
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A general approach to the design of allosteric, transcription factor-regulated DNAzymes
G.
Adornetto
a,
A.
Porchetta
ab,
G.
Palleschi
ab,
K. W.
Plaxco
cd and
F.
Ricci
*ab
aDipartimento di Scienze e Tecnologie Chimiche University of Rome Tor Vergata, Via della Ricerca Scientifica, Rome 00133, Italy. E-mail: Francesco.ricci@uniroma2.it
bConsorzio Interuniversitario Biostrutture e Biosistemi “INBB”, Rome 00136, Italy
cDepartment of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, California 93106, USA
dCenter for Bioengineering, University of California Santa Barbara, Santa Barbara, California 93106, USA
First published on 10th March 2015
Abstract
Here we explore a general strategy for the rational design of nucleic acid catalysts that can be allosterically activated by specific nucleic-acid binding proteins. To demonstrate this we have combined a catalytic DNAzyme sequence and the consensus sequence recognized by specific transcription factors to create a construct exhibiting two low-energy conformations: a more stable conformation lacking catalytic activity and lacking the transcription factor binding site, and a less stable conformation that is both catalytically active and competent to bind the transcription factor. The presence of the target transcription factor pushes the equilibrium between these states towards the latter conformation, concomitantly activating catalysis. To demonstrate this we have designed and characterized two peroxidase-like DNAzymes whose activities are triggered upon binding either TATA binding protein or the microphthalmia-associated transcription factor. Our approach augments the current tool kit for the allosteric control of DNAzymes and ribozymes and, because transcription factors control many key biological functions, could have important clinical and diagnostic applications.
Introduction
DNAzymes and ribozymes, naturally occurring or in vitro selected RNAs or DNAs1 that catalyze specific chemical reactions,2–11 couple the advantages of enzymes (e.g., high turnover and specificity) with those of nucleic acids (e.g., low cost, ready designability) and thus represent a promising set of tools for use in biosensing,1,12–17 synthetic biology18 and bionanotechnology.1,19–22 Catalytic nucleic acids have similarly emerged as a new class of gene silencing agents,23,24 leading to the development of, for example, new cancer therapies.25,26An advantage of nucleic acid catalysts is the ease with which they can be rationally redesigned to introduce allosteric regulation, a mechanism that allows the fine-scale regulation of their activity upon the binding of an effector,27 an effect that has proven of value in a range of applications, including synthetic biology28–32 and biosensing.33 Most commonly, the design of allostery has been achieved via fusion of the catalytic nucleic acid with a regulation domain for the binding of the effector. This could be a specific short oligonucleotide sequence34–36 or a small molecule,37–40 peptide,33 or protein41 target that binds an aptamer domain. This binding event induces a conformational change that, in turn, regulates the catalytic activity.28–33,42 Here we expand on this theme by demonstrating a new class of allosterically regulated, catalytic nucleic acids that employ nucleic-acid-binding proteins as their effectors.
As our test bed nucleic-acid catalyst we have employed a guanine-rich, horseradish peroxidase (HRP)-mimicking DNAzyme.41,43,44 This single-strand DNA adopts a G-quadruplex structure that, in the presence of the cofactor hemin, catalyzes the oxidation of HRP-substrates. To re-engineer this DNAzyme to introduce transcription factor (TF)-regulated allostery we combined this catalytic domain (red domain in Fig. 1) with a consensus sequence recognized by a specific TF (green domain in Fig. 1) in such a way that the fusion populates two low-energy conformations. In the more stable of these, termed the off-state, the catalytic domain and the double-stranded, TF-binding region are “sequestered” and thus inactive (Fig. 1, left). In the less stable conformation, termed the on-state, both the domains are in their functional forms. TF binding thus pushes the equilibrium between these conformations from the former, off-state, towards the latter, on-state,45 activating catalysis (Fig. 1, right).
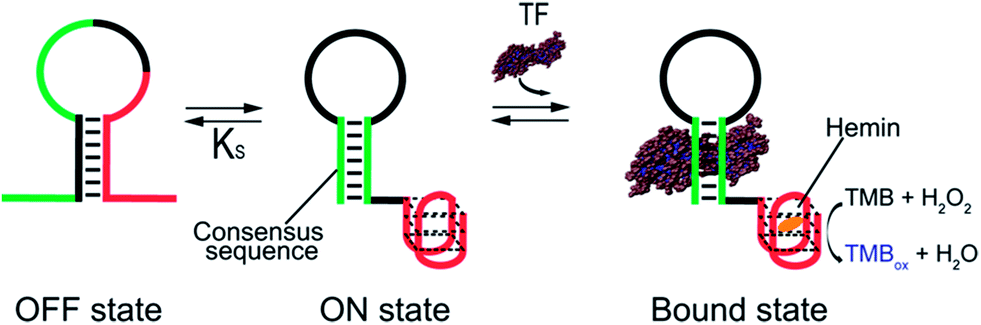 | ||
| Fig. 1 Transcription factor-induced activation of a DNAzyme. Here we demonstrate a DNAzyme allosterically activated by specific transcription factors (TF-regulated DNAzyme). To do so we coupled two functional domains: (i) a catalytic DNAzyme domain (red sequence in the cartoon) and (ii) a double-stranded transcription factor (TF)-binding domain (green). A sequence element complementary to the sequence of the DNAzyme stabilizes an alternative conformation (left) that “sequesters” both domains in an inactive (i.e., non-catalytic and non-TF-binding, respectively) state. This off-state is in equilibrium with a second conformation, the on-state, in which both domains are functional. TF binding shifts this equilibrium towards the on-state, activating catalysis. Here we used the HRP-like G-quadruplex DNAzyme as our model catalytic domain. In the presence of hemin and hydrogen peroxide, this domain catalyzes the oxidation of the HRP substrate 3,3′,5,5′-tetramethylbenzidine (TMB) to give a coloured product which is detectable by absorbance (λmax = 650 nm). | ||
Results and discussion
As our first allosteric effector we employed microphthalmia-associated transcription factor (MITF), a DNA-binding protein associated with melanoma and renal cell carcinoma.46–48 We designed four variants of MITF-regulated DNAzymes, each presenting the same consensus binding sequence46–48 for the TF (Fig. 2A, green). The four differ, however, in the stability of their off-states (Fig. 2B). Specifically, by increasing the number of bases in the double-stranded stem used to stabilize the off-state we obtained variants with estimated49 free energies ranging from −30.5 to −57.7 kJ mol−1. The stability of the on-state, in contrast, is effectively identical in all four variants (predicted stability = −26.9 kJ mol−1), and thus this approach provides a means of tuning the switching equilibrium constant, Ks, between the two states. This, in turn, provides a means of controlling the range of TF concentrations over which the DNAzyme is properly regulated.45,50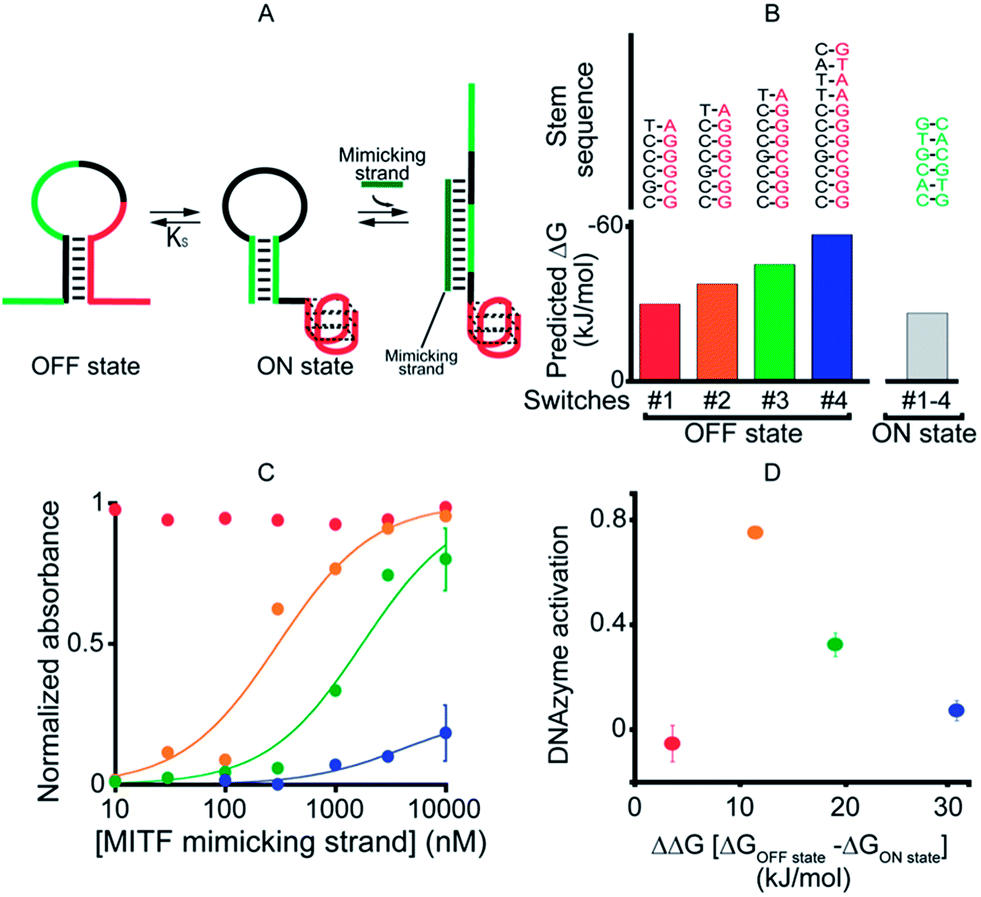 | ||
| Fig. 2 As a first proof-of-principle of transcription factor-induced DNAzyme activation, we designed four DNAzyme variants that are activated by MITF, a transcription factor that is involved in cancer progression. (A) Initially, in order to characterize the different variants, we used a simple DNA strand mimicking the action of the target TF (mimicking strand). (B) In order to modulate the range of the effector's concentration at which activation is observed, the variants were designed with increasingly stable off-states whilst maintaining approximately constant stability for the on-state (indicated are the m-fold predicted free energies). (C) As expected,45 the concentration range over which the allosteric effector causes activation of the DNAzyme is dependent on the stability of the off-state. (D) Optimal DNAzyme activation is observed at intermediate stabilities of the off-state (the y-axis shows the DNAzyme activation, the difference in activity when changing the effector concentration from 10 nM to 1 μm). DNAzyme activation was followed here and in the next figures by measuring absorbance at 650 nm in a solution of 50 nM TF-regulated DNAzyme in the presence of the DNAzyme cofactor hemin (500 nM) 40 min after the addition of the DNAzyme substrates TMB and H2O2. For clarity, error bars have been depicted for only the last point on these binding curves and those in the following figures. These error bars represent the standard deviations of measurements performed on at least three independent replicates. | ||
To avoid wasting the (rather expensive) TF, we first characterized our re-designed DNAzymes using a DNA strand (the “MITF-mimicking strand”) that binds to and thus stabilizes the on-state mimicking the target TF (Fig. 2A). As expected, the range of mimicking-strand concentrations over which activation is observed depends strongly on the switching equilibrium constant (Fig. 2C). Variant #1, for example, which exhibits the least stable off-state (predicted ΔG = −30.5 kJ mol−1), is partially activated even in the absence of its effector. In contrast, variant #4, for which the predicted stability of the off-state conformation is the highest (ΔG = −57.7 kJ mol−1), remains nearly inactive even at the highest effector concentrations we have employed (10 μM). The two variants between these extrema, in contrast, exhibit robustly regulated activation and show the optimal increase in activity upon increasing the effector concentration (Fig. 2D).
Because it is well regulated, we selected variant #2 to further characterize the extent to which the allosterically regulated catalyst is regulated by its specific TF (Fig. 3). For this variant we observe a monotonic increase in activation with increasing TF and an EC50 (the effector concentration at which 50% activation is observed) of 375 ± 105 nM (Fig. 3, left). The activation fold of the DNAzyme activity in the presence of saturating concentration of the target protein is 1.9 which is in good agreement with previous strategies for the allosteric activation of the same G-quadruplex peroxidase-like DNAzyme.1,15,27,41
 | ||
| Fig. 3 (Left) Our MITF-regulated DNAzyme (variant #2) is activated by its target TF in a dose-dependent fashion and is insensitive to other, non-targeted proteins (right). For the specificity test (right) we have used a concentration of target MITF and of non-specific proteins of 500 nM. In the y-axis of the right panel the difference (Δabsorbance) between the absorbance value obtained in the presence of the target and that obtained in a blank solution has been used. | ||
In contrast, the DNAzyme remains inactive when challenged with other, non-effector proteins. Incubation with 500 nM of the transcription factor TATA binding protein, for example, does not produce any significant cross-reactivity (Fig. 3, right).
To demonstrate the generality of our approach we designed a DNAzyme that is, instead, regulated by TATA binding protein (TBP), a TF present in virtually all eukaryotic cells.51 To do so, we engineered three variants differing in Ks (Fig. 4A and B) that, upon switching to the on-state, exhibit the consensus binding sequence of TBP.50,51 We found that the resultant activation profiles (using again a TF-mimicking DNA strand) are consistent with the predicted energy gap between the on- and off-states. From these results we selected variant #2, which exhibits an intermediate Ks and thus robust activation, for further characterization. The 2.6-fold, TBP-induced activation of this DNAzyme occurs with an EC50 of 104 ± 12 nM (Fig. 4C) and, as was true for its MITF-activated counterpart, the DNAzyme is insensitive to other, non-targeted proteins (500 nM) (Fig. 4D).
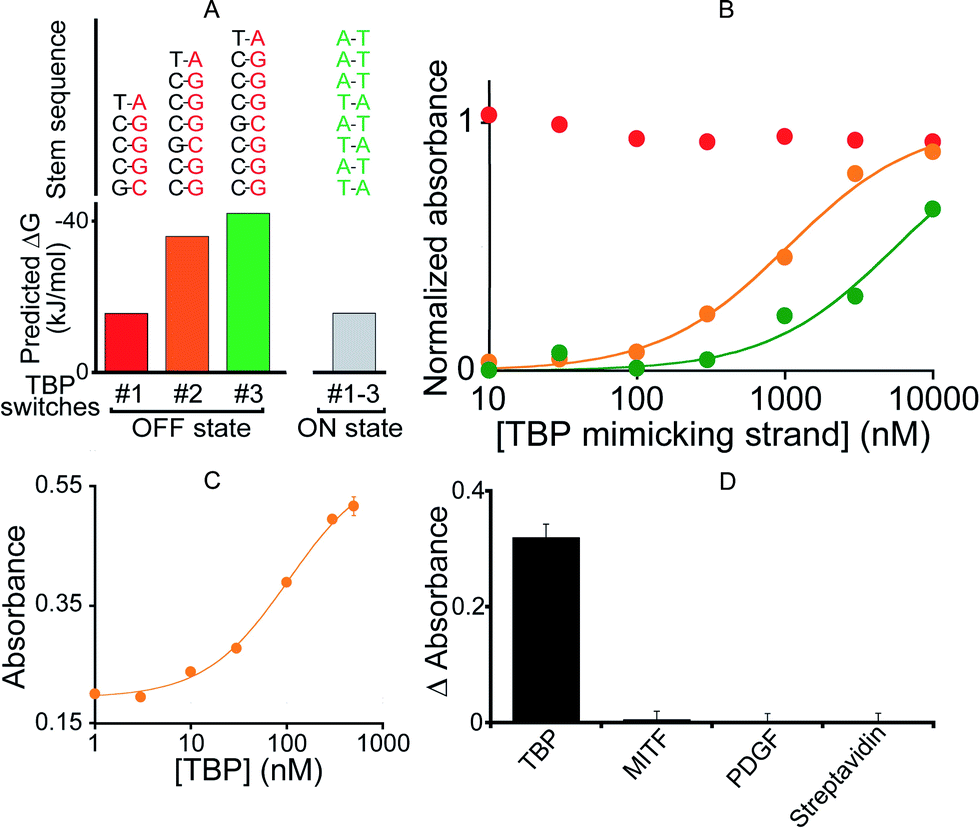 | ||
| Fig. 4 (A) To demonstrate the generality of our strategy we designed variants containing the consensus binding site for TATA binding protein (TBP). (B) Using a DNA strand mimicking the action of the target TF (see Fig. 2a), we find that, as expected, increasing the off-state stability shifts the activation profile to higher effector concentrations. (C) The intermediate variant (variant #2) was selected for further tests with the TF showing the activation of the TF-regulated DNAzyme as the concentration of TBP increases (EC50 = 104 ± 12 nM). (D) This DNAzyme, too, exhibits good specificity against other non-specific proteins. Here, we have used a concentration of target TBP and of non-specific proteins of 500 nM. In the y-axis of panel D the difference (ΔAbsorbance) between the absorbance value obtained in the presence of the target and that obtained in a blank solution has been used. | ||
Conclusions
In nature, the regulation of the activity of an enzyme is usually achieved through allosteric regulation, in which a binding site distal from the active site can recognize a molecule (an allosteric effector) and lead to a conformational switch that affects the enzyme's affinity for the substrate. Through this mechanism the activity of naturally occurring enzymes can be finely regulated by a variety of allosteric effectors in a concentration-dependent fashion. Allostery, also called “the second secret of life” by Perutz,52 can thus be considered as the optimal strategy to regulate the affinities and activities of biomolecules and bioreceptors. Because of this, engineering allostery into artificial systems could greatly improve the functionality of biomolecules employed in biotechnologies. In response, we have demonstrated here a general strategy for the design of nucleic acid catalysts allosterically activated by specific transcription factors.The development of allosterically regulated DNAzymes or ribozymes through both rational design and combinatorial selection strategies has been the subject of several studies starting from the seminal and fundamental work of Breaker and coworkers regarding the design and development of allosteric ribozymes.2,3,8,37,53–55 In the past ten years several efforts have been devoted to the development of nucleic acid catalysts that can be activated or regulated by different effectors including short nucleic acid sequences,21,34–36 and aptamer targets.33,37–41 Allosteric nucleic acid catalysts have thus been demonstrated to be useful tools not only for biosensing purposes56 but also as controllable therapeutic agents for gene therapy strategies23–26 or as molecular tools for controlling gene expression.30–32 Despite this, to the best of our knowledge, there is no report of a rational design of DNAzymes allosterically regulated by transcription factors (TF). We demonstrated here a general strategy to obtain TF-responsive DNAzymes by joining a TF consensus sequence with a G-quadruplex peroxidase-like DNAzyme domain. Our approach is based on the rational design of a conformational switching probe that flips between a non-binding inactive conformation to a second binding-competent active conformation in the presence of the specific TF. Because of this, we can regulate the TF concentration range at which activation of the DNAzyme is observed. We can thus control in a fine-tuned fashion the activity of our TF-regulated DNAzyme not only using different concentrations of the specific TF, but also using variants with different stabilities of the non-binding conformation, over completely different TF concentration ranges.
Compared to similar strategies reported earlier on the allosteric activation of catalytic nucleic acids, our approach appears very versatile. The consensus site recognized by each TF almost invariably consists of a specific double stranded DNA sequence with a length ranging from 6 to 12 base pairs, rendering the rational design of the necessary conformational switch quite straightforward. We also note that our approach can have important biological and clinical implications. While there is only a limited number of molecular targets recognizable via the use of aptamers, more than 10% of the ca. 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 human genes encode DNA-binding proteins suitable for the regulatory role presented here, the majority of which function as transcription factors thus controlling crucial biological mechanisms.57 Despite the limited activation fold achieved with our strategy, we note that this is comparable to other examples of allosterically activated DNAzymes based on the use of the peroxidase-like G-quadruplex.1,15,27,41 Because other examples of DNAzymes and ribozymes activated by different targets show larger activation folds we believe that this difference might be due to the low efficiency and high background signal characteristics of peroxidase-like DNAzyme in comparison to other DNAzymes.1,27,42–44 Finally, our strategy could be applied in the design of RNA/DNA chimeras to produce TF-regulated ribozymes and RNA-binding-protein-regulated DNAzymes that could have potential applications in several fields. Engineering allosteric catalytic nucleic acids that respond to TFs could in fact lead to novel molecular tools for gene expression regulation, gene regulatory mechanisms or novel riboregulators that can allow the development of genetic circuits in the growing fields of synthetic biology and bioengineering.58–60
000 human genes encode DNA-binding proteins suitable for the regulatory role presented here, the majority of which function as transcription factors thus controlling crucial biological mechanisms.57 Despite the limited activation fold achieved with our strategy, we note that this is comparable to other examples of allosterically activated DNAzymes based on the use of the peroxidase-like G-quadruplex.1,15,27,41 Because other examples of DNAzymes and ribozymes activated by different targets show larger activation folds we believe that this difference might be due to the low efficiency and high background signal characteristics of peroxidase-like DNAzyme in comparison to other DNAzymes.1,27,42–44 Finally, our strategy could be applied in the design of RNA/DNA chimeras to produce TF-regulated ribozymes and RNA-binding-protein-regulated DNAzymes that could have potential applications in several fields. Engineering allosteric catalytic nucleic acids that respond to TFs could in fact lead to novel molecular tools for gene expression regulation, gene regulatory mechanisms or novel riboregulators that can allow the development of genetic circuits in the growing fields of synthetic biology and bioengineering.58–60
Experimental section
Unmodified oligonucleotides were synthesized and purified (desalt purification) either by IBA GmBH (Göttingen, Germany) or Sigma-Aldrich (St. Louis, Missouri) and used without further purification. The sequences of the constructs are as follows.MITF variant #1: 5′-CCACGTG ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) CACGTG TCTTGGGT
CACGTG TCTTGGGT ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) GGTTGGG-3′.
GGTTGGG-3′.
MITF variant #2: 5′-CCACGTG CACGTG TCTTGGGT GTTGGG-3′.
MITF variant #3: 5′-CCACGTG CACGTG TCTTGGGT TTGGG-3′.
MITF variant #4: 5′-CCACGTG ![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) ACGTG TCTTGG TTGGG-3′.
ACGTG TCTTGG TTGGG-3′.
TBP variant #1: 5′-CTATATAAA TTTATATA TCTTGGGT GGGTTGGG-3′.
TBP variant #2: 5′-CTATATAAA TTTATATA TCTTGGGT GTTGGG-3′.
TBP variant #3: 5′-CTATATAAA G TTTATATA TCTTGGGT TTGGG-3′.
In the sequences above the underlined bases represent the stem portion of the non-binding conformation (off-state), while the italic bases represent the TF recognition element of the binding conformation (on-state) (see Fig. 1).
Mimicking strand oligonucleotides were purchased either from IBA GmBH (Göttingen, Germany) or from Sigma-Aldrich (St. Louis, Missouri) and below their sequences are reported:
MITF-mimicking strand: 5′-AGACAC GTGAGG-3′.
TBP-mimicking strand: 5′-AGATATA TAAAAGG-3′.
The mimicking strand sequences bind the TF-regulated DNAzyme sequences in a manner that induces the TF-regulated DNAzyme to adopt its catalytically active conformation.
Microphthalmia-associated transcription factor (MITF) was a gift from Prof. Colin Goding's group at the University of Oxford (UK). It is constituted by residues 198–302 of the human MITF-M (melanocyte specific), which encompasses the DNA-binding domain bHLH-LZ.48
TATA-binding protein was obtained by expression of recombinant, His-tagged proteins in Escherichia coli, as described previously.61
All experiments with TFs and mimicking strands were conducted with a DNAzyme concentration of 50 nM in a 25 mM HEPES buffer containing 20 mM KCl, 200 mM NaCl and 1% DMSO at pH 7.4. Absorbance at 650 nm was measured 40 min after the addition of the DNAzyme substrates, 3,3′,5,5′-tetramethylbenzidine (TMB) liquid substrate, supersensitive, for ELISA (ready-to-use solution with H2O2, purchased from Sigma-Aldrich, St. Louis, Missouri) using either a Bio-Rad Model 550 Microplate Reader or a Tecan Infinite M1000 PRO.
Activation curves were measured in microtiter plates. In every well, a concentration of the TF-regulated DNAzyme of 50 nM was used in a total volume of 150 μl and different concentrations of the proper mimicking strand or protein were added and allowed to react for 1 h at 37 °C. After this, the DNAzyme cofactor hemin was added at a final concentration of 500 nM and incubated for 1 h at 37 °C. Finally, 150 μl of the DNAzyme substrates TMB and H2O2 were added and the absorbance was measured at 650 nm after 40 min of incubation. The absorbance of the off-state was set relative to 0 for all normalized figures, while the absorbance obtained in the presence of a saturating target concentration was set relative to 1. The curves were fitted with the following equation:
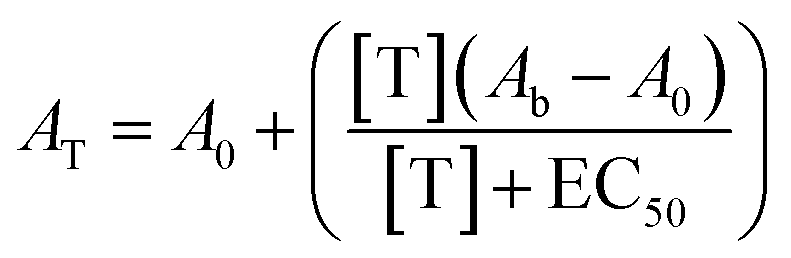
Acknowledgements
This work was supported by the European Research Council, ERC (project no. 336493) (F.R.), by the Associazione Italiana per la Ricerca sul Cancro, AIRC (project no. 14420) (F.R.) and by the Int. Research Staff Exchange Scheme (IRSES). Prof. Goding and his group at the University of Oxford (UK) are acknowledged for kindly providing the protein MITF.Notes and references
- I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37(6), 1153–1165 RSC.
- R. R. Breaker and G. F. Joyce, Trends Biotechnol., 1994, 12, 268–275 CrossRef CAS.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1(4), 223–229 CrossRef CAS.
- M. Illangasekare, G. Sanchez, T. Nickles and M. Yarus, Science, 1995, 267(5198), 643–647 CrossRef CAS.
- A. Jenne and M. Famulok, Chem. Biol., 1998, 5(1), 23–34 CrossRef CAS.
- J. B. Murray, A. A. Seyhan, N. G. Walter, J. M. Burke and W. G. Scott, Chem. Biol., 1998, 5(10), 587–595 CrossRef CAS.
- M. Illangasekare and M. A. Yarus, RNA, 1999, 5(11), 1482–1489 CrossRef CAS.
- J. Tang and R. R. Breaker, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(11), 5784–5789 CrossRef CAS.
- J. Rogers and G. F. Joyce, RNA, 2001, 7, 395–404 CrossRef CAS.
- J. W. Liu, A. K. Brown, X. L. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. A. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056–2061 CrossRef CAS PubMed.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2007, 129, 9838–9839 CrossRef CAS PubMed.
- R. Orbach, F. Remacle, R. D. Levine and I. Willner, Chem. Sci., 2014, 5, 1074–1081 RSC.
- Y. Xiao, V. Pavlov, T. Niazov, A. Dishon, M. Kotler and I. Willner, J. Am. Chem. Soc., 2004, 126, 7430–7431 CrossRef CAS PubMed.
- V. Pavlov, Y. Xiao, R. Gill, A. Dishon, M. Kotler and I. Willner, Anal. Chem., 2004, 76, 2152–2156 CrossRef CAS PubMed.
- T. Li, E. Wang and S. Dong, Chem. Commun., 2008, 3654–3656 RSC.
- R. Orbach, F. Wang, O. Lioubashevski, R. D. Levine, F. Remacle and I. Willner, Chem. Sci., 2014, 5, 3381–3387 RSC.
- X. Liu, R. Freeman, E. Golub and I. Willner, ACS Nano, 2011, 5(9), 7648–7655 CrossRef CAS PubMed.
- D. Balke, C. Wichert, B. Appel and S. Müller, Appl. Microbiol. Biotechnol., 2014, 98(8), 3389–3399 CrossRef CAS PubMed.
- T. A. Lincoln and G. F. Joyce, Science, 2009, 323, 1229–1232 CrossRef CAS PubMed.
- B. J. Lam and G. F. Joyce, J. Am. Chem. Soc., 2011, 133(9), 3191–3197 CrossRef CAS PubMed.
- G. Eckhoff, V. Codrea, A. D. Ellington and X. Chen, J. Syst. Chem., 2010, 1, 13 CrossRef CAS.
- B. Shlyahovsky, Y. Li, O. Lioubashevski, J. Elbaz and I. Willner, ACS Nano, 2009, 3(7), 1831–1843 CrossRef CAS PubMed.
- A. U. Khan, Clin. Chim. Acta, 2006, 367, 20–27 CrossRef CAS PubMed.
- Y. Isaka, Curr. Opin. Mol. Ther., 2007, 9, 132–136 CAS.
- C. R. Dass, P. F. M. Choong and L. M. Khanchigian, Mol. Cancer Ther., 2008, 7, 243 CrossRef CAS PubMed.
- Y. Qu, L. Zhang, M. Mao, F. Zhao, X. Huang, C. Yang, Y. Xiong and D. Mu, Cancer Gene Ther., 2008, 15, 517–525 CrossRef CAS PubMed.
- J. L. Vinkenborg, N. Karnowski and M. Famulok, Nat. Chem. Biol., 2011, 7(8), 519–527 CrossRef CAS PubMed.
- M. Wieland and H. S. Hartig, Angew. Chem., Int. Ed., 2008, 47, 2604–2607 CrossRef CAS PubMed.
- M. A. Aleman-Garcia, R. Orbach and I. Willner, Chem.–Eur. J., 2014, 20, 5619–5624 CrossRef CAS PubMed.
- B. Klauser, J. Atanasov, L. K. Siewert and J. S. Hartig, ACS Synth. Biol. DOI:10.1021/sb500062p.
- P. Ketzer, J. K. Kaufmann, S. Engelhardt, S. Bossow, C. von Kalle, J. S. Hartig, G. Ungerechts and D. M. Nettelbeck, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(5), E554–E562 CrossRef CAS PubMed.
- S. Ausländer, P. Stücheli, C. Rehm, D. Ausländer, J. S. Hartig and M. A. Fussenegger, Nat. Methods, 2014, 11(11), 1154–1160 CrossRef PubMed.
- J. S. Hartig, S. H. Najafi-Shoushtari, I. Grüne, A. Yan, A. D. Ellington and M. Famulok, Nat. Biotechnol., 2002, 20(7), 717–722 CrossRef CAS PubMed.
- D. Y. Wang and D. A. Sen, J. Mol. Biol., 2001, 310, 723–734 CrossRef CAS PubMed.
- D. Y. Wang, B. H. Y. Lai, A. R. Feldman and D. A. Sen, Nucleic Acids Res., 2002, 30, 1735–1742 CrossRef CAS PubMed.
- D. Y. Wang, B. H. Y. Lai and D. A. Sen, J. Mol. Biol., 2002, 318, 33–43 CrossRef CAS.
- J. Tang and R. R. Breaker, Chem. Biol., 1997, 4(6), 453–459 CrossRef CAS.
- M. Levy and A. D. Ellington, Chem. Biol., 2002, 9(4), 417–426 CrossRef CAS.
- W. Chiuman and Y. Li, PLoS One, 2007, 2(11), e1224 Search PubMed.
- B. J. Lam and G. F. Joyce, Nat. Biotechnol., 2009, 27, 288–292 CrossRef CAS PubMed.
- C. Teller, S. Shimron and I. Willner, Anal. Chem., 2009, 81, 9114–9119 CrossRef CAS PubMed.
- M. Famulok, J. S. Hartig and G. Mayer, Chem. Rev., 2007, 107(9), 3715–3743 CrossRef CAS PubMed.
- P. Travascio, Y. Li and D. A. Sen, Chem. Biol., 1998, 5(9), 505–517 CrossRef CAS.
- P. Travascio, P. K. Witting, A. G. Mauk and D. A. Sen, J. Am. Chem. Soc., 2001, 123, 1337–1348 CrossRef CAS PubMed.
- A. Vallée-Bélisle, F. Ricci and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(33), 13802–13807 CrossRef PubMed.
- (a) C. Bertolotto, F. Lesueur, S. Giuliano, T. Strub, M. de Lichy, K. Bille, P. Dessen, B. d'Hayer and H. Mohamdi, et al. , Nature, 2011, 480(7375), 94–98 CrossRef CAS PubMed; (b) J. Vachtenheim and J. Borovanský, Exp. Dermatol., 2010, 19, 617–627 CrossRef CAS PubMed.
- C. R. Goding, Pigm. Cell Melanoma Res., 2014, 27, 7–8 CrossRef PubMed.
- I. Aksan and C. R. Goding, Mol. Cell. Biol., 1998, 18(12), 6930–6938 CAS.
- M. Zuker, Nucleic Acids Res., 2003, 31(13), 3406–3415 CrossRef CAS PubMed.
- A. Vallée-Bélisle, A. J. Bonham, N. O. Reich, F. Ricci and K. W. Plaxco, J. Am. Chem. Soc., 2011, 133(35), 13836–13839 CrossRef PubMed.
- (a) N. Hernandez, Genes Dev., 1993, 7, 1291–1308 CrossRef CAS; (b) A. Bareket-Samish, I. Cohen and T. E. Haran, J. Mol. Biol., 2000, 299(4), 965–977 CrossRef CAS PubMed.
- M. Perutz, Mechanisms of Cooperativity and Allosteric Regulation in Proteins, Cambridge University Press, Cambridge, U.K., 1990 Search PubMed.
- G. A. Soukup and R. R. Breaker, Curr. Opin. Struct. Biol., 2000, 10(3), 318–325 CrossRef CAS.
- G. M. Emilsson and R. R. Breaker, Cell. Mol. Life Sci., 2002, 59(4), 596–607 CrossRef CAS.
- R. R. Breaker, Curr. Opin. Biotechnol., 2002, 13(1), 31–39 CrossRef CAS.
- F. Wang, R. Orbach and I. Willner, Chem.–Eur. J., 2012, 18, 16030–16036 CrossRef CAS PubMed.
- M. M. Babu, N. M. Luscombe, L. Aravind, M. Gerstein and S. A. Teichmann, Curr. Opin. Struct. Biol., 2004, 14, 283–291 CrossRef CAS PubMed.
- A. A. Green, P. A. Silver, J. J. Collins and P. Yin, Cell, 2014, 159(4), 925–939 CrossRef CAS PubMed.
- A. S. Khalil, T. K. Lu, C. J. Bashor, C. L. Ramirez, N. C. Pyenson, J. K. Joung and J. J. Collins, Cell, 2012, 150(3), 647–658 CrossRef CAS PubMed.
- A. S. Khalil and J. J. Collins, Nat. Rev. Genet., 2010, 11(5), 367–379 CrossRef CAS PubMed.
- J. J. Stewart and L. A. Stargell, J. Biol. Chem., 2001, 276, 30078–30084 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |