
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA convergent total synthesis of ouabagenin†
Ken
Mukai
,
Satoshi
Kasuya
,
Yuki
Nakagawa
,
Daisuke
Urabe
and
Masayuki
Inoue
*
Graduate School of Pharmaceutical Sciences, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: inoue@mol.f.u-tokyo.ac.jp
First published on 30th March 2015
Abstract
A convergent total synthesis of ouabagenin, an aglycon of cardenolide glycoside ouabain, was achieved by assembly of the AB-ring, D-ring and butenolide moieties. The multiply oxygenated cis-decalin structure of the AB-ring was constructed from (R)-perillaldehyde through the Diels–Alder reaction and sequential oxidations. The intermolecular acetal formation of the AB-ring and D-ring fragments, and combination of the intramolecular radical and aldol reactions, assembled the requisite steroidal skeleton in a stereoselective fashion. Finally, stereoselective installation of the C17-butenolide via the Stille coupling and hydrogenation led to ouabagenin.
Ouabain (Scheme 1), which belongs to a unique class of steroids known as cardenolides, has been used for the treatment of congestive heart failure for more than two centuries.1,2 Ouabain was isolated from the bark and roots of the ouabaio tree, and was more recently identified as an adrenal hormone that naturally occurs in mammals.3 The cardiac activity of ouabain is attributed to a high affinity inhibitory interaction with the membrane-bound sodium pump, Na+/K+-ATPase. As functional modulation of Na+/K+-ATPase activates multiple downstream signal transduction pathways, ouabain is implicated in the regulation of diverse physiological and pathological states. Importantly, cardenolides have received considerable attention for their preventive actions of proliferative diseases such as cancer.4
 | ||
| Scheme 1 Synthetic plan of ouabagenin (1). | ||
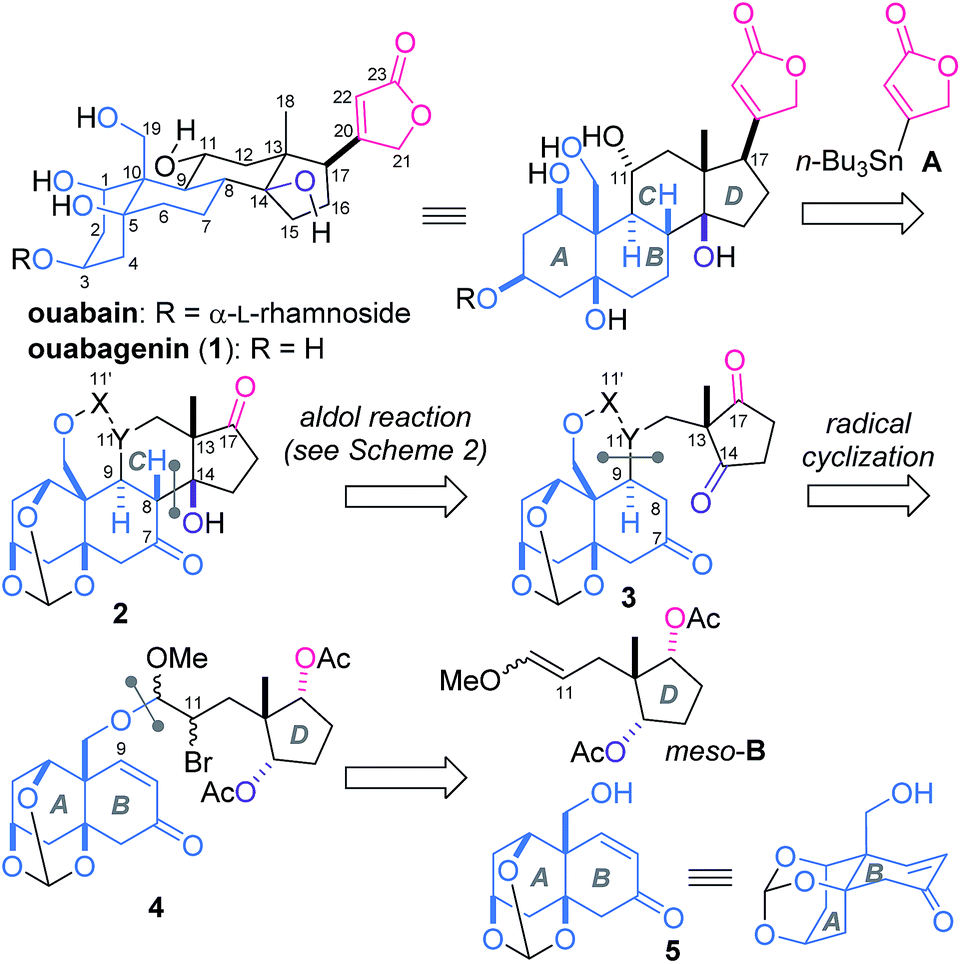
The steroidal skeleton of ouabagenin (1, Scheme 1), an aglycon of ouabain, possesses six hydroxy groups and the β-oriented butenolide. This highly oxygenated structure of 1 has posed a formidable synthetic challenge.5,6 Despite numerous synthetic studies on cardenolides, only two successful total syntheses of 1 have been reported to date. Deslongchamps constructed 1 through a polyanionic cyclization methodology and converted 1 to ouabain,7 while Baran designed the redox relay strategy and efficiently transformed adrenosterone to 1.8 Motivated by the architectural complexity coupled with the valuable bioactivities, we have launched synthetic studies of cardenolides based on a distinctive convergent strategy.9 Here we report the total synthesis of ouabagenin (1) by assembling the AB-ring fragment with the simple achiral D-ring and butenolide moieties.
The three-dimensional structure of 1 is disparate from the classical steroidal skeleton in that all the functional groups at the ring junctions (C5, 10, 13, 14) are β-configured (Scheme 1). This atypical ring fusion gives the nucleus a characteristic ‘U’ shape. The ring framework of 1 is further decorated by one primary (C19) and three secondary (C1, 3 and 11) hydroxy groups and an unsaturated lactone ring (C17). The congested disposition of these functionalities creates synthetic problems. To simplify the overall scheme, we retrosynthetically disassembled 1 into AB-ring 5, D-ring meso-B and butenolide A. According to this plan, achiral A10 and meso-B9 were easily prepared, while introduction of only four out of ten stereocenters of 1 would be necessary for the synthesis of 5. The remaining six centers were planned to be installed en route to 1. Most importantly, the stereochemistry of the four angular positions (C8, 9, 13, 14) was to be controlled by two intramolecular reactions. After tethering 5 and meso-B through the acetal linkage, 6-exo radical cyclization of 4 would establish the C9-stereochemistry. Aldol reaction of 3 would then be employed for simultaneous introductions of the C8-, 13- and 14-centers of 2. The two peripheral functional groups at C11 and 17 were planned to be constructed from 2 at the last stage of the total synthesis.
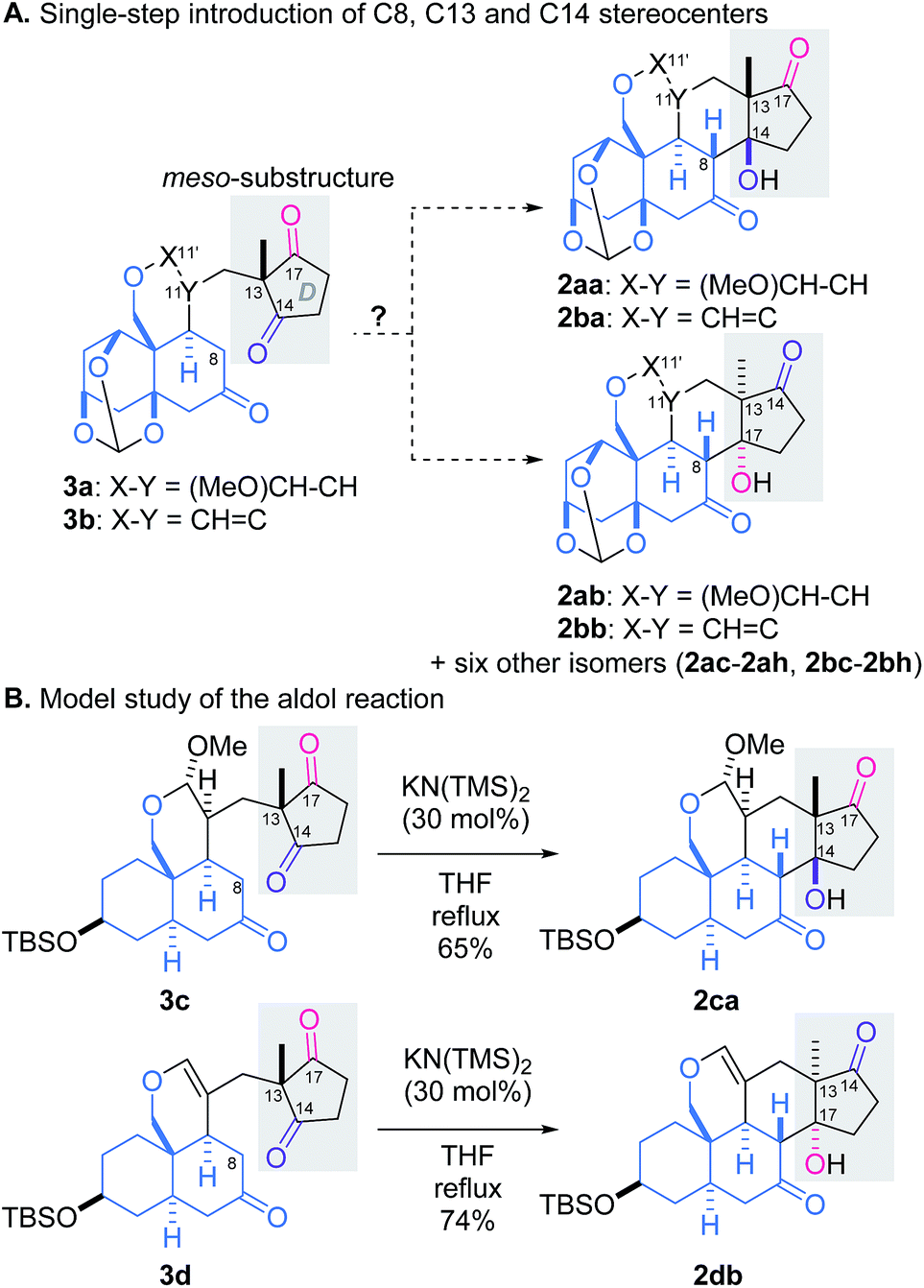
It would be challenging to control the three stereocenters in the reaction from 3 to 2: eight diastereomers would be potentially generated through desymmetrization of the meso-substructure (Scheme 2A).11,12 We envisioned that the linker structure (X–Y) would affect the stereochemical outcome of the aldol reaction, and accordingly designed the two possible intermediates, acetal 3a and vinyl ether 3b, which would be integrated in the planned scheme. To assess suitability of 3a or 3b as the precursor, the relative energies of the eight isomers of each aldol reaction were evaluated in silico. The PM6 calculation indicated that two products (2aa and 2ab from 3a, 2ba and 2bb from 3b) were thermodynamically more stable than the six other adducts.13 However, selectivity between desired 2aa/2ba and undesired 2ab/2bb was not clarified by the simulation due to their small energy difference (<0.5 kcal mol−1).
 | ||
| Scheme 2 Design of the substrates of Aldol reaction. | ||
To experimentally examine the role of the linker (X–Y), the synthetically more accessible models 3c and 3d were prepared and subjected to the aldol reactions (Scheme 2B).13 Despite their abbreviated structures, the trans-decalin rings of 3c and 3d were assumed to mimic the rigid orthoester-protected AB-ring of 3a and 3b, respectively. Treatment of acetal 3c with catalytic KN(TMS)2 in refluxing THF led to formation of desired 2ca as the sole isolable isomer, whereas application of the same conditions to vinyl ether 3d selectively afforded undesired 2db. These results demonstrated the crucial function of the linker in influencing the stereoselectivity, and permitted us to target acetal 3a as the key intermediate for the total synthesis.
Prior to the synthesis of 3a, AB-ring 5 was prepared from (R)-perillaldehyde 614 in 15 steps (Scheme 3). The Diels–Alder reaction between 6 and Rawal's diene C15 proceeded in regio- and diastereoselective manner to afford the cis-decalin, which was treated with aqueous acid to provide 7 as a single product.16 The two carbonyl groups of 7 were simultaneously reduced with LiAlH4 to produce diol 8, the allylic alcohol of which was chemoselectively oxidized with MnO2 to provide ketone 9. Treatment of 9 with TBSOTf and Et3N then introduced the two TBS groups at the C3-enol and C19-hydroxy groups, resulting in 10. Pd(OAc)2-mediated oxidation17 of 10 in turn provided dienone 11.18 For introduction of the β-configured C1- and 5-oxygen functional groups, the C3-β-alcohol was utilized as the directing group. Namely, ketone 11 was stereoselectively reduced by the action of the chiral reductant D19 to produce 12 (dr = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).20 Thus obtained 12 was treated with m-CPBA to induce the requisite bis-β-epoxidation in addition to the epoxidation at C7′, leading to tris-epoxide 13. After Dess–Martin oxidation21 of alcohol 13 to ketone 14, reductive opening of the two epoxides of 14 proximal to the C3-ketone was realized using Al/Hg6g,22 to generate 15. Next, DIBAL-H transformed 15 into tetraol 16 through stereoselective hydride attack on the C3-ketone from the opposite side of the C1,5-hydroxy groups and regioselective attack on the C7′-epoxide.
:1).20 Thus obtained 12 was treated with m-CPBA to induce the requisite bis-β-epoxidation in addition to the epoxidation at C7′, leading to tris-epoxide 13. After Dess–Martin oxidation21 of alcohol 13 to ketone 14, reductive opening of the two epoxides of 14 proximal to the C3-ketone was realized using Al/Hg6g,22 to generate 15. Next, DIBAL-H transformed 15 into tetraol 16 through stereoselective hydride attack on the C3-ketone from the opposite side of the C1,5-hydroxy groups and regioselective attack on the C7′-epoxide.
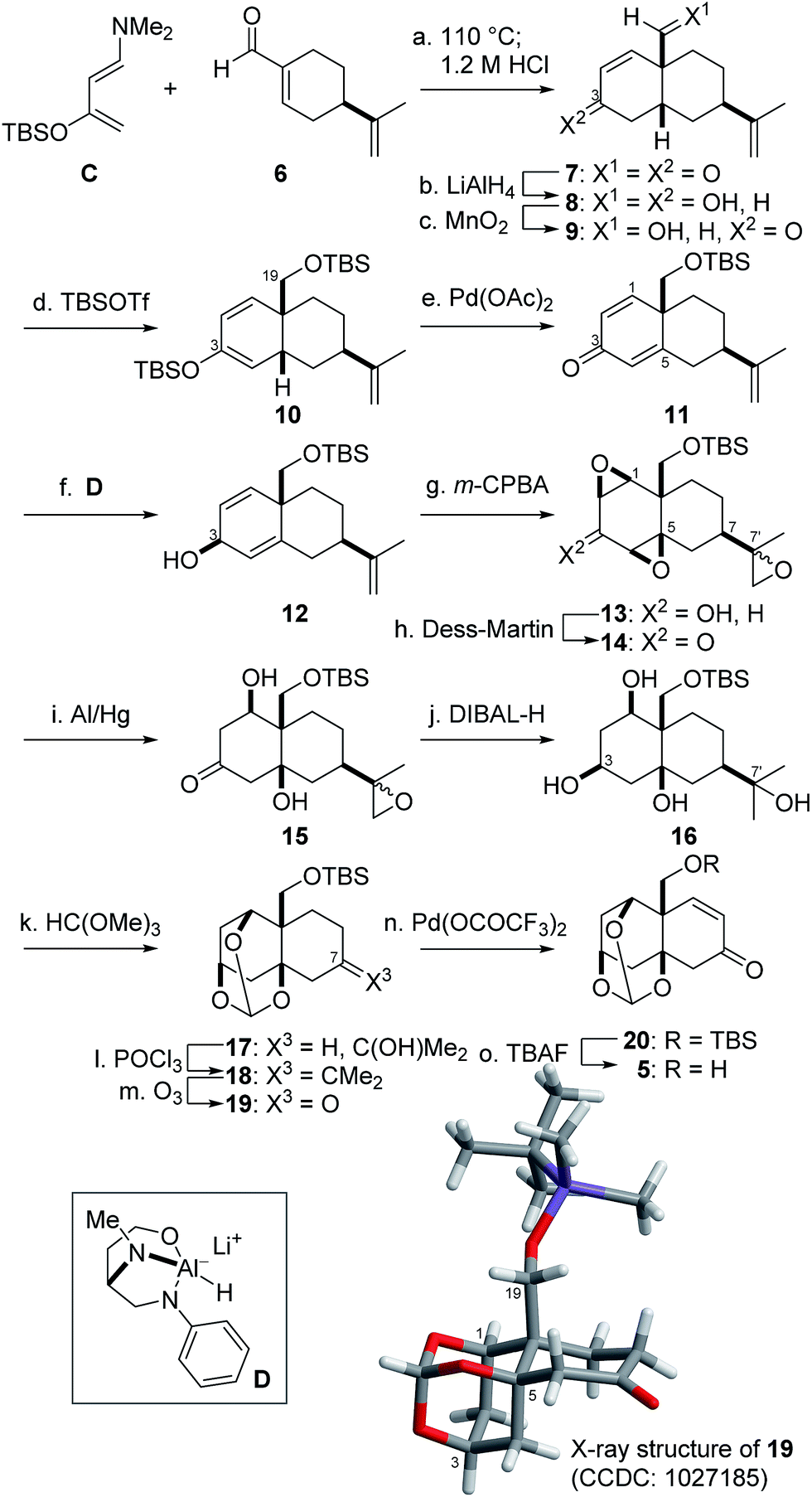 | ||
| Scheme 3 Stereoselective synthesis of AB-ring 5. Reagents and conditions: (a) toluene, 110 °C; 1.2 M aqueous HCl, THF, 67%; (b) LiAlH4, Et2O, −78 °C, 80%; (c) MnO2, CH2Cl2, 89%; (d) TBSOTf, Et3N, CH2Cl2, 0 °C; (e) Pd(OAc)2, CH3CN, 0 °C, 81% (2 steps); (f) D, t-BuOMe, Et2O, −100 to −40 °C, 68% for 12 (dr = 3:1); (g) m-chloroperbenzoic acid (m-CPBA), NaHCO3, CH2Cl2, 0 °C, 82%; (h) Dess–Martin reagent, NaHCO3, CH2Cl2, 0 °C, 100%; (i) Al/Hg, NaHCO3, THF, EtOH, H2O, 0 °C; (j) diisobutylaluminium hydride (DIBAL-H), CH2Cl2, –40 °C; (k) p-tolSO3H·pyridine, HC(OMe)3, DMF, 0 °C; (l) POCl3, pyridine, 0 °C; (m) O3, CH2Cl2, –78 °C; Me2S, 22% (5 steps); (n) Pd(OCOCF3)2, NaOAc, DMSO, 50 °C, 91%; (o) tetra-n-butylammonium fluoride (TBAF), AcOH, THF, 50 °C, 91%. | ||
Having established the C1,3,5-stereocenters, the AB-ring fragment 5 was prepared from 16 through five-step functional group manipulations (Scheme 3). After the three cis-hydroxy groups were protected as the orthoester, the remaining tertiary hydroxy group of 17 was regioselectively dehydrated using POCl3 to produce 18. Ozonolysis of tetrasubstituted olefin 18 gave rise to C7-ketone 19, the X-ray crystallographic analysis of which confirmed β-orientations of the three hydroxy and C19-hydroxy methyl groups. Finally, Pd(OCOCF3)2-promoted dehydrogenation,23 followed by TBAF-treatment, transformed 19 into the requisite 5.
We have thus reached a critical stage in the total synthesis, i.e., unification of the two fragments (5, meso-B) and stereoselective construction of the tetracyclic system by a combination of intramolecular radical and aldol reactions (Scheme 4). Before the coupling, the vinyl ether of D-ring meso-B was converted to dibromide E. The C19-hydroxy group of AB-ring 5 selectively displaced the O-activated C11′-bromo group of E in the presence of an acid scavenger PhNMe2, leading to acetal 4 as a diastereomixture. The remaining C11-bromo group of 4 was in turn utilized as a radical donor by treatment with Et3B/O224 and n-Bu3SnH. The acetal tether of 4 effectively constrained the generated radical to add from the β-face of the C9-olefin to provide 21 (C11,11′-diastereomixture).25 Saponification of the two acetates of 21 and subsequent Dess–Martin oxidation of 22 furnished the diastereomerically pure triketone 3a after chromatographic purification (26% yield from 5). As expected from the computational simulation and the model experiments in Scheme 2, the acetal-tethered 3a was selectively converted into the desired isomer 2aa. Specifically, treatment of 3a with KN(TMS)2 (30 mol%) in refluxing THF delivered desired 2aa (65%) along with its minor diastereomer 2ab (8%). Of note, this mild five-step sequence realized introduction of four new stereocenters (C8, 9, 13 and 14) without affecting the pre-existing oxygen functionalities.
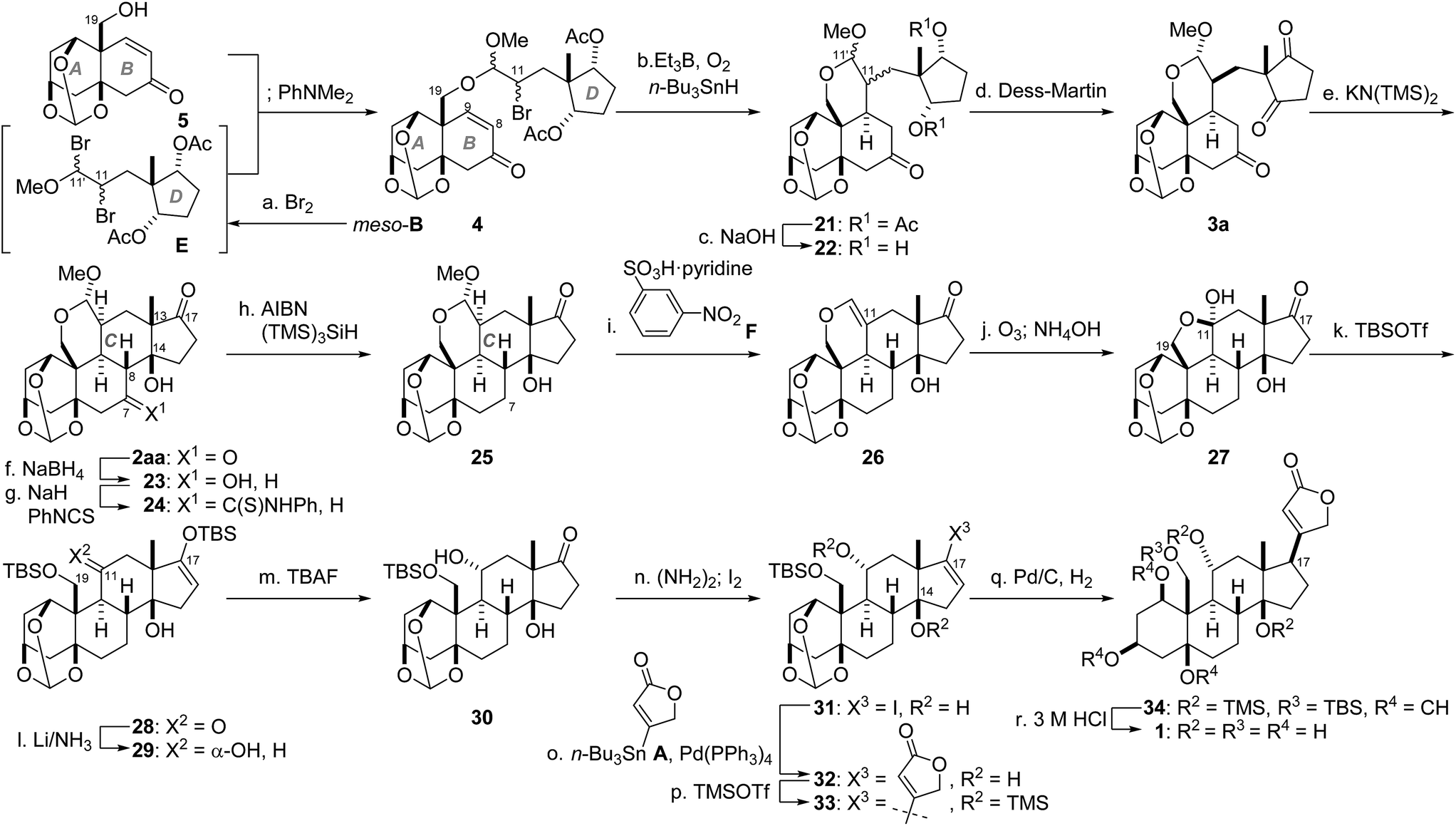 | ||
| Scheme 4 Total synthesis of ouabagenin (1). Reagents and conditions: (a) meso-B (2.5 equiv.), Br2, CH2Cl2, −78 °C; 5 (1.0 equiv.), PhNMe2; (b) Et3B, n-Bu3SnH, O2, toluene; (c) NaOH, MeOH, H2O; (d) Dess–Martin reagent, NaHCO3, CH2Cl2, 26% for 3a (4 steps from 5); (e) KN(TMS)2 (30 mol%), THF, reflux, 65% for 2aa, 8% for 2ab; (f) NaBH4, THF; (g) NaH, H2O, PhNCS, THF; (h) AIBN, (TMS)3SiH, benzene, reflux, 39% (3 steps); (i) F, toluene, reflux, 81%; (j) O3, MeOH, −78 °C; Me2S; NH4OH; (k) TBSOTf, Et3N, CH3CN, −35 to 0 °C, 68% (2 steps); (l) Li, NH3, THF, −78 °C, 96%; (m) TBAF, THF, −78 °C, 99%; (n) (NH2)2, Et3N, EtOH, 50 °C; I2, Et3N, THF, 89%; (o) A (3.0 equiv.), Pd(PPh3)4, LiCl, CuCl, DMSO, 60 °C; (p) TMSOTf, 2,6-lutidine, CH2Cl2; SiO2, 58% (2 steps); (q) H2, Pd/C, EtOAc; (r) 3 M aqueous HCl, MeOH, 56% (2 steps). | ||
Having constructed the properly substituted steroid, ouabagenin (1) was synthesized from 2aa through adjustments of the C7,11-functional groups, attachment of the C17-butenolide and global deprotection. First, the C7-oxygen functionality was removed. Chemoselective NaBH4 reduction at C7 of diketone 2aa afforded alcohol 23, which was derivatized into thiocarbamate 24 by means of thioisocyanate and NaH.26,27 Reductive cleavage of the C7-thiocarbamate of 24 was realized by mixing with AIBN and (TMS)3SiH in refluxing benzene, providing 25. Next, the α-oriented C11-hydroxy group was constructed from the C11-branched carbon linker, which served as the stereocontrolling element. Before doing so, the acetal of 25 was converted to the vinyl ether of 26 by acid-promoted elimination of MeOH.28 Oxidative scission of the resultant C11-olefin of 26 with ozone, reductive workup and the basic deformylation at C19-OH gave hemiacetal 27. TBSOTf and Et3N then introduced TBS groups at both C19- and C17-oxygen atoms of 27, liberating the C11-ketone of 28. Subjection of 28 to Birch reduction conditions resulted in formation of 29 with the thermodynamically favorable equatorial C11-OH group.
The β-oriented C17-butenolide was incorporated through the Stille coupling reaction29 and stereoselective hydrogenation (Scheme 4). Treatment of bis-TBS ether 29 with TBAF at −78 °C resulted in formation of C17-ketone 30, which was in turn transformed to vinyl iodide 31via the hydrazone intermediate.30 The Stille coupling between 31 and A proceeded in the presence of Pd(PPh3)4 and CuCl in DMSO,31 leading to 32 bearing the entire carbon structure of 1. To achieve hydrogenation from the concave α-face of the ‘U’-shape skeleton, the C14-hydroxy group of 32 was capped with the bulky TMS-ether to generate 33. Presumably because the C14-OTMS blocked the approach of reagents from the β-face, Pd/C-catalyzed hydrogenation of 33 occurred selectively from the α-face to yield 34 as the major product. Finally, global deprotection of 34 with 3 M HCl in MeOH removed the two TMS, one TBS and one methine groups, giving rise to ouabagenin (1). The analytical data of synthetic 1 including the optical rotation, IR, 1H-, 13C-NMR and HRMS were identical with those of authentic 1.
In conclusion, we achieved the convergent total synthesis of ouabagenin (1) using highly functionalized AB-ring 5 together with the simple achiral D-ring meso-B and butenolide A (33 steps from (R)-perillaldehyde 6). After preinstalling the four stereocenters of 5, introduction of the six remaining stereocenters and construction of the entire pentacyclic structure of 1 required only 18 steps, demonstrating the efficiency of the strategy. The key transformations that enabled the convergent synthesis include: (i) the mild acetal formation between AB-ring 5 and dibromide E; (ii) the tether-guided 6-exo radical cyclization of 4; (iii) the stereoselective aldol reaction of triketone 3a that was judiciously designed based on in silico and model experiments; (iv) installation of the C17-butenolide by the Stille coupling using A; (v) the stereoselective hydrogenation of C17-double bond; and (vi) the single-step global deprotection. Because of its flexibility and robustness, the present route would be applicable for the synthesis of a variety of bioactive cardenolides, and synthetic and SAR studies of such molecules will be our next focus.
Acknowledgements
This research was financially supported by the Funding Program for a Grant-in-Aid for Scientific Research (A) (JSPS) to M.I., and (C) (JSPS) and on Innovative Areas (MEXT) to D.U. A fellowship to K.M. and S.K. from JSPS is gratefully acknowledged. We thank Naoto Aoki for preparation of 2db, and Dr Kimiko Hasegawa (Rigaku Corporation) for X-ray crystallographic analyses.Notes and references
- L. F. Fieser and M. Fieser, Steroids, Reinhold, New York, 1959, p. 727 Search PubMed.
- S. H. Rahimtoola and T. Tak, Curr. Probl. Cardiol., 1996, 21, 781 CrossRef CAS PubMed.
- A. Y. Bagrov, J. I. Shapiro and O. V. Fedorova, Pharmacol. Rev., 2009, 61, 9 CrossRef CAS PubMed.
- (a) I. Prassas and E. P. Diamandis, Nat. Rev. Drug Discovery, 2008, 7, 926 CrossRef CAS PubMed; (b) R. A. Newman, P. Yang, A. D. Pawlus and K. I. Block, Mol. Interventions, 2008, 8, 36 CrossRef CAS PubMed; (c) J. A. R. Salvador, J. F. S. Carvalho, M. A. C. Neves, S. M. Silvestre, A. J. Leitão, M. M. C. Silva and M. L. Sá e Melo, Nat. Prod. Rep., 2013, 30, 324 RSC.
- For recent reviews on total synthesis of steroids, see: (a) F. C. E. Sarabèr and A. de Groot, Tetrahedron, 2006, 62, 5363 CrossRef; (b) A.-S. Chapelon, D. Moraléda, R. Rodriguez, C. Ollivier and M. Santelli, Tetrahedron, 2007, 63, 11511 CrossRef CAS; (c) B. Heasley, Chem.–Eur. J., 2012, 18, 3092 CrossRef CAS PubMed.
- Total syntheses of digitoxigenin: (a) G. Stork, F. West, H. Y. Lee, R. C. A. Isaacs and S. Manabe, J. Am. Chem. Soc., 1996, 118, 10660 CrossRef CAS; (b) M. Honma and M. Nakada, Tetrahedron Lett., 2007, 48, 1541 CrossRef CAS; total synthesis of rhodexin A: (c) M. E. Jung and D. Yoo, Org. Lett., 2011, 13, 2698 CrossRef CAS PubMed; semi synthesis of digitoxin: (d) K. Wiesner and T. Y. R. Tsai, Pure Appl. Chem., 1986, 58, 799 CrossRef CAS; Semi synthesis of strophanthidin: (e) P. Kočovský and I. Stieborová, Tetrahedron Lett., 1989, 30, 4295 CrossRef; recent synthetic studies of ouabagenin: (f) L. E. Overman and P. V. Rucker, Tetrahedron Lett., 1998, 39, 4643 CrossRef CAS; (g) M. E. Jung and G. Piizzi, Org. Lett., 2003, 5, 137 CrossRef CAS PubMed.
- Total synthesis of 1 from Hajos–Parrish ketone (41 steps): (a) H. Zhang, M. S. Reddy, S. Phoenix and P. Deslongchamps, Angew. Chem., Int. Ed., 2008, 47, 1272 CrossRef CAS PubMed; (b) M. S. Reddy, H. Zhang, S. Phoenix and P. Deslongchamps, Chem.–Asian J., 2009, 4, 725 CrossRef CAS PubMed.
- Total synthesis of 1 from adrenosterone (20 steps): (a) H. Renata, Q. Zhou and P. S. Baran, Science, 2013, 339, 59 CrossRef CAS PubMed; (b) H. Renata, Q. Zhou, G. Dünstl, J. Felding, R. R. Merchant, C.-H. Yeh and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 1330 CrossRef CAS PubMed.
- We recently reported the total synthesis of 19-hydroxysarmentogenin, one of the cardenolides, K. Mukai, D. Urabe, S. Kasuya, N. Aoki and M. Inoue, Angew. Chem., Int. Ed., 2013, 52, 5300 CrossRef CAS PubMed.
- G. J. Hollingworth, G. Perkins and J. Sweeney, J. Chem. Soc., Perkin Trans. 1, 1996, 1913 RSC.
- For use of desymmetrization strategy for total synthesis, see: (a) C. S. Poss and S. L. Schreiber, Acc. Chem. Res., 1994, 27, 9 CrossRef CAS; (b) S. R. Magnuson, Tetrahedron, 1995, 51, 2167 CrossRef CAS; (c) R. W. Hoffmann, Angew. Chem., Int. Ed., 2003, 42, 1096 CrossRef CAS PubMed; (d) M. Vrettou, A. A. Gray, A. R. E. Brewer and A. G. M. Barrett, Tetrahedron, 2007, 63, 1487 CrossRef CAS; (e) M. Inoue and D. Urabe, Strategies Tactics Org. Synth., 2013, 9, 149 Search PubMed; (f) M. Wang, M. Feng, B. Tang and X. Jiang, Tetrahedron Lett., 2014, 55, 7147 CrossRef CAS.
- Although related intramolecular reactions using the meso-D-ring substructure were reported, one-step construction of the cardenolide ring system had been only achieved in our total synthesis of 19-hydroxysarmentognin (ref. 9). For representative examples, see: (a) S. N. Ananchenko and I. V. Torgov, Tetrahedron Lett., 1963, 4, 1553 CrossRef; (b) A. R. Daniewski, M. M. Kabat, M. Masnyk, J. Wicha, W. Wojciechowska and H. Duddeck, J. Org. Chem., 1988, 53, 4855 CrossRef CAS; (c) S. P. Douglas, J. F. Sawyer and P. Yates, Tetrahedron Lett., 1985, 26, 5955 CrossRef CAS; (d) J.-F. Lavallée and P. Deslongchamps, Tetrahedron Lett., 1988, 29, 6033 CrossRef; (e) R. Ruel and P. Deslongchamps, Can. J. Chem., 1990, 68, 1917 CrossRef CAS; (f) D. Chapdelaine, J. Belzile and P. Deslongchamps, J. Org. Chem., 2002, 67, 5669 CrossRef CAS PubMed; (g) D.-H. Jhuo, B.-C. Hong, C.-W. Chang and G.-H. Lee, Org. Lett., 2014, 16, 2724 CrossRef CAS PubMed; (h) S. Prévost, N. Dupré, M. Leutzsch, Q. Wang, V. Wakchaure and B. List, Angew. Chem., Int. Ed., 2014, 53, 8770 CrossRef PubMed.
- See ESI†.
- M. A. Tius and M. A. Kerr, Synth. Commun., 1988, 18, 1905 CrossRef CAS.
- S. A. Kozmin, J. M. Janey and V. H. Rawal, J. Org. Chem., 1999, 64, 3039 CrossRef CAS PubMed.
- K. Tiefenbacher and J. Mulzer, Angew. Chem., Int. Ed., 2008, 47, 6199 CrossRef CAS PubMed.
- Y. Ito, T. Hirao and T. Saegusa, J. Org. Chem., 1978, 43, 1011 CrossRef CAS.
- Double epoxidation of the two conjugated olefins of 11 led to decomposition, presumably because the six-membered dienone is prone to aromatization. For related reactions, see: (a) M. F. Plano, G. R. Labadie, M. G. Sierra and R. M. Cravero, Tetrahedron Lett., 2006, 47, 7447 CrossRef CAS; (b) Y. Jing, C.-G. Xu, K. Ding, J.-R. Lin, R.-H. Jin and W.-S. Tian, Tetrahedron Lett., 2010, 51, 3242 CrossRef CAS.
- T. Sato, Y. Goto and T. Fujisawa, Tetrahedron Lett., 1982, 23, 4111 CrossRef CAS.
- Reduction of 11 with achiral reagents (e.g. NaBH4 and CeCl3, LiAlH4, AlH3, and DIBAL-H) generated the α-oriented alcohol or 1,4-reduced product as the major product.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
- E. J. Corey and H. E. Ensley, J. Org. Chem., 1973, 38, 3187 CrossRef CAS.
- (a) R. J. Theissen, J. Org. Chem., 1971, 36, 752 CrossRef CAS; (b) T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566 CrossRef CAS PubMed.
- (a) K. Nozaki, K. Oshima and K. Utimoto, J. Am. Chem. Soc., 1987, 109, 2547 CrossRef CAS; for a review, see: (b) C. Ollivier and P. Renaud, Chem. Rev., 2001, 101, 3415 CrossRef CAS PubMed.
- (a) Y. Ueno, K. Chino, M. Watanabe, O. Moriya and M. Okawara, J. Am. Chem. Soc., 1982, 104, 5564 CrossRef CAS; (b) G. Stork, R. Mook Jr, S. A. Biller and S. D. Rychnovsky, J. Am. Chem. Soc., 1983, 105, 3741 CrossRef CAS; for a review, see: (c) X. J. Salom-Roig, F. Dénès and P. Renaud, Synthesis, 2004, 1903 CAS; for recent applications of the methodology in natural product synthesis, see: (d) M. Inoue, T. Sato and M. Hirama, J. Am. Chem. Soc., 2003, 125, 10772 CrossRef CAS PubMed; (e) K. C. Nicolaou, P. K. Sasmal, A. J. Roecker, X.-W. Sun, S. Mandal and A. Converso, Angew. Chem., Int. Ed., 2005, 44, 3443 CrossRef CAS PubMed; (f) J. Wegner, S. V. Ley, A. Kirschning, A.-L. Hansen, J. M. Garcia and I. R. Baxendale, Org. Lett., 2012, 14, 696 CrossRef CAS PubMed; (g) M. Bian, Z. Wang, X. Xiong, Y. Sun, C. Matera, K. C. Nicolaou and A. Li, J. Am. Chem. Soc., 2012, 134, 8078 CrossRef CAS PubMed.
- For a review on radical-mediated deoxygenation, see: S. W. McCombie, W. B. Motherwell and M. J. Tozer, Org. React., 2012, 77, 161 CrossRef CAS.
- M. Oba and K. Nishiyama, Tetrahedron, 1994, 50, 10193 CrossRef CAS.
- D. Nakagawa, M. Miyashita and K. Tanino, Tetrahedron Lett., 2010, 51, 2771 CrossRef CAS.
- V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1 CrossRef CAS.
- D. H. R. Barton, R. E. O'Brien and S. Sternhell, J. Chem. Soc., 1962, 470 RSC.
- X. Han, B. M. Stoltz and E. J. Corey, J. Am. Chem. Soc., 1999, 121, 7600 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data for all new compounds, experimental procedures. CCDC 1027184 and 1027185. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00212e |
| This journal is © The Royal Society of Chemistry 2015 |