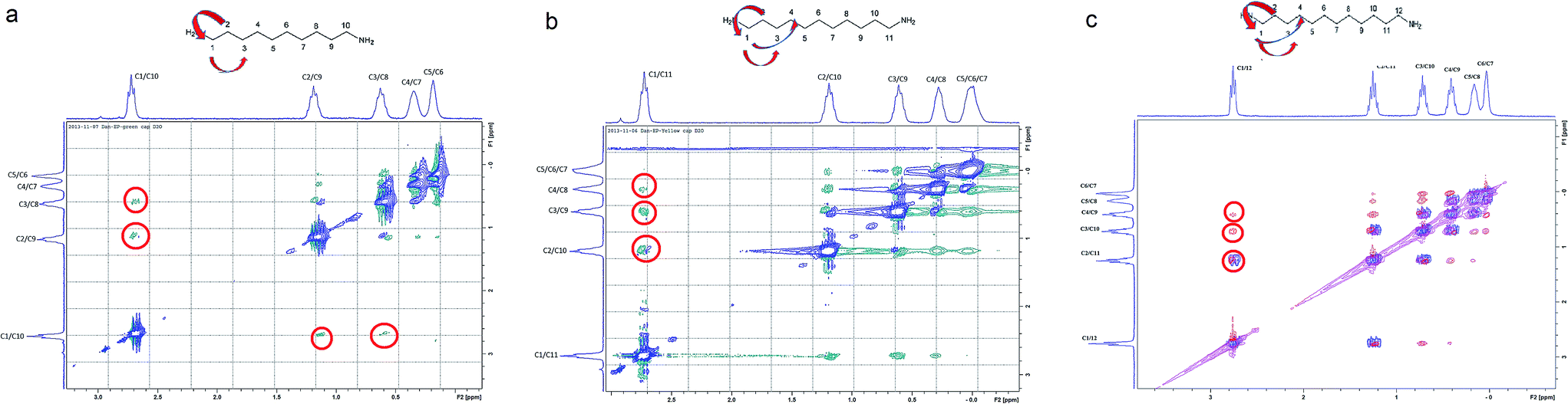 | ||
| Fig. 3 Stack between the COSY (blue) and ROESY (a and b) green, (c) (red) spectra of (a) PTSG{1}, (b) PTSG{2} and (c) PTSG{3}. Long range spatial interactions can be observed between C1–C2, C1–C3, C1–C4 carbon atoms away. | ||
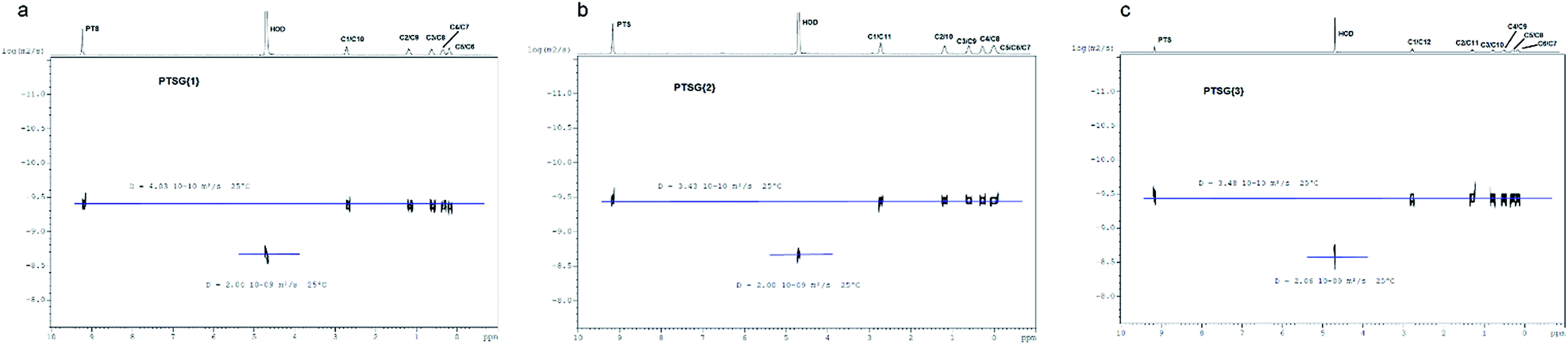 | ||
| Fig. 4 DOSY spectra of (a) PTSG{1}, (b) PTSG{2} and (c) PTSG{3}. | ||
| PTSG{1} | PTSG{2} | PTSG{3} | |
|---|---|---|---|
| Complex diffusion coeff. (m2 s−1) | 4.03 × 10−10 | 3.43 × 10−10 | 3.48 × 10−10 |
| HOD diffusion coeff. (m2 s−1) | 2.04 × 10−9 | 2.00 × 10−9 | 2.06 × 10−9 |
| Weighed complex diffusion coeff. (m2 s−1) | 3.95 × 10−10 | 3.43 × 10−10 | 3.38 × 10−10 |
On the basis of these NMR data one may conclude that the molecular alkane strands of 1–3 are confined within PTSG{1–3} cages, which are stable in water. Based on long distance interactions observed in COSY/ROESY experiments, alkane strand of 1 is stable in the elongated and slightly compressed conformations, while alkane strands of 2 and 3 adopt in aqueous solution the compressed conformations.
X-ray crystal structures of PTSG{1–3}2
In the crystal, the “Pyrene box” results from the self-assembly via H-bonding (dO⋯H–N of 2.00 Å) and charge interactions of two sulfonate moieties from PTS4− anions and of two G+ cations. This network is reinforced by two bridging water molecules, which are simultaneously H-bonded to G+ (dO⋯H–N of 2.00 Å) and to PTS4− (dS⋯H–O of 1.98 Å), playing a critical role in organizing the partners via three sulfonate moieties on the faces of the “Pyrene box”. The 1,ω-diammonium-alkanes, 1–3 are confined within inner space of parallelepipedic boxes defined between two G+ and two PTS4, while each ammonium moieties of 1,ω-diammoniumalkanes form two anchoring H-bonds (dN–O = 1.93 Å) with two other sulfonate moieties themselves non bonded to G+ cations (Fig. 5–7). It results that the 1,ω-diammonium-alkanes, 1–3 practically immobilized via synergetic H-bonds and hydrophobic interactions with the pyrene walls, have well-defined electron density sites in the Fourier maps, with any observed motional disorder for carbon atoms. All combined H-bonding and hydrophobic host–guest interactions are vital for the encapsulation and insulation as well as for the low mobility of the alkane guest chains in the “Pyrene Box”. The average distance between the two planes of non H-bonded sulfonate groups is 13.80 Å. 1,10-Diammonium-decane, 1 fits perfectly the length of the “Pyrene box”, with each ammonium moiety bonding to a sulfonate moiety of PTS4−. The chain adopts a classical ‘zig–zag’ elongated conformation, with an average sp3carbon-sp3carbon length of 1.52 Å in the expected range (Fig. 5).14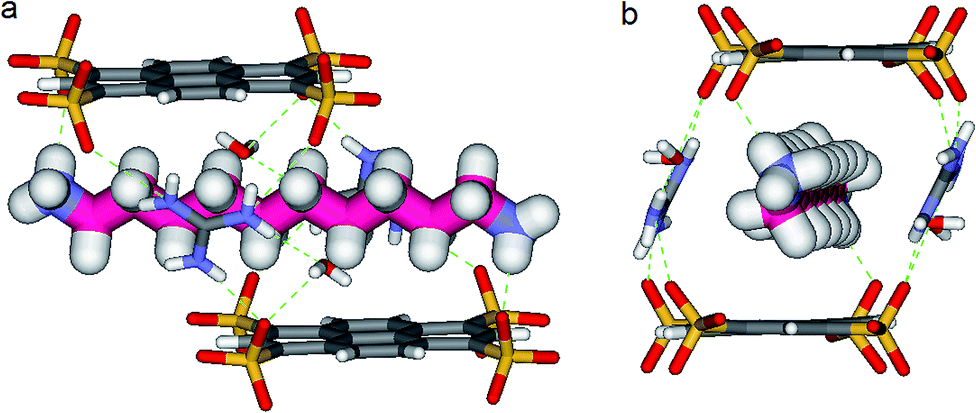 | ||
| Fig. 5 X-ray structure of PTSG{1}: (a) Side and (b) Front view of the PTSG{1} inclusion complex. 1,10-diammonium-decane guest, 1 (magenta) presents inside the PTSG box, an uncompressed zig–zag architecture. | ||
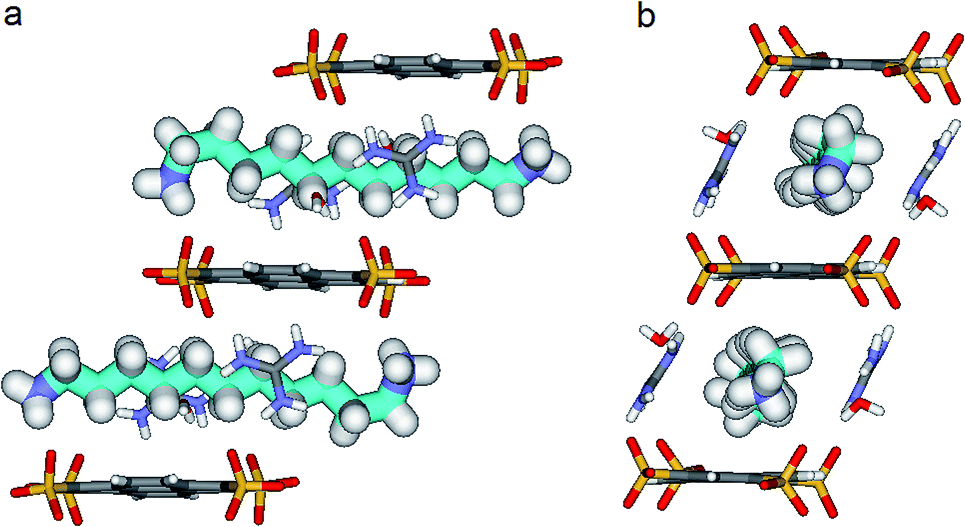 | ||
| Fig. 6 X-ray structure of PTSG{2}: (a) Side and (b) Front view of the PTSG{2} inclusion complex. 1,11-Diammonium-undecane guest 2 (cyan), presents inside the PTSG box, an slightly compressed architecture with a gauche conformation at the terminal part of the chain. | ||
 | ||
| Fig. 7 X-ray structure of PTSG{3}: (a) Side and (b) Front view of the PTSG{3} inclusion complex. 1,12-diammoniumdodecane, 3 presents inside the PTSG box, two highly compressed architectures 3a and 3b having 6 gauche (orange) and having 4 gauche (red-violet) conformations, respectively. | ||
By increasing the alkane chain length by only one carbon atom, the 1,11-diammonium-undecane chain becomes longer than the length of the box and adopts an asymmetrical conformation, 2 having one gauche conformation at the end of the chain (Fig. 6). X-ray structure determinations at variable temperature were performed in order to better understand the dynamic behavior of the compressed chains of 2 within the box. The undecane chain in PTSG{2} show dynamic behaviors at temperatures higher than 140 K and becomes disordered and symmetrical at room temperature. We interpreted this reversible flipping change as the rapid movement of the gauche conformation at room temperature from one end of the chain to the other.12
Another incremental increase to 1,12-diammonium-dodecane, 3 leads to an even more compressed alkane chain in PTSG{3} (Fig. 7). Careful crystallographic modelling allowed us to identify two co-existing conformers 3a and 3b of the chain in a 2:1 ratio. Both cases show high compression, with conformer 3a having 6 gauche conformations and 3b having 4 gauche, respectively.
Interestingly, no dynamic behaviors, leading to the reversible change between conformations, were observed in the case of both PTSG{1} and PTSG{3}, even at temperatures as high as 340 K. This is consistent with the “natural” stability of the extended decane chain of 1, perfectly fitting inside the “Pyrene box”, but totally unexpected for the two similarly disordered dodecane chains 3a and 3b, probably too constrained to move.
The compression under confinement results in a decrease of the overall length of the natural uncompressed ‘zig–zag’-type 1,12-diammonium-dodecane, 3 and 1,11-diammonium-undecane 2 chains: from 16.534 Å to 14.465 Å (3a and b) and from 15.036 Å to 14.239 Å (2a and b), respectively.
Non-Covalent Interactions (NCI) plots15 showed that the confined alkane chains present strong van der Waals contacts with all the inner surfaces of the “Pyrene box” and the compressed states present attractive hydrogen–hydrogen contacts inside the gauche conformations. A closer examination of these ‘crenel’ conformations 3a and 3b revealed a distance of only 2.24 Å between the hydrogens inside (i.e. the hydrogens in positions 1 and 4, 3 and 6 etc.), shorter than the sum of their van der Waals radius (approx. 2.5 Å)-see ref. 12 for details.16
With all this in mind, it remains to determine how strong are the stabilizing interactions and how much does they contribute to the overall structure and crystallization process? The combination of H-bonding and hydrophobic interactions is not immediately apparent, as one cannot estimate quantitatively the synergetic interaction strengths.
We know that the Pyrene box is always forming in the presence of PTS4−, G+ and 1 eq. of 1,ω-diammonium-alkanes 1–3. Exchanging the G+ with diammonium cations 1–3 by increasing their ratio to 2 or 4 eq., only PTS{1}2 single crystals are obtained, while the corresponding to PTS{2}2 and PTS{3}2 are precipitating. The absence of the guanidinium cations induce a new “face to face” relative arrangement of the PTS4− platforms into the novel crystalline-“pseudoPyrene box” so that the average distance between the two planes of the sulfonate groups is drastically reduced to 7.11 Å. The 1,10-diammonium-decane, 1 much longer to fit the new constraints is H-bonded to two PTS4− platforms from neighboring “pseudo-Pyrene boxes” resulting in the formation of polymeric superstructures of H-bonded alkane chains bridging the “pseudoPyrene boxes” (Fig. 8). The “pseudoPyrene box” is reinforced by two bridging water molecules, which are simultaneously H-bonded to ammonium cations (dO⋯H–N of 1.97 Å) and to PTS4− (dO⋯H–N of 1.98 Å), playing a critical role in organizing the PTS4− anions in the face-to-face configuration of the “pseudoPyrene box”. This is inducing a small compression behavior of the decane chain adopting an asymmetrical conformation, having one gauche conformation between C2–C3 atoms of the chain (Fig. 8).
 | ||
| Fig. 8 X-ray structure of PTS{1}2: (a) side and (b) front view of the PTS(1)2(H2O)4 inclusion complex. 1,11-diammonium-dodecane guest 2 (magenta) presents inside the “pseudoPyrene box” a slightly compressed architecture with the gauche conformation present between the C2–C3 of the chain. | ||
Competitive crystallization experiments
Finally, we decided to perform competitive crystallization experiments in the presence of PTS4−:G+ (1/4, mol/mol) and equimolar mixtures 1 + 2, 2 + 3 and 1 + 3 and (1 + 2 + 3), respectively (Table 2). They are excellent candidates for the generation of dynamic supramolecular libraries in solution, resulting in the formation of interchanging equilibrating mixture of components of Pyrene or pseudoPyrene boxes of different compositions, which might converts into unique/specific solid state species via a kinetically irreversible crystallization process.17,18
:G+ (1:4, mol/mol) components
| Mixture of 1,ω-diammoniumalkanes | Resulted crystallized structure |
|---|---|
| 1 + 2 | PTS{1}2 |
| 2 + 3 | Precipitate |
| 1 + 3 | PTSG{1}0.62{3}0.38 |
| 1 + 2 + 3 | PTSG{1}0.62{3}0.38 |
Crystals of PTS{1}2*, a polymorph of the original PTS{1}2 suitable for X-ray analysis were formed from a mixture of PTS4− + 1 + 2, while the mixture PTS4− + 2 + 3 forms a precipitate within minutes. Interestingly, in the PTS{1}2* structure the 1,10-diammonium-decane, 1 present the same conformation with a gauche conformation between the C2–C3 of the chain (Fig. 8), but the packing behaviors are different (Fig. S1†). The selective crystallization of PTS{1}2 against PTSG{1} can be explained by lower solubility of – “pseudoPyrene boxes” in PTS{1}2, kinetically selected in competition with the more soluble monomeric PTSG{1} containing the alkane chains included in the “Pyrene box” capsule.
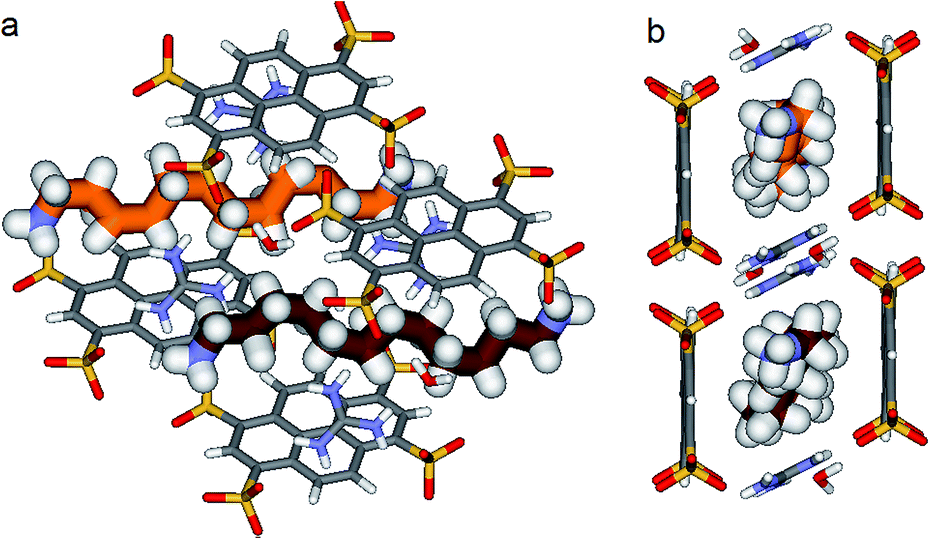
Things became interesting when a hybrid PTSG{1}0.62{3}0.38 is obtained as a unique crystallizing component from the mixtures of PTS4−:G+ (1:4, mol mol−1) and 1 + 3 or 1 + 2 + 3. The “Pyrene box” encapsulate simultaneously both non-compressed 1 (62%) and highly compressed 3a (19%) and 3b (19%) at the same time and in the same crystal (Fig. 9). This is based on the relative similar constitutional stabilities of un-compressed natural form of 1 and of the highly compressed form of 3, strongly interacting the “Pyyrene box” host via coupled H-bonding/hydrophobic interactional algorithms, as well as several stabilizing dihydrogen contacts. This confirms the previously observed high stability of PTSG{3} over a large temperature domains. The N1–Nω distance is 14.031 Å, a mean of the distance in the case of PTSG{1} (13.803 Å) and the case of PTSG{3} (14.467 Å). Dimensional anchoring and host–guest complex interaction, coupled with hydrogen–hydrogen interactions, are robust enough to induce simultaneous encapsulation of 1 and 3.
 | ||
| Fig. 9 X-ray structure of PTSG(1)0.62(3)0.38 resulted as unique crystallizing component from the reaction mixtures of PTS4−:G+ (1:4, mol/mol) and 1 + 3 or 1 + 2 + 3. 1,10-Diammonium, 1 (62%) and 1,12-diammonium, 3 (2 × 19%) guests are present inside the PTSG box as an elongated zig–zag 1 architecture and the two highly compressed architectures 3a and 3b, respectively. | ||
Conclusion
Molecular encapsulation of the alkane chains has long intrigued chemists on account of their strained geometries and variable adaptive behaviors to form dynamic molecular capsules, but these compounds or their derivatives have most often eluded crystallographic characterization.In this paper we highlighted that the alkane chains may be successfully constrained/anchored inside a rigid crystalline capsules. They adopt specific conformations at different levels of compression depending on their dimensional behaviors that are sufficiently kinetic stable under the confined conditions, to allow a conventional structure determination by X-ray diffraction. These results not only shows that chemists are able to tune, from inside, physical properties (pressure) at the molecular scale, but also provides new hints for the, so far, not fully understood conformations of long alkane chains under confined conditions. The undecane chain in PTSG{2} shows an interesting movement of the gauche conformation from one end of the chain to the other, while the decane chain of PTSG{1} and the dodecane chain of PTSG{3}, are stable in an elongated zig–zag and highly compressed crenel-type conformations, respectively.
Variable relative spatial disposition of the PTS4− platforms within the crystal induce a different dimensional disposition of the sulfonate anchoring points used for the 1,ω-diammonium-alkanes binding. The presence of G+ cations induce a slightly slipped relative position of the PTS4− platforms in the “Pyrene box”, compared with their “face to face” relative arrangement in the “pseudoPyrene box”. These different capsules can select between multiple configurations of alkane chains showing different compression behaviors and a specific adaptation, through different specific confinement mechanisms. The uncompressed and compressed conformations of the 1,10-diammonium-decane, 1 under different confinement conditions in the two crystalline boxes, shed light on the constitutional adaptivity of alkane chain to environmental factors, in order to maximize the multivalent interacting/stabilizing contacts with the host capsule. Moreover, a chemical selection can be obtained from mixtures of alkane chains via the encapsulation of kinetically stable conformations observed during the encapsulation of pure components.
The synergetic selection of 1,10-diammonium-decane 1 and 1,12-diammonium-dodecane 3 in the same crystal structure can lead to several interesting discussions. First, under confined conditions hydrogen–hydrogen interactions might be considered not as weak repulsive as previously envisaged. Further calculations are underway to quantify this speculation. Secondly this work may form a basis for future work explaining the relatively weak selectivity of natural hosts, such as enzymes.
Furthermore, this contribution adds several new behaviors to the systematic rationalization and prediction of hydrophobic clustering in the capsules; it unlocks the door to the new compounds world paralleling that of biology.
Experimental
All the compounds were purchased from Sigma-Aldrich and were used without further purification. Suitable crystals for all the structures were readily obtained in large quantities by slow evaporation of aqueous solutions obtained by dissolving: 50 mg 1,3,5,8-pyrenetetrasulfonate tetrasodium salt, 100 mg guanidinium hydrochloride, 30 mg alkane diamine and 50 μL HCl 37% in 1 mL H2O. After 1 week the crystals were filtered and dried under mild vacuum for 2 h.NMR experiments
1H NMR experiments were performed on an AVANCE 300 MHz Bruker spectrometer in D2O with the use of the residual solvent peak as reference. NMR titration experiments were performed by adding successive aliquots of 0.1 M D2O solutions of 1,ω-diammonium alkane dichloride and guanidinium to 0.4 ml 0.01 M solution of sodium 1,3,5,8-pyrene tetrasulfonate. In the case of the 2D-NMR experiments 10 mg of the corresponding crystals obtained by the method described above were dissolved in 0.5 mL D2O.2D-ROESY (Rotating-frame Overhauser Effect Spectroscopy) NMR experiments were performed at 298 K applying the 180–90° selective-spinlock—FID pulse sequence. The mixing time was recorded with 300 ms. A typical proton-proton COSY45 experiment was performed (cosygpqf). Spectra were acquired with 2 Ko data points in F2 Frequency axis and 128 experiments.
2D-DOSY (Diffusion-Ordered SpectroscopY) NMR
2D-DOSY (Diffusion-Ordered SpectroscopY) NMR experiments were performed at 298 K with a Bruker Dual z-gradient probe head capable of producing gradients in the z direction with strength 55 G cm−1. The DOSY spectra were acquired with the ledbpgp2s pulse program (2D sequence for diffusion measurement using echo and led with bipolar gradient pulse).19 All spectra were recorded with 8 Ko time domain data points in the F2 Frequency axis and 32 experiments (F1). The gradient strength was logarithmically incremented in 32 steps from 2% up to 95% of the maximum gradient strength. All measurements were performed with a diffusion delay D (D2O) of 80 ms in order to keep the relaxation contribution to the signal attenuation constant for all samples. The gradient pulse length d (2*P30) was 5 ms in order to ensure full signal attenuation. The diffusion dimension of the 2D DOSY spectra was processed by means of the Bruker Topspin software (version 2.1).X-ray diffraction
All the structures have been measured on an Agilent Technologies Gemini-S four circle diffractometer using Mo-Kα radiation (λ = 0.71073 Å) and equipped with a Sapphire3 detector at 175 K at the joint X-ray scattering facility of the Pôle Balard at the University of Montpellier, France. The structures have been solved using the ab initio charge flipping method as implemented in SUPERFLIP.20 Hydrogen atom positions were determined using Fourier differential maps in the case of PTSG{1} and the structure of PTSG{2a}. Hydrogens in the case of PTSG{2b} and PTSG{3} were added geometrically. It should be noted that in all cases the ammonium hydrogens were found using Fourrier maps. All structures were initially refined using non-linear least-squares methods as implemented in CRYSTALS,21 in which the hydrogen atoms were treated as riding on their parent atoms and with Uiso(H) constrained to in general 1.2–1.5 times Ueq(H) that of the parent atom. The final difference Fourier maps showed in many cases residual peaks due to bonding effects (in the pyrene planes and on the sulfonate moieties), because of the in general very good resolution of the experimental data (mostly around 0.7 Å or better, where the experimental resolution is defined according to Dauter).22Disorder models and temperature dependence: in the case of 1,10-diammonium decane and 1,12-diammonium dodecane the asymmetric unit contains 1/2 of a pyrene and 1/2 of a chain. This is due to a local inversion center at the middle of the alkanedyil chain. At 175 K, in the case of 1,11-diammonium undecane, this inversion center disappears and the unit cell is doubled over the c axis. However, at room temperature (298 K), the unit cell is again halved (i.e. the same as for 1 and 3). The structure of 2 below room temperature can thus be considered as a commensurate superstructure of the room temperature structure with wave vector q = 0.5c*RT where c*RT is the room-temperature reciprocal c-vector. Reflections hkl: l = 2n+1 are in the low-temperature phase in general much weaker than reflections hkl: l = 2n. The superstructure is only due to the disorder in the alkandyl chains, the PTS4− and G+ moieties remaining exactly in place in neighboring unit cells. The disappearance of the low-temperature superstructure with rising temperature was monitored by plotting the ratio of the mean 〈I/σ(I)〉l=odd over 〈I/σ(I)〉l=even. Structurally this means that the low-temperature superstructure has a pseudo inversion center at the middle of the undecane chain, which becomes a real inversion center, i.e. belonging to the space group symmetry, only at room temperature where the rapid movement of the gauche conformation from one end of the chain to the other makes that the two confirmations are even likely. This process was found to be completely reversible. The room temperature structure of PTSG{2} can be alternatively refined in the doubled unit cell as well as in the small unit cell (the same as for PTSG{1}, since the disorder is nearly complete, but because of the very faint intensity of the commensurate super reflections, the refinement is not very stable in the doubled unit cell. In the small unit cell the site occupancy factors of the two disordered parts is necessarily 0.5, because of the presence of the space group inversion center at the middle of the odd-numbered chain. The site occupancy factors of the two disordered conformers of 3 were also refined as a function of temperature, but no significant temperature dependence was observed, showing that the movement of 3 is most possibly impossible due to a too high confinement in the Pyrene box.
Acknowledgements
This work was conducted within the framework of DYNANO, PITN-GA-2011-289033 (http://www.dynano.eu) and ANR “LABEX Chemisyst” (D.D.) awarded by “Pole Balard” Montpellier.Notes and references
- (a) G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049 CrossRef CAS PubMed; (b) S. J. Dalgarno, P. K. Thallapally, L. J. Barbour and J. L. Atwood, Chem. Soc. Rev., 2007, 36, 236 RSC; (c) M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161 RSC; (d) D. J. Cram and J. M. Cram, Container Molecules and Their Guests, Royal Society of Chemistry, Cambridge, 1994 Search PubMed.
- (a) L. Trembleau and J. Rebek, Science, 2003, 301, 1219 CrossRef CAS PubMed; (b) D. Ajami and J. Rebek, Nat. Chem., 2009, 1, 87 CrossRef CAS PubMed; (c) J. Rebek, Chem. Commun., 2000, 637 RSC.
- T. Jacobs, V. J. Smith, L. H. Thomas and L. J. Barbour, Chem. Commun., 2014, 50, 85 RSC.
- A. Muller and P. Gouzerh, Chem. Soc. Rev., 2012, 41, 7431 RSC.
- Y.-M. Legrand, A. van der Lee and M. Barboiu, Science, 2010, 329, 299 CrossRef CAS PubMed; Y.-M. Legrand, A. Gilles, E. Petit, A. van der Lee and M. Barboiu, Chem.–Eur. J., 2011, 17, 10021 CrossRef PubMed.
- (a) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS PubMed; (b) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251 CrossRef CAS PubMed; (c) K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703 RSC; (d) Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349 CrossRef CAS PubMed.
- (a) D. Voet and J. D. Voet, Biochemistry, Wiley, New York, 2nd edn, 1995 Search PubMed; (b) B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland, New York, 4th edn, 2002 Search PubMed; (c) T. M. Devlin, Textbook of Biochemistry: With Clinical Correlations, Wiley-Liss, Hoboken, NJ, 6th edn, 2006 Search PubMed.
- (a) O. Hayashida, L. Sebo and J. Rebek, J. Org. Chem., 2002, 67, 8291 CrossRef CAS PubMed; (b) O. Hayashida, A. Shivanyuk and J. Rebek, Angew. Chem., Int. Ed., 2002, 41, 3423 CrossRef CAS; (c) A. Asadi, D. Ajami and J. Rebek, J. Am. Chem. Soc., 2011, 133, 10682 CrossRef CAS PubMed.
- (a) B. C. Gibb, in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, Wiley-VCH, 2008, p. 291 Search PubMed; (b) H. Y. Gan and B. C. Gibb, Chem. Commun., 2012, 48, 1656 RSC; (c) S. M. Liu, D. H. Russell, N. F. Zinnel and B. C. Gibb, J. Am. Chem. Soc., 2013, 135, 4314 CrossRef CAS PubMed; (d) C. L. D. Gibb and B. C. Gibb, Chem. Commun., 2007, 1635 RSC.
- (a) F. Kotzybahibert, J. M. Lehn and K. Saigo, J. Am. Chem. Soc., 1981, 103, 4266 CrossRef CAS; (b) F. Kotzybahibert, J. M. Lehn and P. Vierling, Tetrahedron Lett., 1980, 21, 941 CrossRef CAS.
- A. Grego, A. Muller and I. A. Weinstock, Angew. Chem., Int. Ed., 2013, 52, 8358 CrossRef CAS PubMed.
- D. Dumitrescu, Y. M. Legrand, E. Petit, A. van der Lee and M. Barboiu, Chem. Commun., 2014, 50, 14086 RSC.
- V. Kieweg, F. G. Krautle, A. Nandy, S. Engst, P. Vock, A. G. AbdelGhany, P. Bross, N. Gregersen, I. Rasched, A. Strauss and S. Ghisla, Eur. J. Biochem., 1997, 246, 548 CAS.
- C. Pascard, C. Riche, M. Cesario, F. Kotzybahibert and J. M. Lehn, J. Chem. Soc., Chem. Commun., 1982, 557 RSC.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. T. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed.
- (a) C. F. Matta, J. Hernandez-Trujillo, T. H. Tang and R. F. W. Bader, Chem.–Eur. J., 2003, 9, 1940 CrossRef CAS PubMed; (b) J. Poater, M. Sola and F. M. Bickelhaupt, Chem.–Eur. J., 2006, 12, 2889 CrossRef CAS PubMed; (c) R. F. W. Bader, Chem.–Eur. J., 2006, 12, 2896 CrossRef CAS PubMed.
- (a) F. Dumitru, E. Petit, d. L. A. van and M. Barboiu, Eur. J. Inorg. Chem., 2005, 4255 CrossRef CAS; (b) Y.-M. Legrand, d. L. A. Van, N. Masquelez, P. Rabu and M. Barboiu, Inorg. Chem., 2007, 46, 9083 CrossRef CAS PubMed; (c) F. Dumitru, Y. M. Legrand, A. van der Lee and M. Barboiu, Chem. Commun., 2009, 2667–2669 RSC; (d) F. Dumitru, Y.-M. Legrand, E. Petit, A. van der Lee and M. Barboiu, Dalton Trans., 2012, 41, 11860 RSC.
- (a) S. Mihai, A. Cazacu, C. Arnal-Herault, G. Nasr, A. Meffre, d. L. A. van and M. Barboiu, New J. Chem., 2009, 33, 2335 RSC; (b) M. Barboiu, M. Ruben, G. Blasen, N. Kyritsakas, E. Chacko, M. Dutta, O. Radekovich, K. Lenton, D. J. R. Brook and J.-M. Lehn, Eur. J. Inorg. Chem., 2006, 784 CrossRef CAS; (c) M. Barboiu, G. Vaughan, R. Graff and J.-M. Lehn, J. Am. Chem. Soc., 2003, 125, 10257 CrossRef CAS PubMed.
- D. Wu, A. Chen and C. S. Johnson Jr, J. Magn. Reson., Ser. A, 1995, 115, 260–264 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- Z. Dauter, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 1703 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1040388–1040390. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03945a |
| ‡ Present address: “C.D. Nenitzescu” Center for Organic Chemistry, Romanian Academy, Spl. Independetei 202b, Bucharest, Romania and SARA Pharm Solutions, 266-268 Calea Rahovei, 050912, Bucharest, Romania. |
| This journal is © The Royal Society of Chemistry 2015 |