
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective thioacetylation of chitosan end-groups for nanoparticle gene delivery systems†
V. D.
Pickenhahn
 a,
V.
Darras
a,
F.
Dziopa
a,
K.
Biniecki
b,
G.
De Crescenzo
a,
M.
Lavertu
*a and
M. D.
Buschmann
*a
a,
V.
Darras
a,
F.
Dziopa
a,
K.
Biniecki
b,
G.
De Crescenzo
a,
M.
Lavertu
*a and
M. D.
Buschmann
*a
aDept. Chemical Engineering and Inst. Biomedical Engineering, Ecole Polytechnique, Montreal, QC, Canada. E-mail: marc.lavertu@polymtl.ca; michael.buschmann@polymtl.ca
bANRis Pharmaceuticals Inc., Kirkland, QC, Canada
First published on 7th May 2015
Abstract
Chitosan (CS) end-group chemistry is a conjugation strategy that has been minimally exploited in the literature to date. Although the open-chain form of the CS reducing extremity bears a reactive aldehyde moiety, the most common method to generate a reactive end-group on CS is nitrous acid depolymerization, which produces a 2,5-anhydro-D-mannose unit (M-Unit) bearing also an aldehyde moiety. However, the availability of the latter might be low, since previous literature suggests that its hydrated and non-reactive form, namely the gem-diol form, is predominant in acidic aqueous conditions. Oxime-click chemistry has been used to react on such aldehydes with various degrees of success, but the use of a co-solvent and additional chemical reagents remain necessary to obtain the desired and stable covalent linkage. In this study, we have assessed the availability of the aldehyde reactive form on chitosan treated with nitrous acid. We have also assessed its reactivity towards thiol-bearing molecules in acidic conditions where CS amino groups are fully protonated and thus unreactive towards aldehyde. LC-MS and NMR spectroscopy methods (1H and DOSY, respectively) confirmed the regioselective thioacetylation of the reactive aldehyde with conversion rates between 55 and 70% depending on the thiol molecule engaged. The stabilization of the hemithioacetal intermediates into the corresponding thioacetals was also found to be facilitated upon freeze-drying of the reaction medium. The PEGylation of the CS M-Unit aldehyde by thioacetylation was also performed as a direct application of the proposed conjugation approach. CS-b-PEG2 block copolymers were successfully synthesized and were used to prepare block ionomer complexes with plasmid DNA, as revealed by their spherical morphology vs. the rod-like/globular/toroidal morphology observed for polyplexes prepared using native unmodified chitosan. This novel aqueous thiol-based conjugation strategy constitutes an alternative to the oxime-click pathway; it could be applicable to other polymers.
Introduction
Chitosan (CS), a linear and cationic polysaccharide composed of D-glucosamine (GlcNH2) and N-acetyl-D-glucosamine (GlcNHAc) units, is derived from chitin by deacetylation. This non-toxic polyelectrolyte holds great interest due to its biocompatibility, biodegradability and mucoadhesive properties.1 Chitosan and its derivatives have been proposed for applications including gene and drug delivery, tissue repair, water purification and cosmetics.2–6 Two general approaches have been explored to chemically modify CS: lateral “graft” and “block” modifications. The former involves conjugation to CS lateral functional groups (amine or hydroxyl) whereas the latter relies on conjugation to CS end-groups.Several strategies for grafting onto CS amines (N-2-graft) have been proposed in the literature. For example, PEG and other graft-copolymers have been proposed to enhance CS solubility at physiological pH and increase the colloidal stability of CS-based polyelectrolyte complexes,7,8 while ligands for specific cell targeting9,10 or fluorescent dyes11,12 have also been grafted onto CS amines. However, lateral grafting can potentially compromise the ability of CS to bind nucleic acid and thus limit the stability and efficiency of chitosan/nucleic acid polyelectrolyte complexes for gene delivery applications. Indeed, lateral grafting can impede the ability of CS to electrostatically bind to negatively charged species by reducing its effective charge density and by potentially creating steric hindrance with bulky moieties.4 Alternatively, the O-6 grafting approach has been proposed to overcome the charge density reduction issue, although grafting of a bulky moiety at this position is likely to create steric hindrance and hence limit complexation with oppositely charged polymers or molecules as well. Additionally, O-6 grafting is technically challenging as it necessitates protection–deprotection steps for the CS amine moieties.13
To overcome these limitations, CS block conjugation strategies (e.g., branched CS,14,15 PEGylation,16 CS-PEI block-copolymer formation,17 CS labeling,18etc.) have recently been proposed as a means to modify the CS properties without compromising its ability to bind oppositely charged macro-ions such as nucleic acids. Two different CS attachment sites have been explored to date: the first is formed after CS depolymerization by nitrous acid (HONO) where a 2,5-anhydro-D-mannose unit (M-Unit) is formed at the reducing end of the cleaved polymer (Fig. 1, reaction 1), while the second site is available on the open-chain form, present in trace amounts, of the CS reducing extremity (either GlcNH2 or GlcNHAc units) and allows mutarotation between the alpha and beta anomers. These coupling strategies thus rely on the reaction of the aldehyde moiety with nucleophilic species. However, in both cases, the aldehyde moiety appears to be mostly present in its hydrated and unreactive form (Fig. 1, reaction 2), also referred to as the geminal- or gem-diol form, under acidic aqueous conditions.19–21
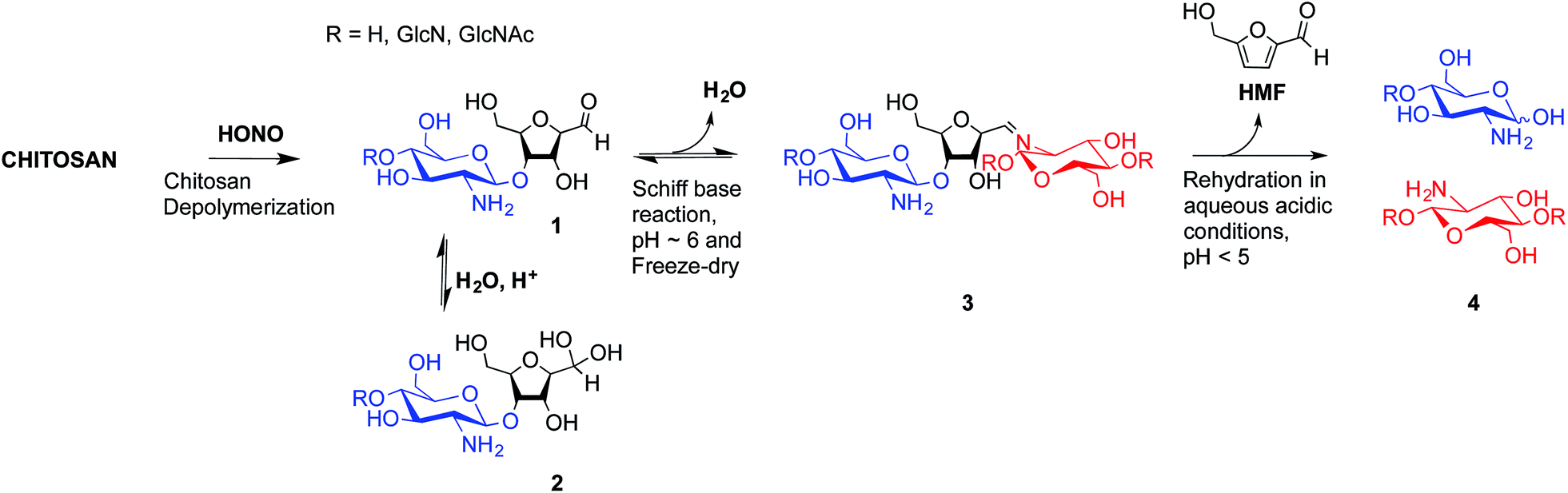 | ||
| Fig. 1 Production of 2,5-anhydro-D-mannose unit (M-Unit) at the reducing end of chitosan by depolymerization in nitrous acid (HONO): chitosan depolymerization with nitrous acid (HONO) is a rapid, well-understood, and easily controlled method for producing chitosan harbouring a 2,5-anhydro-D-mannose unit (M-Unit) at the reducing end of the cleaved polymer.22 A free aldehyde group (electrophile) is then potentially accessible (1) for reaction with nucleophilic moieties (e.g., CS amine groups, thiols, oxyamines, etc.). Tømmeraas et al.20 demonstrated that the M-Unit aldehyde also exists in its gem-diol hydrated form (2). The neutralization of CS and subsequent freeze-drying of the depolymerization medium induces a Schiff base formation between CS neutralized amines that react with the CS M-Unit aldehyde (3). The rehydration of the imino-adducts in acidic aqueous conditions cleaves the imino linkage between CS chains, transforming the M-Unit into hydroxymethylfurfural (HMF) (4). | ||
The amines of CS in their neutral form are strong nucleophiles that can react with the aldehyde of CS's reducing end (Fig. 1 – reaction 3). Therefore block conjugation to the CS end-group requires that the proportion of CS amines in their reactive form be minimized, for example by performing reactions at pH significantly lower than the chitosan pKa, typically near 6.5. However, chitosan pKa varies with both ionic strength and CS charge density and can reach values as low as about 5.5 at high charge density and in the absence of added salt.23 To date, all CS end-group conjugation reactions that have been implemented rely on oxime-click chemistry.16,18,24,25 The oxyamine moieties involved in these studies have a pKa value around 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 and are therefore only slightly more reactive than CS amines in acidic aqueous conditions. Additionally, although the carbon–nitrogen double bond resulting from oxime-click chemistry is more hydrolytically stable than standard imino linkages,27 a conjugate stabilization by an external chemical reagent (e.g., hydrides) is necessary to stabilize the structure.28 Moreover, it appears that CS conjugations with such chemistry usually require a polar aprotic co-solvent addition such as acetonitrile, DMF or DMSO to improve the reaction efficiency.29
26 and are therefore only slightly more reactive than CS amines in acidic aqueous conditions. Additionally, although the carbon–nitrogen double bond resulting from oxime-click chemistry is more hydrolytically stable than standard imino linkages,27 a conjugate stabilization by an external chemical reagent (e.g., hydrides) is necessary to stabilize the structure.28 Moreover, it appears that CS conjugations with such chemistry usually require a polar aprotic co-solvent addition such as acetonitrile, DMF or DMSO to improve the reaction efficiency.29
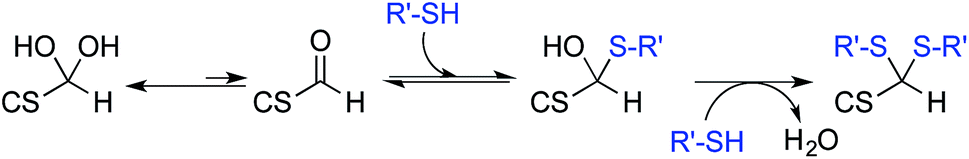
The only slightly higher reactivity of oxyamine moieties towards CS aldehyde as compared to CS amines, along with the necessity of an external chemical treatment to stabilize the products and the requirement of an organic co-solvent addition, constitute limitations of the oxime-click pathway. These limitations could be overcome by a thiol-based chemistry. Indeed, thiol moieties are highly reactive towards double bonds as well as towards carbonyl groups in aqueous conditions at pH values as low as 1 where CS amines are present only in the ionized and non-reactive form.30 Moreover, many equilibrium measurements have demonstrated the ability of thiols to add to the carbonyl group more efficiently than other nucleophiles (e.g., hydroxyls or amines) in both acid- and base-catalyzed pathways.31 Whereas amines produce Schiff base compounds (Fig. 1, reaction 3), thiols react with either aldehydes or ketones to produce hemithioacetals through a double equilibrium (Fig. 2). It is worth mentioning that the reactive species is the dehydrated carbonyl compound so that dehydration and hemithioacetal formation represent the rate-limiting steps of this pH-dependent process.30,32
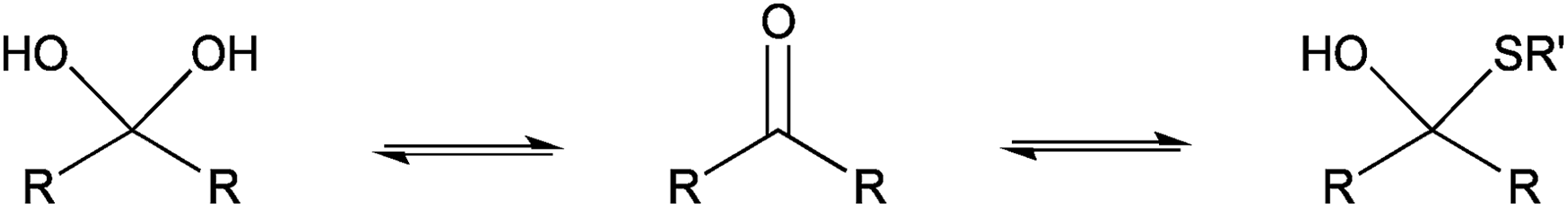 | ||
| Fig. 2 Schematic representation of the equilibria involved in thiol-carbonyl additions. | ||
Indeed, acid-catalyzed hemithioacetal formation takes place optimally below pH 330 and the final product is unstable under alkaline conditions, since the attack of hydroxide ions readily reverts the product to the starting reactants.33,34
By analogy with Schiff base formation where amines and carbonyls react to give an imino linkage (Fig. 1, reaction 3) that needs to be stabilized by reduction, hemithioacetals can be stabilized by thioacetal formation via a second thiol nucleophilic attack (intra- or inter-molecular) associated with the release of water.35 This chemical process is widely used in organic synthesis as a carbonyl group protection strategy; it is more conveniently performed in anhydrous organic solvent.36 To the best of our knowledge, such a strategy has not been implemented yet in aqueous conditions for polymer derivatization.
The main objectives of the present study were to determine which form of the aldehyde predominates on the CS end-group (i.e. hydrated vs. dehydrated form for a CS depolymerized using HONO) and to assess its reactivity towards thiol moieties in aqueous conditions. NMR spectroscopy experiments were performed in order to assess the availability of the CS aldehyde end-group after HONO depolymerization, since this issue has not been clearly addressed. The mechanism of stabilization of hemithioacetals by conversion to their corresponding thioacetals was also investigated by liquid chromatography-mass spectrometry (LC-MS) analysis of the products of the reaction between mannose and two small thiol-bearing molecules, namely 3-mercaptopropionic acid (MPA) and β-mercaptoethanol (BME). This process was then examined by reacting MPA and BME with a CS bearing an M-Unit end in aqueous conditions. The conjugation efficiency was determined by a combination of NMR and Ellman assays. Finally, the PEGylation of the CS M-Unit aldehyde by thioacetylation was examined as a direct application of this conjugation strategy. Fig. 3 summarizes the objectives and the hypotheses of our study. Of interest, although the unreactive hydrated gem-diol M-Unit aldehyde moieties are predominant in acidic aqueous conditions, the thiol species react preferentially with this M-Unit versus CS amines post-HONO depolymerization, therefore avoiding the M-Unit cleavage after rehydration of the freeze-dried product. The conjugation between the M-Unit and thiol species is followed by stabilization of the hemithioacetal intermediate into the corresponding thioacetal by a second thiol nucleophilic attack. By analogy with the Schiff base formation, freeze-drying can thus be implemented to favour the present reaction by water removal.
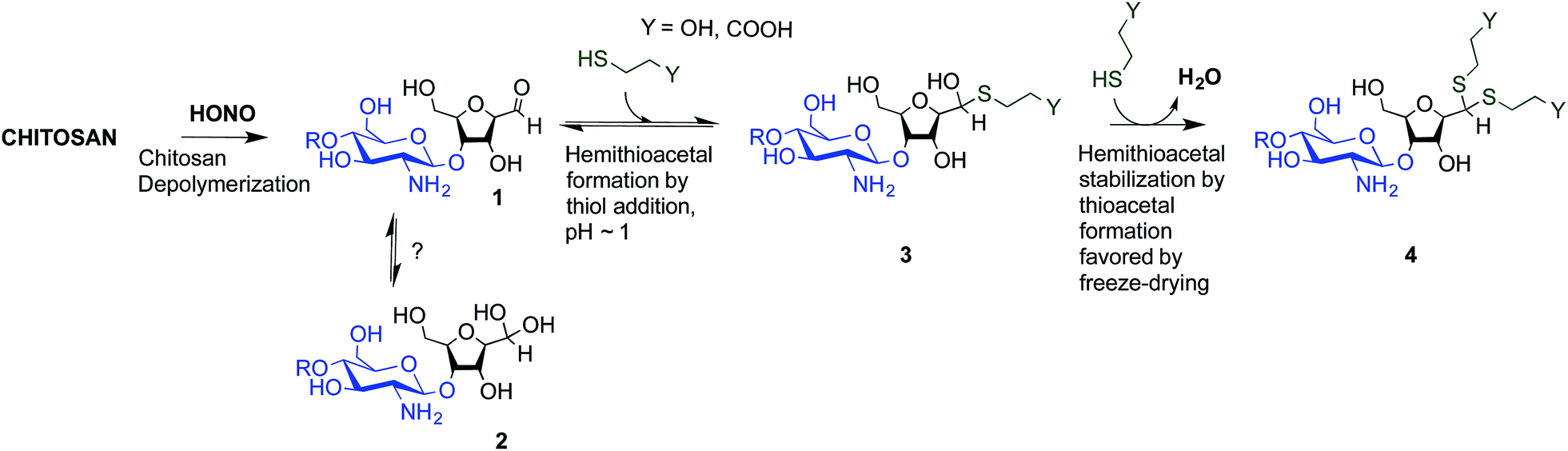 | ||
| Fig. 3 Thioacetal conjugation to the chitosan M-Unit formed post-HONO depolymerization: the first objective of this study was to assess the availability of the reactive form of the unhydrated M-Unit aldehyde (2). Although there could be a strong displacement of the equilibrium towards the unreactive aldehyde-hydrated or gem-diol form in water, we hypothesized that efficient nucleophilic conjugation to the M-Unit was possible in acidic aqueous conditions. The second objective was to assess the M-Unit reactivity towards thiol moieties in aqueous conditions. The proposed reaction pathways between CS end-groups and thiols include the M-Unit CS aldehyde reacting directly with a thiol-bearing model molecule (β-mercaptoethanol and 3-mercaptopropionic acid, BME and MPA respectively) to form a hemithioacetal intermediate (3) through a pH-dependent equilibrium. By analogy with Schiff base formation where the equilibrium displacement occurs by water removal, the hemithioacetal can be stabilized into the corresponding thioacetal (4) by freeze-drying. | ||
Materials and methods
Each chemical reaction was performed on at least three independent occasions (N = 3), in Ar degassed ddH2O and fresh reactants to minimize disulfide bond formation.Reagents, materials
Chitosan with a degree of deacetylation (DDA) of 91.7%, Mn = 193 kg mol−1 (PDI = 1.256) and 99.5%, Mn = 0.8 kg mol−1 (PDI = 1.245) was provided by Marinard Biotech Inc. Deuterium oxide (Cat #151882), deuterium chloride 35 wt% in deuterium oxide (Cat #543047), sodium nitrite (Cat #431605), hydrochloric acid standard solution 1.0 M in H2O (Cat #31894-9), hydrochloric acid 37% (Cat #320331), sodium hydroxide solution 1.0 M (Cat #319511), sodium acetate (Cat #241245), DTNB (5,5′-dithiobis-(2-nitrobenzoic acid)) (Cat #D8130), GlcNH2D-(+)-glucosamine hydrochloride 99% (Cat #C-1276), MPA (3-mercaptopropionic acid) ≥99% (Cat #63768), BME (β-mercaptoethanol) (Cat #M6250), sodium acetate trihydrate BioXtra (Cat #S7670), Dowex® 50WX8-100 [H+] (Cat #217506), Dowex® 1X8-50 [Cl−] (Cat #217417) and sodium azide (Cat #S2002) were purchased from Sigma-Aldrich. UltraPure™ TRIS (Cat #15504-020), glacial acetic acid (Cat #351271-212) and Spectra/Por®6 dialysis membrane (MWCO = 1000 Da, Cat #132640) were purchased from Life Technologies, Fisher Scientific and Spectrum Labs respectively. mPEG–SH 2 kDa and the plasmid DNA (pDNA) pEGFPLuc were purchased from JenKem Technology USA and from Clontech Laboratories, respectively.Aldehyde availability
:HONO molar ratio of 3. The mixture was stirred for 3 h at 50 °C. The pD (pD = pH + 0.4)37 of the depolymerization medium was ca. 1.9 at the end of the reaction.
1H NMR (ESI S1†) (500 MHz, D2O/DCl, 70 °C, ns = 2000, d1 = 6 s, acquisition time = 2 s) δ 2.06 (s, 1.38H, NHAc), 3.13–3.19 (br, 4.5H, H2D), 3.49–3.51 (br, 1H, H2A), 3.73–3.95 (m, 27H, H3–H6), 4.12–4.13 (q, J = 5.1 Hz, 1H, H5M), 4.22 (t, J = 3.9 Hz, 1H, H4M), 4.44 (t, J = 3.9 Hz, 1H, H3M), 4.58 (br, 0.5H, H1A), 4.79–4.88 (m, 4.5H, H1D), 5.09 (d, J = 5.3 Hz, 0.98H, H1M gem-diol).
SEC-MALLS: Mn = 823 (±41) g mol−1; Mw = 1024 (±28) g mol−1; PDI = 1.245 (±0.027).
Thiol reactivity towards M-Unit CS aldehyde
2,5-Anhydro-D-mannose (M-Unit) synthesis. 2,5-Anhydro-D-mannose was synthesized according to Claustre et al.38 Briefly, GlcNH2·HCl (5 mmol, 1 g) was dissolved in 25 mL degassed ddH2O and was allowed to stir overnight at room temperature. The colorless reaction medium was cooled down to 0 °C and NaNO2 (12.5 mmol, 862 mg) was added. Dowex® 50WX8-100 [H+] resin (42.5 mmol, 8.85 g dried, 25 mL) was added slowly under stirring and the heterogeneous mixture stirred for 4 h at 0–5 °C. The H+ resin was removed by filtration and the filtrate was neutralized with Dowex® 1X8-50 [CO32−] resin (60 mmol, 17.14 g dried, 50 mL), flash-frozen and freeze-dried to give the expected yellowish solid with 85% yield.
1H NMR (500 MHz, D2O, 25 °C, ns = 64, d1 = 6 s, acquisition time = 2 s) δ 3.36–3.40 (m, 2H, H6), 3.91–3.95 (m, 2H, H2 & H5), 4.05–4.08 (t, J = 5.6 Hz, 1H, H4), 4.18–4.21 (t, J = 5.8 Hz, 1H, H3), 5.09–5.10 (d, J = 5.4 Hz, 0.88H, H1 gem-diol), 8.46 (s, 0.12H, H1 aldehyde).
MS (ESI+): [M + H+] = 163.0625; [M + Na+] = 185.0460 (expected: [M + H+] = 163.0601; [M + Na+] = 185.0420).
2,5-Anhydro-D-mannose (M-Unit) conjugation with thiol-bearing molecules. The synthesized 2,5-anhydro-D-mannose M-Unit (0.1 mmol, 16.2 mg) was dissolved in 5 mL degassed ddH2O. The pH of the solution was adjusted to 1 with 3 M HCl solution prior to the addition of the thiol-bearing molecule (0.5 mmol, 53.2 μL for MPA and 35.1 μL for BME). The pH was readjusted to 1 with 3 M HCl solution. The reaction mixture was stirred for 72 h at 25 °C, under Ar atmosphere and covered with aluminum foil. The reaction mixture turned clear pink-orange after 72 h and was split into 3 parts (Methods I, II and III): the first was dedicated to the direct LC-MS analysis of the reaction medium in order to determine the thioacetal proportion in resulting conjugates that formed in situ; the second one was directly flash-frozen and then freeze-dried prior to LC-MS analyses to assess the effect of FD (freeze-drying) on the thioacetal proportion in resulting conjugates and to ascertain that no by-products appear post-FD, whereas the third was treated with 1 M acetate buffer pH 4 before flash-freezing and freeze-drying in order to determine by LC-MS the effect of an increase in pH on the resulting conjugates. It is worth mentioning that Method III was included to prevent any CS acid hydrolysis that could occur when Method II, i.e. FD at pH 1, would be transposed to the polymer.
:water (60:40 v/v) was used after each injection to reduce carry-over. Mass spectra were acquired for m/z ranging from 50 to 1200.
Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) experiments were performed using a Thermo Scientific Quantum Ultra triple quadrupole mass spectrometer operated in positive electrospray ion mode, equipped with a Thermo Scientific Surveyor liquid chromatography system. Xcalibur software (Thermo Scientific) was used to process the data. Separations were carried out on an XSELECT CSH™ C18 column (4.6 × 100 mm, 5 μm particles) from Waters operated under the same chromatographic gradients as those described above. MS/MS spectra were acquired on m/z values for protonated [M + H]+ and sodium adduct [M + Na]+ species of targeted compounds.
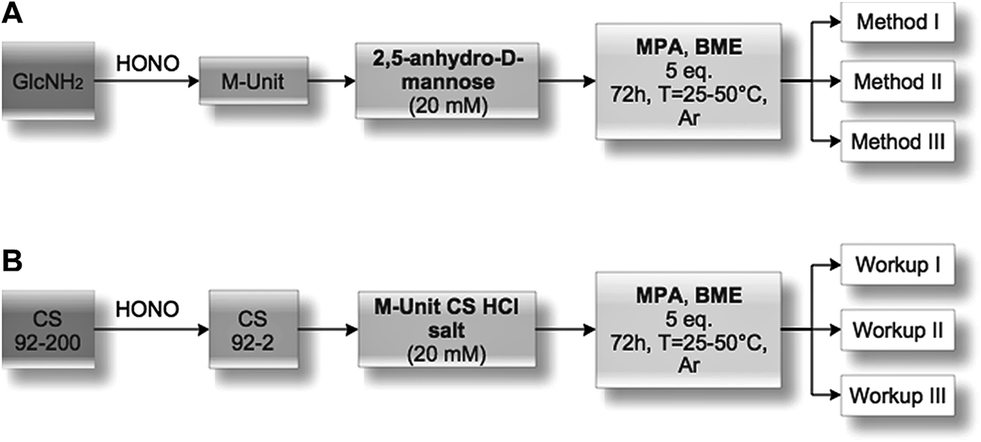 | ||
| Fig. 4 Experimental design flowchart. (A) Mechanistic studies. Glucosamine (GlcNH2) was treated with nitrous acid to form the 2,5-anhydro-D-mannose (M-Unit) that was reacted with 2 thiol-bearing molecules (β-mercaptoethanol and 3-mercaptopropionic acid, BME and MPA, respectively). The reaction products were treated using one of 3 methods, i.e. Method I: direct LC-MS analyses to determine to which extent thioacetal formation occurs in situ; Method II: freeze-drying (FD) + LC-MS analyses to assess the effect of FD on the thioacetal proportion and to ascertain that no by-products appear post-FD; Method III: acetate buffer pH 4 + FD + LC-MS analyses to determine the effect of an increase in pH prior to FD (this pH increase was included here to prevent any CS acid hydrolysis that could occur when Method II, i.e. FD at pH 1, would be transposed to the polymer). (B) Chitosan M-Unit reactivity. CS 92-200 was depolymerized with nitrous acid to produce CS 92-2 HCl salt bearing the M-Unit at the cleaved end of the polymer. M-Unit CS 92-2 HCl salt was reacted with MPA and BME and the reaction products treated with one of 3 workups: Workup I: dialysis vs. HCl 1 mM solution + FD to remove all thiol model excess and to determine the in situ thioacetal formation rate; Workup II: FD + dialysis vs. HCl 1 mM solution + FD to determine the effect of FD on the functionalization rate; Workup III: acetate buffer pH 4 + FD + dialysis vs. HCl 1 mM solution + FD to determine the effect of an increase in pH prior to FD on the functionalization rate (this pH increase was included to prevent any CS acid hydrolysis that could occur during FD at pH 1 in Workup II). The degree of functionalization of the CS conjugates was determined by 1H NMR, whereas covalent conjugation was assessed by DOSY NMR experiments and Ellman assays in order to rule out the possibility of a simple physical mixture of reagents. | ||
M-Unit CS 92-2 HCl salt synthesis. Chitosan was depolymerized using nitrous acid to achieve specific number-average molar mass targets (Mn) of 2 kg mol−1. For depolymerization, chitosan (1 g) was dissolved in 184.5 mL ddH2O and 9.54 mL HCl 1 M solution at 50 °C. Then 5.975 mL of fresh sodium nitrite solution (10 mg mL−1 in ddH2O obtained by solubilization of 76.5 mg NaNO2 in 7.65 mL ddH2O) were added to the completely dissolved CS to reach 0.5% (w/v) chitosan concentration. These conditions correspond to a GlcNH2
:HONO molar ratio of 6. The viscous colorless mixture was stirred for 3 h at 50 °C. The reaction medium was then dialyzed 5× against 4 L of an aqueous solution of HCl at pH 3 (HCl 1 mM solution) over 2 days. The resulting colorless solution was flash-frozen with liquid nitrogen and freeze-dried over 3 days to give the desired white powder with 60–70% yield.
1H NMR (500 MHz, D2O, 70 °C, ns = 64, d1 = 6 s, acquisition time = 2 s) δ 2.06 (s, 3.16H, NHAc), 3.14–3.21 (br, 13H, H2D), 3.51–3.56 (br, 1H, H2A), 3.68–3.95 (m, 70H, H3–H6), 4.12 (br, 1H, H5M), 4.21–4.31 (br, 1H, H4M), 4.43 (br, 1H, H3M), 4.61 (br, 1H, H1A), 4.87–4.89 (m, 13H, H1D), 5.08 (d, J = 5.0 Hz, 1H, H1M gem-diol).
SEC-MALLS: Mn = 2342 (±11) g mol−1; Mw = 3117 (±4) g mol−1; PDI = 1.332 (±0.008).
M-Unit CS 92-2 HCl salt conjugation with thiol-bearing molecules. CS 92-2 HCl salt (0.035 mmol, 70 mg) and thiol-bearing model molecules (0.175 mmol, 25.4 μL for MPA, 12.3 μL for BME) were solubilized in 1.73 mL degassed ddH2O. The pH of the reaction medium was adjusted to 1 with 3 M HCl. The reaction medium was stirred for 72 h at either 25 or 50 °C under Ar atmosphere. The resultant colorless liquid was directly flash-frozen with liquid nitrogen and then freeze-dried over 3 days. The freeze-dried white solid was solubilized in 5 mL ddH2O and dialyzed 5× against 2 L HCl 1 mM solution to remove unreacted thiols. The colorless solution was flash frozen and freeze-dried to give the expected white solid with typically 70–80% yield.
Addition of BME (ESI S2†): 1H NMR (500 MHz, D2O/DCl, 70 °C, ns = 64, d1 = 6 s, acquisition time = 2 s) δ 2.06 (s, 5.89H, NHAc), 2.91–2.95 (br, 2.78H, BME_CH2S), 3.17–3.21 (br, 20H, H2D), 3.51–3.53 (br, 1H, H2A), 3.69–3.95 (m, 105H, H3–H6), 4.12–4.14 (br, 1H, H5M), 4.24–4.25 (br, 1H, H4M), 4.57–4.59 (br, 1H, H3M), 4.61–4.62 (br, 1H, H1A), 4.91–4.92 (m, 20H, H1D), 5.08–5.09 (d, J = 5.0 Hz, 0.30H, H1M gem-diol).
SEC-MALLS: Mn = 3177 (±57) g mol−1; Mw = 3680 (±66) g mol−1; PDI = 1.160 (±0.003).
Addition of MPA (ESI S3†): 1H NMR (500 MHz, D2O/DCl, 70 °C, ns = 64, d1 = 6 s, acquisition time = 2 s) δ 2.06 (s, 4.18H, NHAc), 2.74–2.77 (t, J = 7.1 Hz, 2.33H, MPA_CH2–CO), 2.97–3.01 (q, J = 6.8 Hz, 1.78H, MPA_CH2S), 3.15–3.24 (br, 17H, H2D), 3.51–3.56 (br, 1H, H2A), 3.69–3.95 (m, 91H, H3–H6), 4.11 (br, 1H, H5M), 4.21–4.23 (br, 1H, H4M), 4.55 (br, 1H, H3M), 4.62 (br, 1H, H1A), 4.87–4.92 (m, 17H, H1D), 5.08 (d, J = 5.0 Hz, 0.47H, H1M gem-diol).
SEC-MALLS: Mn = 3053 (±81) g mol−1; Mw = 3564 (±48) g mol−1; PDI = 1.182 (±0.016).
Ellman assays. Thiol-derivatized CSs were analyzed using the Ellman assay to assess the presence of free thiols within the products. Ellman stock solutions (50 mM sodium acetate, 2 mM DTNB) were prepared by dissolving 39.7 mg of Ellman reagent and 205.1 mg of sodium acetate in 50 mL double deionized water (ddH2O). Tris 1 M dilution buffer was prepared dissolving 6.1 g of Tris in 50 mL ddH2O and adjusting the pH to 8.0 using HCl 1.0 M standard solution. Thiol concentrations were measured in triplicate by mixing 50 μL of Ellman stock solution with 100 μL of Tris dilution buffer and 10 μL of sample solution. After 15 min the mixture was diluted by the addition of 840 μL of ddH2O and the absorbance at 412 nm read using a microplate reader Tecan Infinite® M200. Thiol concentrations were calculated from a standard curve prepared using either MPA or BME and measurements were performed in triplicates in a 96-well plate using 150 μL sample volumes. The CS used as starting material was dissolved at the appropriate concentration for each sample and used as a blank. Both NaOH and Zn/HCl treatments of the CS adduct solutions were implemented on separate samples to determine the presence of hemithioacetal intermediates and any disulfide bond formation within the final product by the Ellman assay, respectively. Concentrated 1 M NaOH and 1 M HCl solutions were used to minimize changes in CS concentration. After 45–60 min constant agitation of the reaction media, Ellman assays were performed using 10 μL of alkali sample solution for NaOH treatment. Zn/HCl treated samples were obtained by adding few μL of 1 M HCl (to reach pH 1) and 5 equivalents of Zn dust per CS; the supernatants were analyzed after centrifugation (1000g for 1 min).
Characterization: NMR and SEC-MALLS. The deacetylation degree (DDA) of chitosan was determined by 1H NMR spectroscopy as previously described40 using a Bruker Avance 500 spectrometer equipped with a Bruker 5 mm BBFO probe. Cross-polarization magic-angle spinning (CPMAS) and Bloch-decay (BD) 13C NMR spectra were collected on a Bruker Avance 600 instrument equipped with a Bruker 4 mm BL4 CPMAS probe and samples were spun at the magic angle (54.7°) at a rate of 10–12 kHz. Diffusion ordered spectroscopy experiments (DOSY) were conducted on a Bruker II 400 equipped with a Bruker Diff30 probe, using 32 gradients between 11.2 and 358.4 gauss per cm with a gradient pulse (δ) of 1 ms, a diffusion time (Δ) of 60 ms.
Molar mass of starting 92% DDA chitosan was determined by size-exclusion chromatography (SEC) as previously described.41 Measurements were acquired using a gel permeation chromatography system equipped with an LC-20AD isocratic pump, SIL-20AC HT autosampler, and CTO-20AC oven (Shimadzu). This setup was coupled to the following detectors: Dawn HELEOS II multiangle laser light scattering, Viscostar II viscosimeter and Optilab rEX interferometric refractometer (Wyatt Technology Co.). The starting materials were eluted through two Shodex OHpak columns (SB-806M HQ and SB-805 HQ) connected in series with a mobile phase composed of 0.15 M acetic acid, 0.1 M sodium acetate, 0.4 mM sodium azide, 0.1 M NaCl, pH 4.5.42 A dn/dc value of 0.214 (DDA = 92%) was used and the number and weight average molar masses (Mn and Mw) of the CS starting materials were found to be 193 kg mol−1 and 242.5 kg mol−1 respectively.
Modified CS (depolymerized CS and thiol-coupled CSs) were analyzed in SEC using the same conditions but with columns SB-806M HQ and SB-803 HQ that are more suitable for the analysis of low molecular weight chitosans.
Quantitation of CS derivatization efficiency: functionalization degree (F) calculations. The functionalization degrees (F) of each conjugation were calculated according to the following equations:
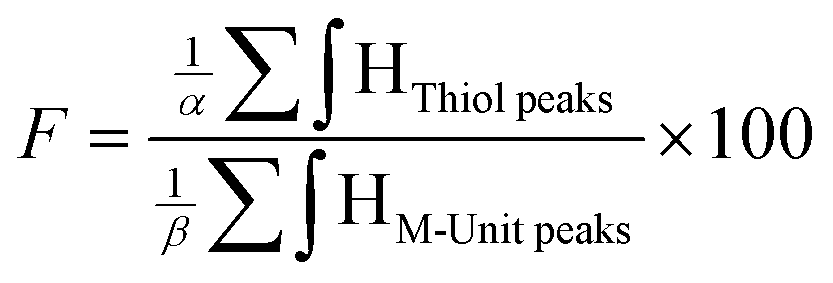 | (1) |
According to the mechanistic studies on the M-Unit model presented below, the hemithioacetal intermediate is fully stabilized into the corresponding thioacetal (as shown in Fig. 3, reaction 4, and Fig. 5) after freeze-drying of the reaction mixture in acidic conditions, thus two thiol-bearing molecules per M-Unit CS salt were considered for the calculation of the functionalization degree (F). For MPA adducts, two well-defined peaks corresponding to –CH2–S– and –CH2–CO– protons (i.e. 8 protons) appear on the NMR spectra. However, for BME adducts, only the –CH2–S– peak is visible on the spectra, in agreement with NMR spectrum simulation43 that predicts that the –CH2–CO– peak is hidden by the CS H3–H6 broad peaks.40 Thus, α values of 4 and 8 in eqn (1) where used for BME and MPA, respectively. For the M-Unit, the well-defined peaks corresponding to H4M and H5M protons were used for integration and a β value of 2 was thus used in the equation. From the above considerations, eqn (1) can be rewritten as eqn (2) and eqn (3) for BME and MPA conjugates, respectively:
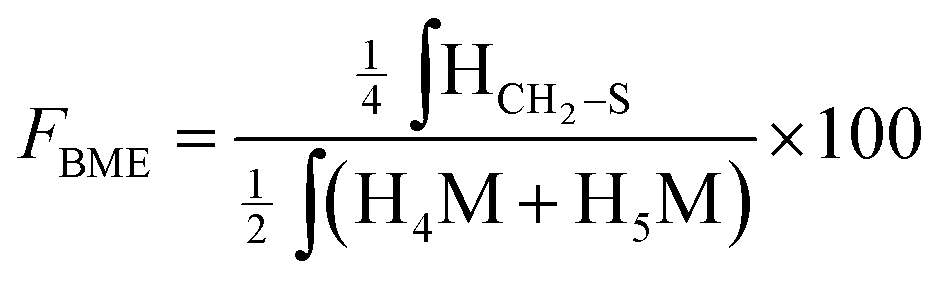 | (2) |
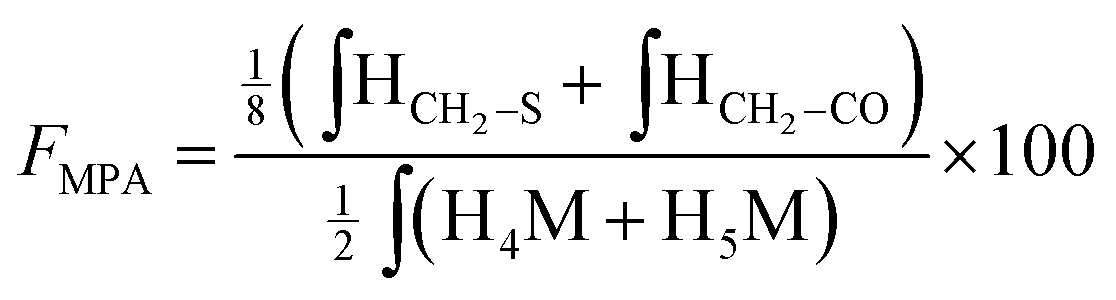 | (3) |
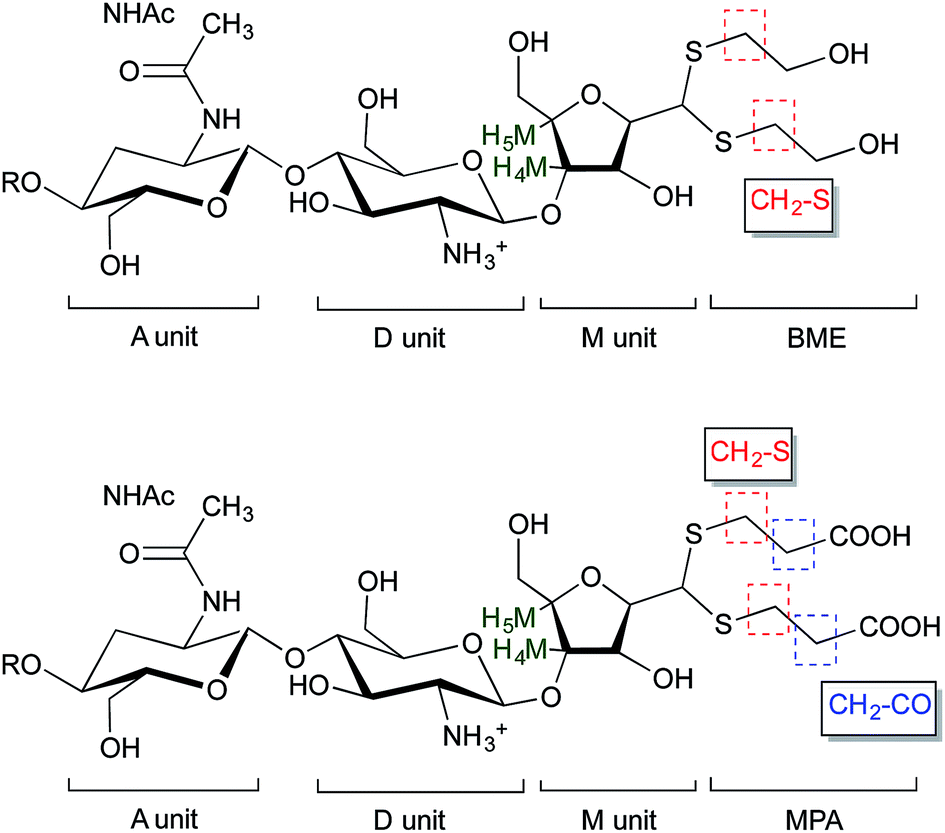 | ||
| Fig. 5 Structure of BME (top) and MPA (bottom) chitosan adducts. The protons corresponding to the 1H NMR peaks used for the calculations of the functionalization degree in eqn (2) and eqn (3) are highlighted. | ||
Similarly, the CS PEGylation efficiency (FPEG) was also calculated by adapting eqn (1) with the PEG characteristic peak integrations:
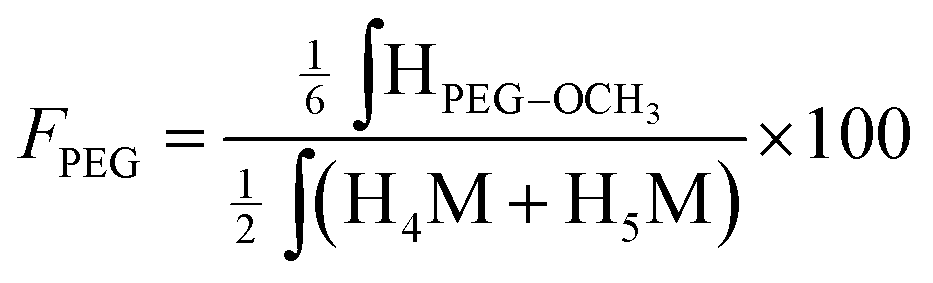 | (4) |
CS-b-PEG2 block-copolymer
M-Unit CS 92-10 HCl salt (m = 100 mg, 0.01 mmol, 5 mM aldehyde) was added to the reduced mPEG–SH solution and the pH of the reaction medium was adjusted to 1 with HCl 3 M solution. The reaction medium was stirred for 72 h at 50 °C, under Ar atmosphere. At the end of the reaction, the reaction medium was flash-frozen and freeze-dried. Unreacted mPEG–SH was discarded by reprecipitation in 5 × 45 mL CH2Cl2. The remaining white pellet was dried under reduced pressure overnight.
1H NMR (Fig. 8) (500 MHz, D2O, 70 °C, ns = 64, d1 = 6 s, acquisition time = 2 s) δ 2.06 (s, 11H, NHAc), 3.14–3.22 (br, 46H, H2D), 3.37 (s, 3.67H, PEG-OCH3), 3.51–3.56 (br, 3H, H2A), 3.69 (s, 181H, PEG Chain –O–CH2–CH2), 3.75–3.95 (m, 238H, H3–H6), 4.12–4.14 (br, 1H, H5M), 4.21–4.23 (br, 1H, H4M), 4.61 (br, 3H, H1A), 4.88–4.90 (m, 46H, H1D), 5.08 (d, J = 5.0 Hz, 0.49H, H1M gem-diol).
:HCl ratio of 1:1. Polymer stock solutions were diluted with ddH2O to reach the amine to phosphate ratio of 3.7 (N/P = 3.7) when equal volumes of chitosan and pDNA (100 μg mL−1) solutions would be mixed. Both CS-b-PEG2/pDNA and CS/pDNA polyplexes were prepared at room temperature, by adding 100 μL of the diluted polymer solution to 100 μL of the pDNA solution followed by immediate mixing by pipetting up and down. The polyplexes were analyzed for their size and morphology by dynamic light scattering (DLS) and environmental scanning electron microscopy (ESEM) 1 h after their formation.
Environmental scanning electron microscopy (ESEM) imaging of the polyplexes was performed as previously described45 using an environmental scanning electron microscope, Quanta 200 FEG (FEI Company Hillsboro, OR), operated in high vacuum mode with accelerating voltage = 20.0 kV; spot size = 3 and working distance = 5 mm.
Results and discussion
Aldehyde availability
Since hemithioacetal formation requires the dehydrated aldehyde as reactive species (referred to as aldehyde in this manuscript), the CS aldehyde availability was assessed by NMR spectroscopy.It has been reported that hydration of an aldehyde in the gas-phase can be observed at a relative humidity (RH%) level as low as 5%.48 The relative humidity of the laboratory where the experiments were performed was between 20 and 50%, and it could be that all aldehyde groups were transformed into gem-diols during the sample transfer and preparation. To eliminate the exposure to air humidity that might favor this formation of the gem-diol, an inert atmosphere solid-state NMR experiment was implemented on an extra-dried CS 99-1 salt (freeze-dried over 3 days and then dried using a Speed-Vac Plus centrifuge at 60 °C, overnight under reduced pressure). Sample preparation was performed within an Ar glove box to verify if air humidity transforms the CS terminal aldehyde into its corresponding hydrate. The solid-state NMR analysis was conducted under an inert atmosphere as well (constant N2 flow). Neither the aldehyde peak (expected around 190 ppm)49 nor the gem-diol peak (expected around 90 ppm)20 were visible on the spectrum. It is worth mentioning that the expected chemical shift of the gem-diol falls within the range of chemical shifts corresponding to C3–C5 peaks and the former is most probably hidden by the latter (ESI S4†). In order to confirm that the absence of the gem-diol in the spectrum was not due to an unexpected side reaction occurring in the preparation of the chitosan sample, the dried sample was subsequently dissolved in D2O and analyzed by standard 1H NMR. This analysis revealed that the hydrated aldehyde form was present at the expected quantitative proportion, as established from CS Mn and DDA values (data not shown).
Mechanisms of conjugation of 2,5-anhydro-D-mannose (M-Unit) and thiol-bearing molecules
The reactivity of aldehydes toward thiols in aqueous conditions was assessed semi-quantitatively by LC-MS using the 2,5-anhydro-D-mannose as an aldehyde model.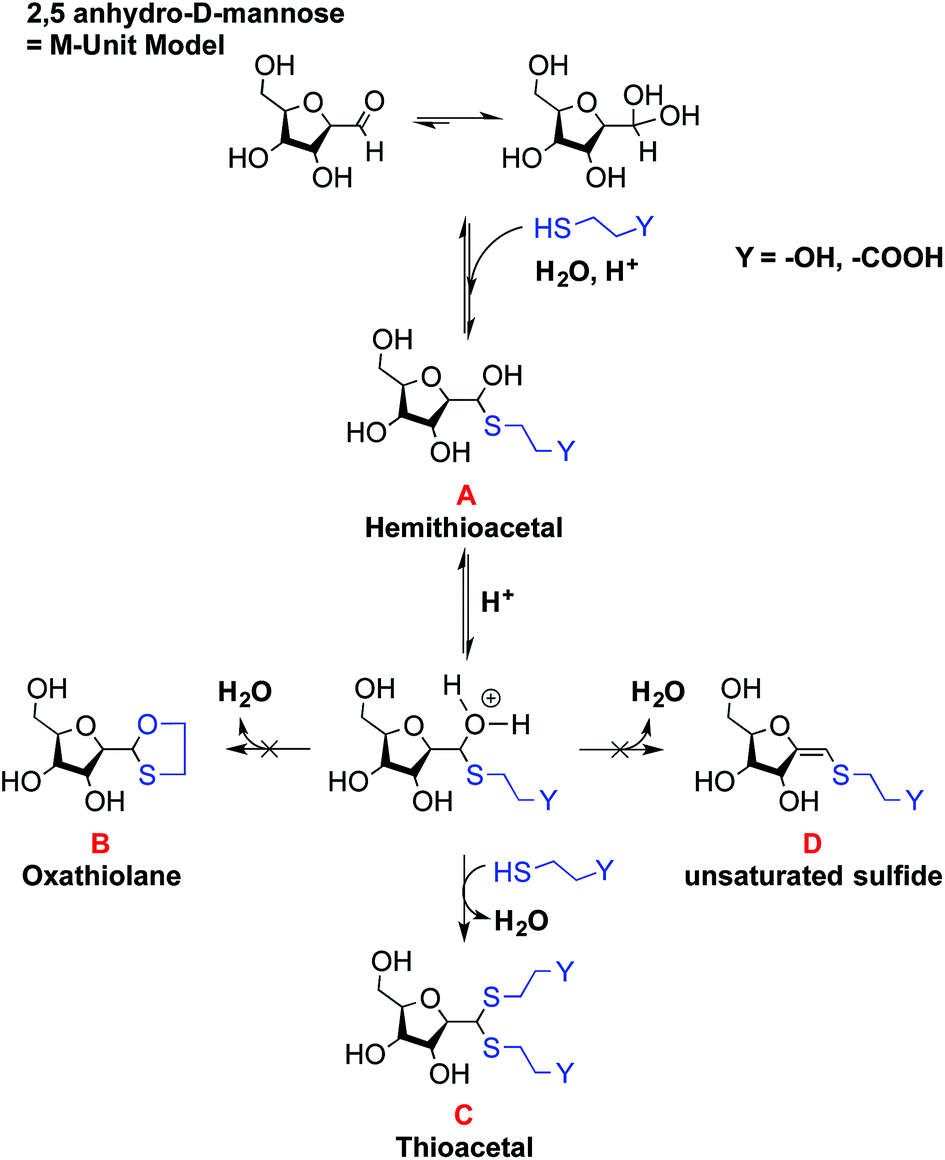 | ||
| Fig. 6 Schematic representation of potential reactions occurring during conjugation of 2,5-anhydro-D-mannose (M-Unit) and 2 thiol-bearing models (3-mercaptopropionic acid and β-mercaptoethanol, MPA and BME respectively) giving the following expected products: product A is the hemithioacetal intermediate that is in equilibrium with its corresponding oxonium, whereas products B and C correspond to the oxathiolane (for BME reactions only) and thioacetal, respectively. Molecule D represents the α,β-unsaturated sulfide. The results of this study suggest that the thioacetal C corresponds to the only stable form observed after freeze-drying. | ||
:1 ratio of hemithioacetal to thioacetal was observed for all thiol models (BME and MPA) tested. Thus the stabilization to the thioacetal intermediate A seems to occur with a second thiol nucleophilic attack to form the corresponding thioacetal C with the release of water. However, our results suggest that this stabilization occurs only to a relatively low extent in aqueous medium.
| Model | Final product (see Fig. 6) | Chemical formula | Expected m/z | Observed m/z | Relative proportion (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| [M + H]+ | [M + Na]+ | [M + H]+ | [M + Na]+ | Method I | Method II | Method III | |||
| M-Unit + BME | A | C8H16O6S | 241.0740 | 263.0560 | — | 263.0550 | 75 ± 13 | — | 4 ± 3 |
| B and D | C8H14O5S | 223.0635 | 245.0454 | — | (245.0450) | — | — | — | |
| C | C10H20O6S2 | 301.0774 | 323.0590 | 301.0884 | 323.0577 | 25 ± 13 | 100 | 96 ± 3 | |
| M-Unit + MPA | A | C9H16O7S | 269.0689 | 291.0509 | — | 291.0502 | 76 ± 3 | — | 18 ± 7 |
| C | C12H20O8S2 | 357.0672 | 379.0492 | — | 379.0483 | 24 ± 3 | 100 | 82 ± 7 | |
| D | C9H14O6S | 251.0584 | 273.0403 | (251.0563) | (273.0386) | — | — | — | |
It is worth mentioning that the LC-MS analyses only provide the relative proportion of observed species so that similar results obtained with both Methods II and III do not necessarily corresponds to equivalent absolute conversion rates. For instance, since the hemithioacetal formation equilibrium is pH-sensitive33 (increase in pH is known to displace the equilibrium towards the starting materials), the increased relative proportion of thioacetal C observed with Method III vs. Method I could be due to a reduction of the absolute amount of hemithioacetal A in the reaction mixture. The conversion degrees or functionalization degrees are calculated below by 1H NMR of the purified conjugated polymers.
The oxathiolane B and α,β-unsaturated sulfide products D appeared as traces in both Methods II and III (Table 1). LC-MS chromatograms revealed the same elution time as for thioacetals C, suggesting an in-source decomposition of B/C into their respective D form. The hypothesis that the oxathiolane B was formed within the MS apparatus by the ionization of the thioacetal C was confirmed by LC-MS/MS analyses of the C adduct obtained from the reaction of the M-Unit and MPA: the fragmentation of C produced compound D (data not shown).
These experiments suggest that the oxonium intermediate (which is in equilibrium with the hemithioacetal intermediate) is stable enough to favor the thioacetal formation notwithstanding the unsaturated compound D formation. The freeze-drying step apparently orients the reaction towards the stable thioacetal formation, more likely due to an increase in concentration by water removal to facilitate the second nucleophilic attack.
M-Unit chitosan HCl salt reactivity
The covalent nature of the conjugation of the CS HCl salt M-Unit to thiol-bearing molecules was confirmed by the Ellman assay where no free thiol moieties were detected after rehydration of the modified polymers. Note that free thiol moieties were not detected after Zn/HCl treatment that would have reduced any disulfide bond potentially formed in the course of the conjugation reaction and/or post-reaction workup. The absence of any hemithioacetal intermediate (base-sensitive) was also confirmed by performing the Ellman assay on the product after exposure to 1 M sodium hydroxide solution. Purified CS-thiol adducts were also analyzed by diffusion ordered spectroscopy (DOSY), a spectroscopic method that distinguishes compounds according to their respective translation diffusion coefficient (ESI S5†), shows that both CS and thiol-bearing models have the same diffusion coefficient in D2O at 25 °C, despite significant molar mass differences (2300 g mol−1vs. 106 g mol−1, for M-Unit CS HCl salt and MPA respectively). Altogether, the aforementioned controls confirmed the presence of the thioacetal linkage between the CS HCl salt M-Unit and both thiol-bearing model species. The results of the conjugation efficiencies between CS and BME or MPA were calculated using eqn (2) and (3) respectively and are summarized in Table 2.
| Thiol-bearing molecule | Temperature (°C) | Workup I | Workup II | Workup III | |||
|---|---|---|---|---|---|---|---|
| F (%) | F (%) | F (%) | F (%) | F (%) | F (%) | ||
| eqn (2) and (3) | eqn (5) | eqn (2) and (3) | eqn (5) | eqn (2) and (3) | eqn (5) | ||
| BME | 25 | 2 (±1) | 3 (±1) | 18 (±2) | 18 (±1) | 11 (±2) | 11 (±1) |
| 50 | 26 (±2) | 24 (±1) | 42 (±2) | 42 (±3) | 24 (±1) | 24 (±0) | |
| 68 (±1)* | 69 (±1)* | 70 (±1)* | 70 (±1)* | — | — | ||
| MPA | 25 | 10 (±1) | 11 (±1) | 18 (±2) | 19 (±2) | 15 (±1) | 13 (±2) |
| 50 | 14 (±1) | 13 (±1) | 54 (±5) | 55 (±2) | 18 (±1) | 17 (±1) | |
| 56 (±1)* | 55 (±1)* | 59 (±1)* | 58 (±1)* | — | — | ||
Despite the inability of these 2D NMR experiments to reveal the expected correlations, the combined NMR and LC-MS analysis indicated that two thiol-bearing molecules react regioselectively with the aldehyde of the terminal M-Unit of chitosan. As discussed above, the MS experiments performed with the mannose monomer indicated clearly that the stabilized form is the thioacetal form, so that, two thiols are expected to react similarly with the M-Unit of chitosan. This expected stoichiometry and regioselectivity for thiol-bearing molecules reacting on chitosan was validated by monitoring the relative proportion of gem-diol. Indeed, the gem-diol signal should decrease concomitantly with the conjugation of thiols onto the M-Unit of chitosan (one gem-diol consumed for two conjugated thiols). The calculated conjugation efficiencies obtained with either eqn (2) (BME) or eqn (3) (MPA) and the following equation should therefore be the same if the two thiols react regioselectively onto the terminal aldehyde function of chitosan:
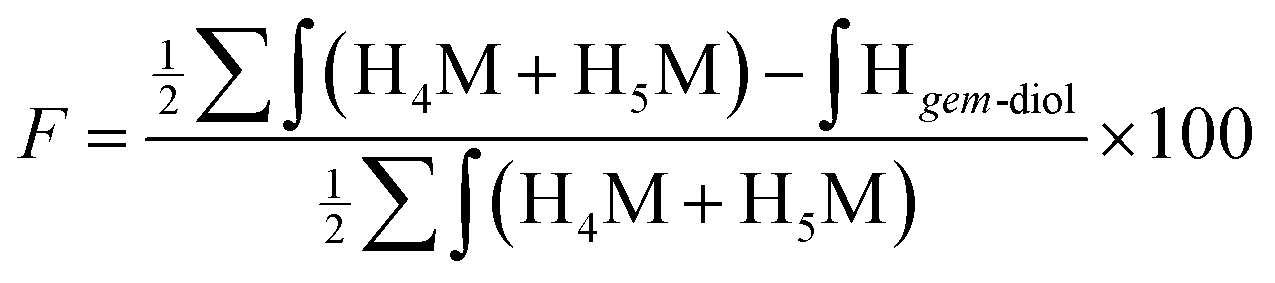 | (5) |
For all conjugation reactions performed in this study, the conjugation efficiencies calculated with both approaches, namely with eqn (2) (BME) or eqn (3) (MPA), which both rely on the reaction stoichiometry, or eqn (5) that is independent from stoichiometry and relies only on the relative proportion of gem-diol vs. M-Unit, were found to be in very close agreement (Table 2). These results indicate that (1) thiol-bearing molecules react selectively with the terminal aldehyde functional group of chitosan, and (2) the thioacetal is the only stable form of product observed.
 | ||
| Fig. 7 Thiol addition to the aldehyde group of the M-Unit CS HCl salt under acidic aqueous conditions: despite the fact that the aldehyde is only present in trace amounts within the reaction medium, the pH-dependent hemithioacetal intermediate formation equilibrium can be displaced by the intermediate stabilization into the corresponding thioacetal at low thiol concentration. | ||
Effective CS PEGylation by thioacetylation of the CS M-Unit aldehyde
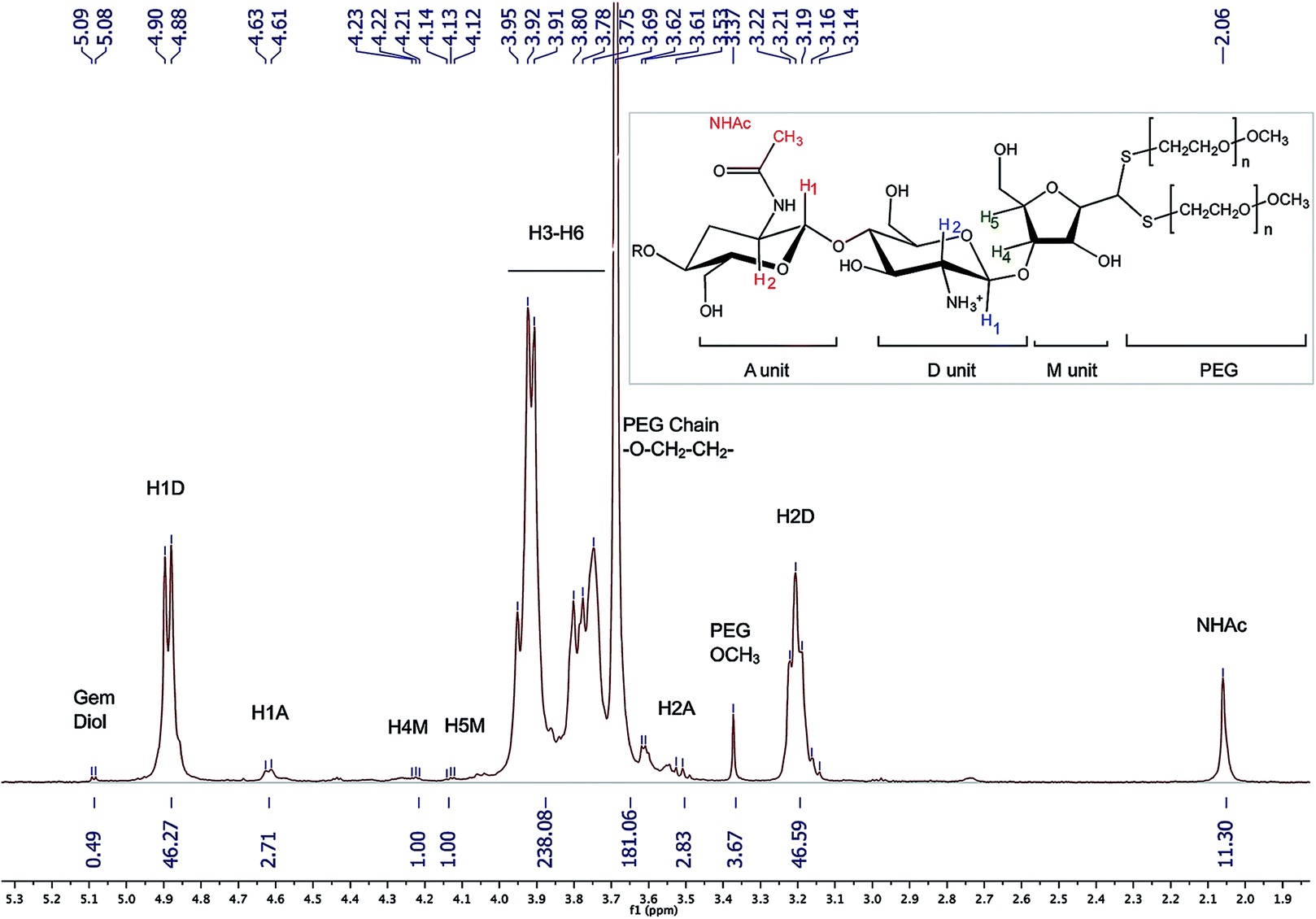 | ||
| Fig. 8 1H NMR spectrum of the CS-b-PEG2 block-copolymer after workup II (D2O, T = 70 °C, HOD peak was presaturated, number of scans (ns) = 64, relaxation period (d1) = 6 s, acquisition time = 2 s, exponential apodization = 1 Hz). Integration of gem-diol proton peak was used to calculate the functionalization degree (in this particular case, F = 51% according to eqn (5)). | ||
The slight discrepancy between these two values could possibly be the due to the presence of residual mPEG–SH post-purification. This hypothesis was confirmed by SEC analysis of the conjugates, where a small residual peak identified as mPEG–SS–PEGm was detected. Because PEG and CS molecular weights are close to each other, the DOSY NMR processing used to validate covalent conjugation of MPA and BME to CS was found to be inefficient for the block-copolymer (data not shown).
 | ||
| Fig. 9 Environmental scanning electron microscopy (ESEM) pictures (high vacuum mode, accelerating voltage = 20.0 kV; spot size = 3 and working distance = 5 mm) of polyplexes formed with pDNA and unmodified CS or CS-b-PEG2 block-copolymer (amine to phosphate ratio = 3.7, N/P = 3.7). (A and B) (×80000 and ×160000, respectively): polyplexes formed with CS 92-10 are heterogeneous in size and present various morphologies (globular, rod-like and toroidal). Pictures C and D (×80000 and ×160000, respectively): polyplexes formed with CS-b-PEG2 (CS 92-10 and mPEG–SH 2 kDa), are uniformly spherical. | ||
| Samples | Z-Average diameter/nm | PDI | Intensity-weighted mean diameter/nm |
|---|---|---|---|
| Unmodified CS polyplexes | 106 (±8) | 0.19 (±0.00) | 131 (±11) |
| CS-b-PEG2 polyplexes | 76 (±5) | 0.23 (±0.02) | 96 (±11) |
Since the PEGylated polyplexes are uniformly spherical and show a narrower size as compared to those prepared with corresponding homopolyions, these observations are consistent with the formation of micellar structures called “Block Ionomer Complexes” (BICs).57–59
Conclusions
This study revealed that the aldehyde present on chitosan mannose (M-Unit) end-group is displaced completely towards its hydrated and unreactive form (gem-diol) in aqueous conditions. The ubiquity of the unreactive gem-diol form in aqueous conditions revealed by 1H NMR (dehydrated reactive form not detected) could be due to both H-bonding and hydration effects. Despite the fact that the aldehyde reactive moiety is only present in trace amounts, the development and optimization of a thiol-based chemistry allowed efficient conjugation to the CS terminal M-Unit in aqueous conditions (F = 55–70% depending on the thiol-bearing molecule). A combination of mass spectrometry and NMR analyses revealed that two thiol-bearing molecules react regioselectively with the terminal aldehyde of the polymer to form a thioacetal. The stabilization of the hemithioacetal intermediate was found to be facilitated by freeze-drying (Fig. 10). As a direct application of this novel conjugation strategy, a CS-b-PEG2 block-copolymer was successfully synthesized by thioacetylation of the CS 92-10 M-Unit aldehyde with a 2 kDa mPEG–SH. This block-copolymer was used to prepare polyplexes with pDNA that were found to be uniformly spherical and more homogeneous as compared to those prepared with native CS.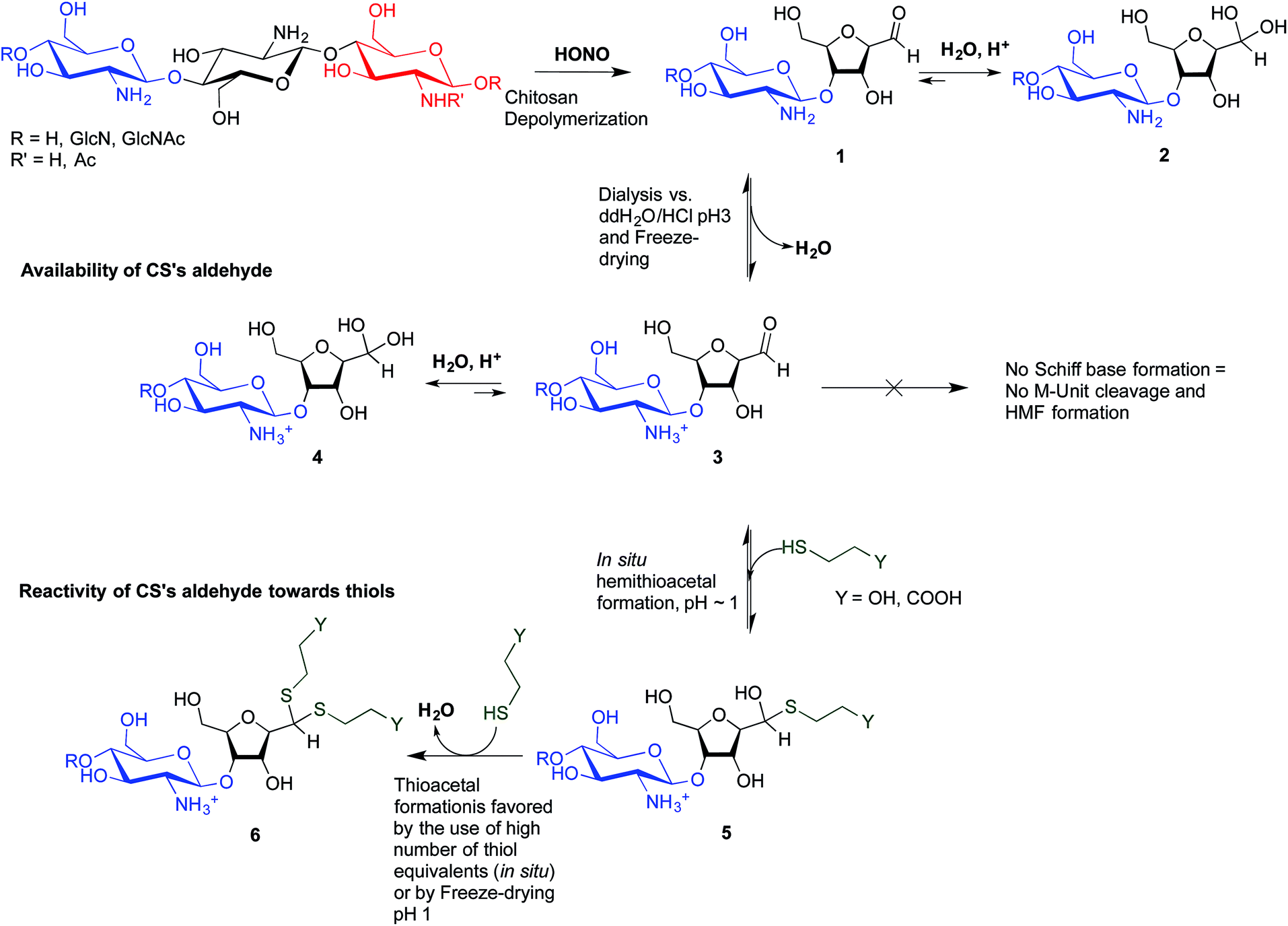 | ||
| Fig. 10 Summary of mechanisms elucidated in this study for thiol-based end-group derivatization of chitosans: CS nitrous acid depolymerization induces the formation of M-Unit that carries an aldehyde moiety at the end of the cleaved polymer (1). The equilibrium between the M-Unit aldehyde and its hydrated form (gem-diol) is strongly displaced towards the latter (2). If the CS depolymerization medium is freeze-dried at pH well below the CS pKa (i.e. pH ∼ 3–4 or below), all the CS amines are protonated and are therefore unable to react with any aldehyde group, maintaining the CS M-Unit integrity at the end of the cleaved polymer (3). Nevertheless, the equilibrium between the M-Unit aldehyde and the corresponding gem-diol is still displaced towards the hydrated form (4). Despite the undetectable aldehyde moieties, thiol molecules and the M-Unit CS aldehyde are engaged in a pH dependent equilibrium with the corresponding hemithioacetal intermediate (5). The stabilization of the latter into its thioacetal form (6) occurs either by increasing the amount of thiol-bearing reactants in the medium (in situ stabilization), or by freeze-drying the reaction medium when low amounts of thiol are engaged. | ||
The new CS end-group thioacetylation process that was developed in this study presents several advantages in comparison to the oxime-click method developed previously.16,18,24 That is (1) it can be used for CS derivatization without interfering with amine groups that are fully protonated and thus unreactive, (2) it is efficient in aqueous media, and (3) there is no need for an external chemical treatment to stabilize the adducts. It is worth mentioning that the stabilization of the hemithioacetal intermediate by a second nucleophilic attack could be sterically hindered by the presence of the first external group for large thiol-bearing substituents. In order to circumvent this issue and to further improve the conjugation efficiency, studies are ongoing where a molecule bearing two thiol groups (a thiol-based “hook”) is used for conjugation to the CS M-Unit. The presence of two thiol moieties along with their adequate positioning on the molecule to be conjugated may allow for an intramolecular stabilization of the hemithioacetal, which is expected to rule out any steric hindrance issues and to occur in situ at significantly lower thiol concentrations vs. the intermolecular stabilization studied herein.
CS end-group modifications such as PEGylation and the formation of other types of block-copolymers as well as CS grafting onto surfaces via a single covalent bond are a few applications of our proposed green chemistry protocol. These could be advantageously applied to various biomedical research fields including gene delivery and tissue engineering. Additionally, we expect this thiol-based chemistry to be applicable to other polymers bearing aldehydes or ketones.
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and ANRis Pharmaceuticals. The authors would like to acknowledge Monica Iliescu Nelea (Polytechnique Montréal), Alexandra Furtos (Université de Montréal) and Cédric Malveau (Université de Montréal) for the ESEM pictures and helpful discussions on mass spectrometry and NMR spectroscopy, respectively.Notes and references
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS PubMed.
- H. Sashiwa and S.-I. Aiba, Prog. Polym. Sci., 2004, 29, 887–908 CrossRef CAS PubMed.
- I. Aranaz, R. Harris and A. Heras, Curr. Org. Chem., 2010, 14, 308–330 CrossRef CAS.
- L. Casettari, D. Vllasaliu, E. Castagnino, S. Stolnik, S. Howdle and L. Illum, Prog. Polym. Sci., 2012, 37, 659–685 CrossRef CAS PubMed.
- M. Garcia-Fuentes and M. J. Alonso, J. Controlled Release, 2012, 161, 496–504 CrossRef CAS PubMed.
- M. D. Buschmann, A. Merzouki, M. Lavertu, M. Thibault, M. Jean and V. Darras, Adv. Drug Delivery Rev., 2013, 65, 1234–1270 CrossRef CAS PubMed.
- I. K. Park, T. H. Kim, Y. H. Park, B. A. Shin, E. S. Choi, E. H. Chowdhury, T. Akaike and C. S. Cho, J. Controlled Release, 2001, 76, 349–362 CrossRef CAS.
- O. Germershaus, S. Mao, J. Sitterberg, U. Bakowsky and T. Kissel, J. Controlled Release, 2008, 125, 145–154 CrossRef CAS PubMed.
- C. Zhang, Q. Ping, Y. Ding, Y. Cheng and J. Shen, J. Appl. Polym. Sci., 2004, 91, 659–665 CrossRef CAS PubMed.
- J. You, F. Q. Hu, Y. Z. Du and H. Yuan, Biomacromolecules, 2007, 8, 2450–2456 CrossRef CAS PubMed.
- J. H. Na, H. Koo, S. Lee, K. H. Min, K. Park, H. Yoo, S. H. Lee, J. H. Park, I. C. Kwon, S. Y. Jeong and K. Kim, Biomaterials, 2011, 32, 5252–5261 CrossRef CAS PubMed.
- H. Prichystalova, N. Almonasy, A. M. Abdel-Mohsen, R. M. Abdel-Rahman, M. M. Fouda, L. Vojtova, L. Kobera, Z. Spotz, L. Burgert and J. Jancar, Int. J. Biol. Macromol., 2014, 65, 234–240 CrossRef CAS PubMed.
- F. Lebouc, I. Dez, J. Desbrières, L. Picton and P.-J. Madec, Polymer, 2005, 46, 639–651 CrossRef CAS PubMed.
- K. Tommeraas, M. Koping-Hoggard, K. M. Varum, B. E. Christensen, P. Artursson and O. Smidsrod, Carbohydr. Res., 2002, 337, 2455–2462 CrossRef CAS.
- M. Morimoto, M. Nakao, N. Ishibashi, Y. Shigemasa, S. Ifuku and H. Saimoto, Carbohydr. Polym., 2011, 84, 727–731 CrossRef CAS PubMed.
- S. P. McManus, A. Kozlowski and P. D. Youso, Monoconjugated Chitosans as Delivery Agents for Small Interfering Nucleic Acids, US Pat., US 8916693, 2010 Search PubMed.
- S. K. Tripathi, R. Goyal, M. P. Kashyap, A. B. Pant, W. Haq, P. Kumar and K. C. Gupta, Biomaterials, 2012, 33, 4204–4219 CrossRef CAS PubMed.
- B. E. Benediktsdottir, K. K. Sorensen, M. B. Thygesen, K. J. Jensen, T. Gudjonsson, O. Baldursson and M. Masson, Carbohydr. Polym., 2012, 90, 1273–1280 CrossRef CAS PubMed.
- J. M. Los, L. B. Simpson and K. Wiesner, J. Am. Chem. Soc., 1956, 78, 1564–1568 CrossRef CAS.
- K. Tømmeraas, K. M. Vårum, B. E. Christensen and O. Smidsrød, Carbohydr. Res., 2001, 333, 137–144 CrossRef.
- E. P. Azevedo, S. V. Santhana Mariappan and V. Kumar, Carbohydr. Polym., 2012, 87, 1925–1932 CrossRef CAS PubMed.
- G. G. Allan and M. Peyron, Carbohydr. Res., 1995, 277, 257–272 CrossRef CAS.
- D. Filion, M. Lavertu and M. D. Buschmann, Biomacromolecules, 2007, 8, 3224–3234 CrossRef CAS PubMed.
- R. Novoa-Carballal and A. H. E. Muller, Chem. Commun., 2012, 48, 3781–3783 RSC.
- R. Novoa-Carballal, C. Silva, S. Moller, M. Schnabelrauch, R. L. Reis and I. Pashkuleva, J. Mater. Chem. B, 2014, 2, 4177–4184 RSC.
- T. O. Eloranta, A. R. Khomutov, R. M. Khomutov and T. Hyvonen, J. Biochem., 1990, 108, 593–598 CAS.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- T. S. Zatsepin, D. A. Stetsenko, A. A. Arzumanov, E. A. Romanova, M. J. Gait and T. S. Oretskaya, Bioconjugate Chem., 2002, 13, 822–830 CrossRef CAS PubMed.
- J. Shao and J. P. Tam, J. Am. Chem. Soc., 1995, 117, 3893–3899 CrossRef CAS.
- G. E. Lienhard and W. P. Jencks, J. Am. Chem. Soc., 1966, 88, 3982–3995 CrossRef CAS.
- E. G. Sander and W. P. Jencks, J. Am. Chem. Soc., 1968, 90, 6154–6162 CrossRef CAS.
- M. P. Schubert, J. Biol. Chem., 1936, 114, 341–350 CAS.
- R. E. Barnett and W. P. Jencks, J. Am. Chem. Soc., 1967, 89, 5963–5964 CrossRef CAS.
- R. Caraballo, H. Dong, J. P. Ribeiro, J. Jiménez-Barbero and O. Ramström, Angew. Chem., 2010, 122, 599–603 CrossRef PubMed.
- E. Campaigne, in Organic Sulfur Compounds, ed. N. Kharasch, Pergamon, Oxford, 1961, pp. 134–145 Search PubMed.
- T. W. G. Peter and G. M. Wuts, in Protective groups in organic synthesis, ed. Wiley, 2006, ch. 4, pp. 477–500 Search PubMed.
- R. Lumry, E. L. Smith and R. R. Glantz, J. Am. Chem. Soc., 1951, 73, 4330–4340 CrossRef CAS.
- S. Claustre, F. Bringaud, L. Azéma, R. Baron, J. Périé and M. Willson, Carbohydr. Res., 1999, 315, 339–344 CrossRef CAS.
- M. Erlandsson and M. Hällbrink, Int. J. Pept. Res. Ther., 2005, 11, 261–265 CrossRef CAS PubMed.
- M. Lavertu, Z. Xia, A. N. Serreqi, M. Berrada, A. Rodrigues, D. Wang, M. D. Buschmann and A. Gupta, J. Pharm. Biomed. Anal., 2003, 32, 1149–1158 CrossRef CAS.
- M. Lavertu, V. Darras and M. D. Buschmann, Carbohydr. Polym., 2012, 87, 1192–1198 CrossRef CAS PubMed.
- S. Nguyen, F. M. Winnik and M. D. Buschmann, Carbohydr. Polym., 2009, 75, 528–533 CrossRef CAS PubMed.
- D. Banfi and L. Patiny, Chimia, 2008, 62, 280–281 CrossRef CAS.
- M. Lavertu, S. Méthot, N. Tran-Khanh and M. D. Buschmann, Biomaterials, 2006, 27, 4815–4824 CrossRef CAS PubMed.
- Y. Niebel, M. D. Buschmann, M. Lavertu and G. De Crescenzo, Biomacromolecules, 2014, 15, 940–947 CrossRef CAS PubMed.
- R. Bell, Adv. Phys. Org. Chem., 1966, 4, 1–29 CAS.
- L. Heux, J. Brugnerotto, J. Desbrières, M. F. Versali and M. Rinaudo, Biomacromolecules, 2000, 1, 746–751 CrossRef CAS.
- J. L. Axson, K. Takahashi, D. O. De Haan and V. Vaida, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6687–6692 CrossRef CAS PubMed.
- H. Saito, R. Tabeta and K. Ogawa, Macromolecules, 1987, 20, 2424–2430 CrossRef CAS.
- E. M. Schulman, O. D. Bonner, D. R. Schulman and F. M. Laskovics, J. Am. Chem. Soc., 1976, 98, 3793–3799 CrossRef CAS.
- D. P. N. Satchell and R. S. Satchell, Chem. Soc. Rev., 1990, 19, 55–81 RSC.
- L. Fournier, G. Lamaty, A. Natat and J. P. Roque, Tetrahedron, 1975, 31, 1025–1029 CrossRef CAS.
- T. D. Claridge, High-resolution NMR techniques in organic chemistry, Newnes, 2008 Search PubMed.
- D. Pinto, C. M. Santos and A. M. Silva, Recent Research Developments in Heterocyclic Chemistry, Pinho e Melo, Research Signpost, Kerala, India, 2007 Search PubMed.
- A. Bax, K. A. Farley and G. S. Walker, J. Magn. Reson., Ser. A, 1996, 119, 134–138 CrossRef CAS.
- J. Furrer, Chem. Commun., 2010, 46, 3396–3398 RSC.
- A. V. Kabanov and V. A. Kabanov, Bioconjugate Chem., 1995, 6, 7–20 CrossRef CAS.
- I. K. Voets, A. de Keizer and M. A. Cohen Stuart, Adv. Colloid Interface Sci., 2009, 147–148, 300–318 CrossRef CAS PubMed.
- D. V. Pergushov, A. H. E. Muller and F. H. Schacher, Chem. Soc. Rev., 2012, 41, 6888–6901 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc00038f |
| This journal is © The Royal Society of Chemistry 2015 |