
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed intermolecular C(sp3)–H bond functionalization towards the synthesis of tertiary carbamates†‡
Prasanna Kumara
Chikkade
a,
Yoichiro
Kuninobu
*ab and
Motomu
Kanai
*ab
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: kuninobu@mol.f.u-tokyo.ac.jp; kanai@mol.f.u-tokyo.ac.jp
bERATO (Japan) Science and Technology Agency (JST), Kanai Life Science Catalysis Project, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
First published on 23rd March 2015
Abstract
We describe the development of an intermolecular unactivated C(sp3)–H bond functionalization towards the direct synthesis of tertiary carbamates. The transformation proceeded using a readily available, abundant first-row transition metal catalyst (copper), and isocyanates as the source of the amide moiety. This is a novel strategy for direct transformation of a variety of unactivated hydrocarbon feedstocks to N-alkyl-N-aryl and N,N-dialkyl carbamates without pre-functionalization or installation of a directing group. The reaction had a broad substrate scope with 3° > 2° > 1° site selectivity. The reaction proceeded even on a gram scale, and a corresponding free amine was directly obtained when the reaction was performed at high temperature. Kinetic studies suggested that radical-mediated C(sp3)–H bond cleavage was the rate-determining step.
Introduction
Carbamates are ubiquitous in nature, and are key functional and structural motifs in a broad range of important compounds, such as pharmaceuticals, agrochemicals, natural products, and functional materials. Molecules with N-alkyl-N-aryl and N,N-dialkyl motifs exhibit various biological properties (Fig. 1).1,2 The biological significance of carbamates has inspired the development of novel and efficient carbamation reactions.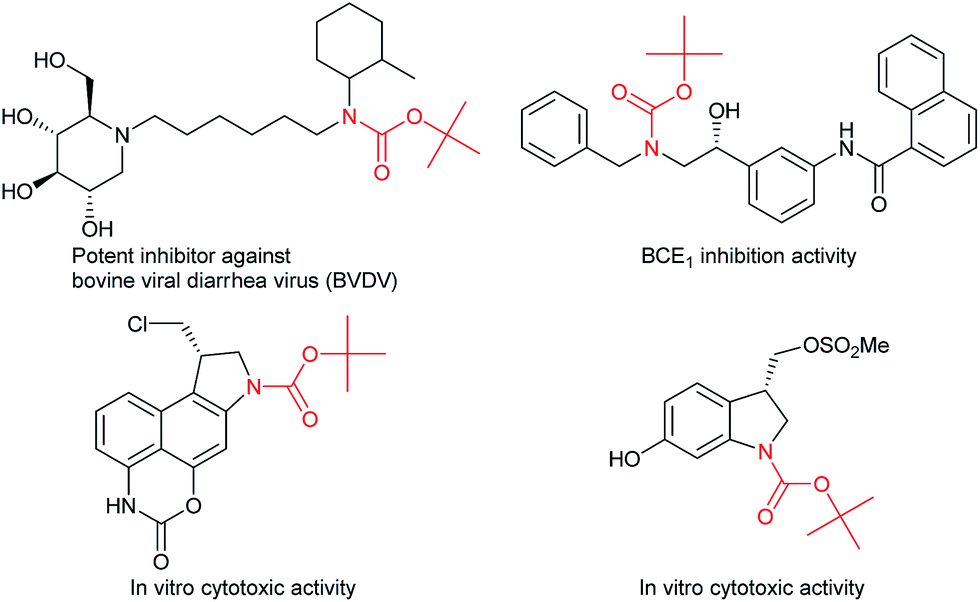 | ||
| Fig. 1 Biologically active compounds with tertiary carbamate motifs. | ||
Catalytic C(sp3)–H amidation, amination, and carbamation of inert hydrocarbons is a challenging and attractive strategy for preparing nitrogen-containing compounds. Reactions such as these that do not require typical functional group manipulations have enormous economic benefits. The notable transformation of a C–H bond to a C–N bond occurs in nitrene chemistry (Scheme 1, route a).3 However, intermolecular amidations using nitrenes are limited to reactions which produce secondary amides and functionalize benzylic or allylic positions. A more efficient copper-catalyzed Ritter-type C–H amidation was explored using a fluorine-based oxidant and acetonitrile as the nitrogen source (Scheme 1, route b); however, this reaction was limited to the synthesis of N-monoalkyl acetamides.4 Hartwig’s and Warren's copper-amide-based strategies constitute powerful synthetic methods (Scheme 1, route c); albeit, N-alkyl-N-aryl and N,N-dialkyl amides (tertiary amides) were obtained in low yields.5 Some other intermolecular and intramolecular C–H amination reactions with or without the use of metal catalysts have also been reported.6
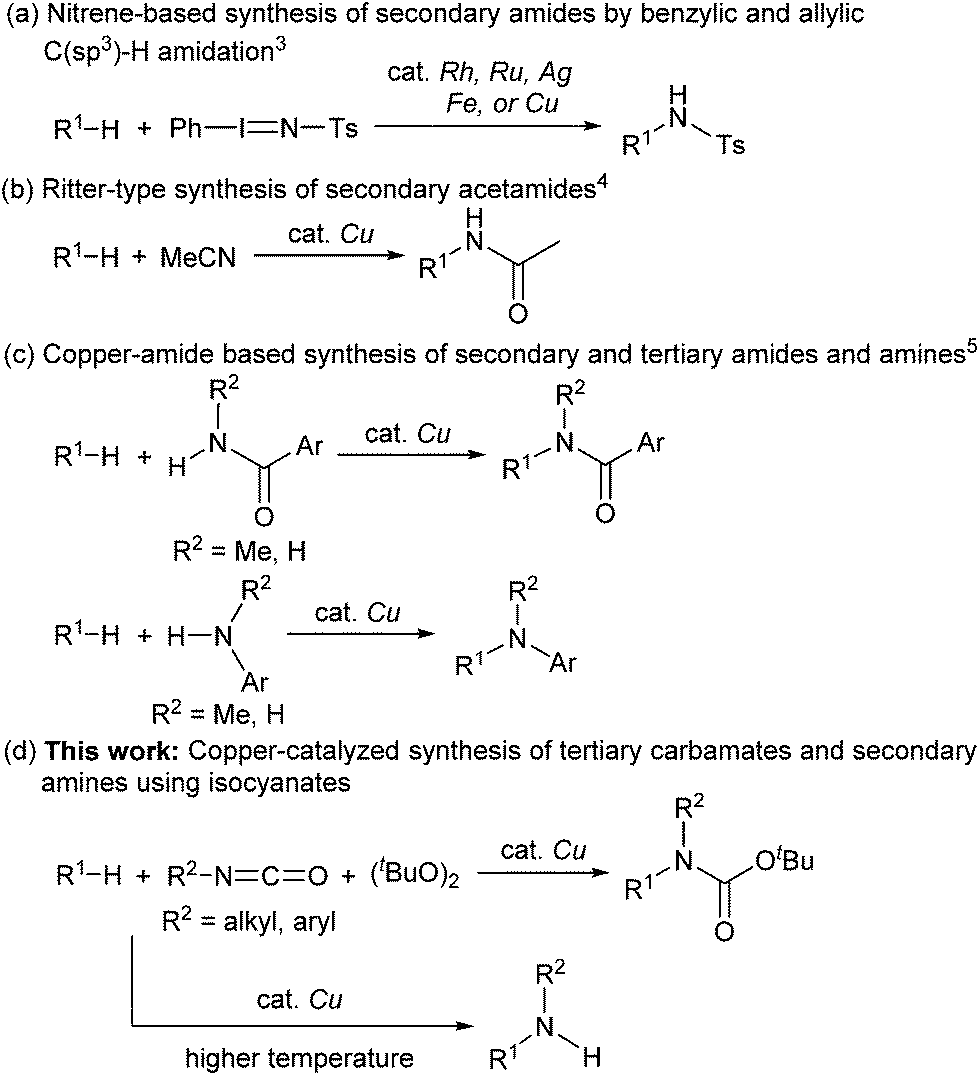 | ||
| Scheme 1 Transition metal-catalyzed C(sp3)–H bond transformations to construct C(sp3)–N bonds. | ||
Despite remarkable progress in C–H amidation and amination reactions, the formation of tertiary carbamates from inert hydrocarbons remains underdeveloped and the synthesis of N-alkyl-N-aryl or N,N-dialkyl tertiary carbamates remains a challenge. Although several reagents are effective as nitrogen atom sources in amidation and amination reactions, isocyanates have not been used to synthesize carbamates. Herein we report a novel and efficient unactivated C(sp3)–H bond functionalization that produces N-alkyl-N-aryl and N,N-dialkyl tertiary carbamates and the corresponding secondary amines using a copper catalyst and isocyanates as carbamation reagents (Scheme 1, route d).
Results and discussion
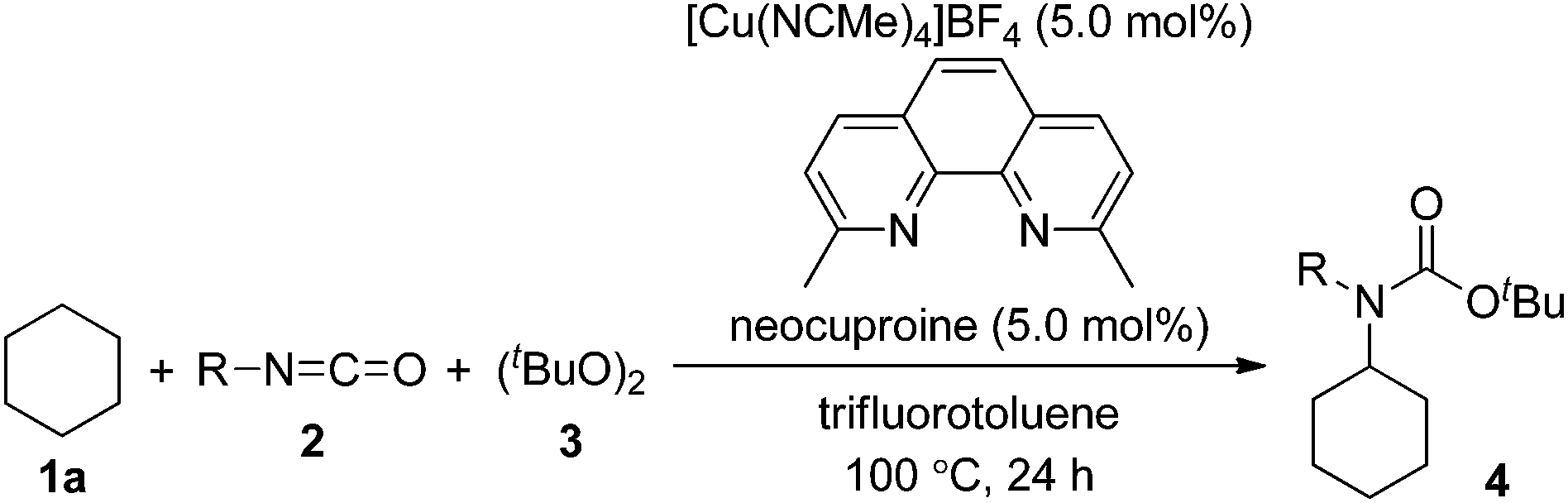
First, we investigated transition metal complexes and oxidants for the reaction between cyclohexane (1a) and phenyl isocyanate (2a) (Table 1). In the presence of a copper(I) iodide–phenanthroline complex (10 mol%) and oxidant 3 in benzene solvent, tert-butyl N-cyclohexyl-N-phenylcarbamate (4a) was obtained in 30% yield (entry 1). Other copper salts were comparably active regardless of their counterions and oxidation states (entries 2–4). The cationic copper complex [Cu(NCMe)4]BF4 proved to be the best catalyst, giving 4a in 47% yield (entry 5). Other peroxides, such as tert-butyl hydroperoxide (TBHP) and tert-butyl perbenzoate (TBPB), did not produce 4a, and di-tert-amyl peroxide (DTAP) had lower reactivity (entries 6–8). Further screening of bipyridyl-type and phenanthrolyl-type ligands revealed that neocuproine was the best ligand, affording the desired product 4a in 67% yield (entries 9–13). Extensive evaluation revealed that a reaction in the presence of 5 mol% of [Cu(NCMe)4]BF4 and 2.5 equiv. of oxidant 3 in trifluorotoluene as a co-solvent gave 4a in 75% yield (entry 14). Other transition metal salts, however, such as Fe(OAc)2, Co(OAc)2, Ni(OAc)2·4H2O, and AgOAc, did not promote the carbamation reaction. In addition, the reaction in the absence of a ligand led to low yield (32%).|
|
|||
|---|---|---|---|
| Entry | Catalyst | Ligand | Yieldb (%) |
| a Reaction conditions: 1a (5.00 mmol), 2a (0.500 mmol), 3 (1.00 mmol), catalyst (0.0500 mmol), ligand (0.0500 mmol), C6H6 (1.0 mL), 100 °C, 36 h. b 1H NMR yield using 1,1,2,2-tetrachloroethane as an internal standard. c TBHP (2.0 equiv.) was used as an oxidant. d TBPB (2.0 equiv.) was used as an oxidant. e DTAP (2.0 equiv.) was used as an oxidant. f Reaction in trifluorotoluene, Cu catalyst (5.0 mol%) and ligand (5.0 mol%), 3 (2.5 equiv.), 100 °C, 24 h. 1,10-Phen = 1,10-phenanthroline, (MeO)2phen = 4,7-dimethoxyphenanthroline, Cl2phen = 4,7-dichlorophenanthroline, tBu2phen = 4,7-di(tert-butyl)phenanthroline, bathocuproine = 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline, neocuproine = 2,9-dimethyl-1,10-phenanthroline. | |||
| 1 | CuI | 1,10-Phen | 30 |
| 2 | CuCl | 1,10-Phen | 36 |
| 3 | CuCl2 | 1,10-Phen | 35 |
| 4 | CuOAc | 1,10-Phen | 32 |
| 5 | [Cu(NCMe)4]BF4 | 1,10-Phen | 47 |
| 6c | [Cu(NCMe)4]BF4 | 1,10-Phen | 0 |
| 7d | [Cu(NCMe)4]BF4 | 1,10-Phen | 0 |
| 8e | [Cu(NCMe)4]BF4 | 1,10-Phen | 20 |
| 9 | [Cu(NCMe)4]BF4 | (MeO)2phen | 60 |
| 10 | [Cu(NCMe)4]BF4 | Cl2phen | 51 |
| 11 | [Cu(NCMe)4]BF4 | t Bu2bipy | 55 |
| 12 | [Cu(NCMe)4]BF4 | Bathocuproine | 53 |
| 13 | [Cu(NCMe)4]BF4 | Neocuproine | 67 |
| 14f | [Cu(NCMe)4]BF4 | Neocuproine | 75 |
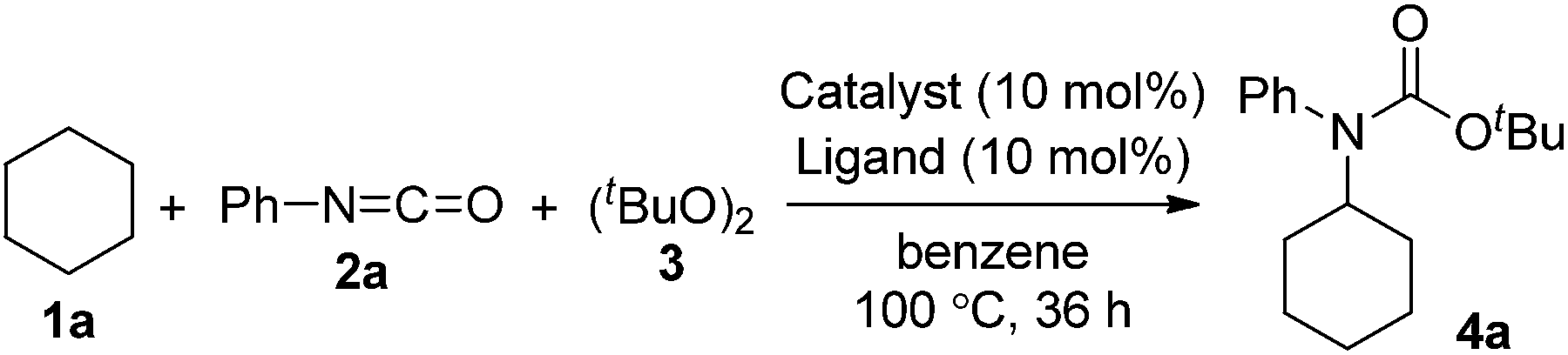
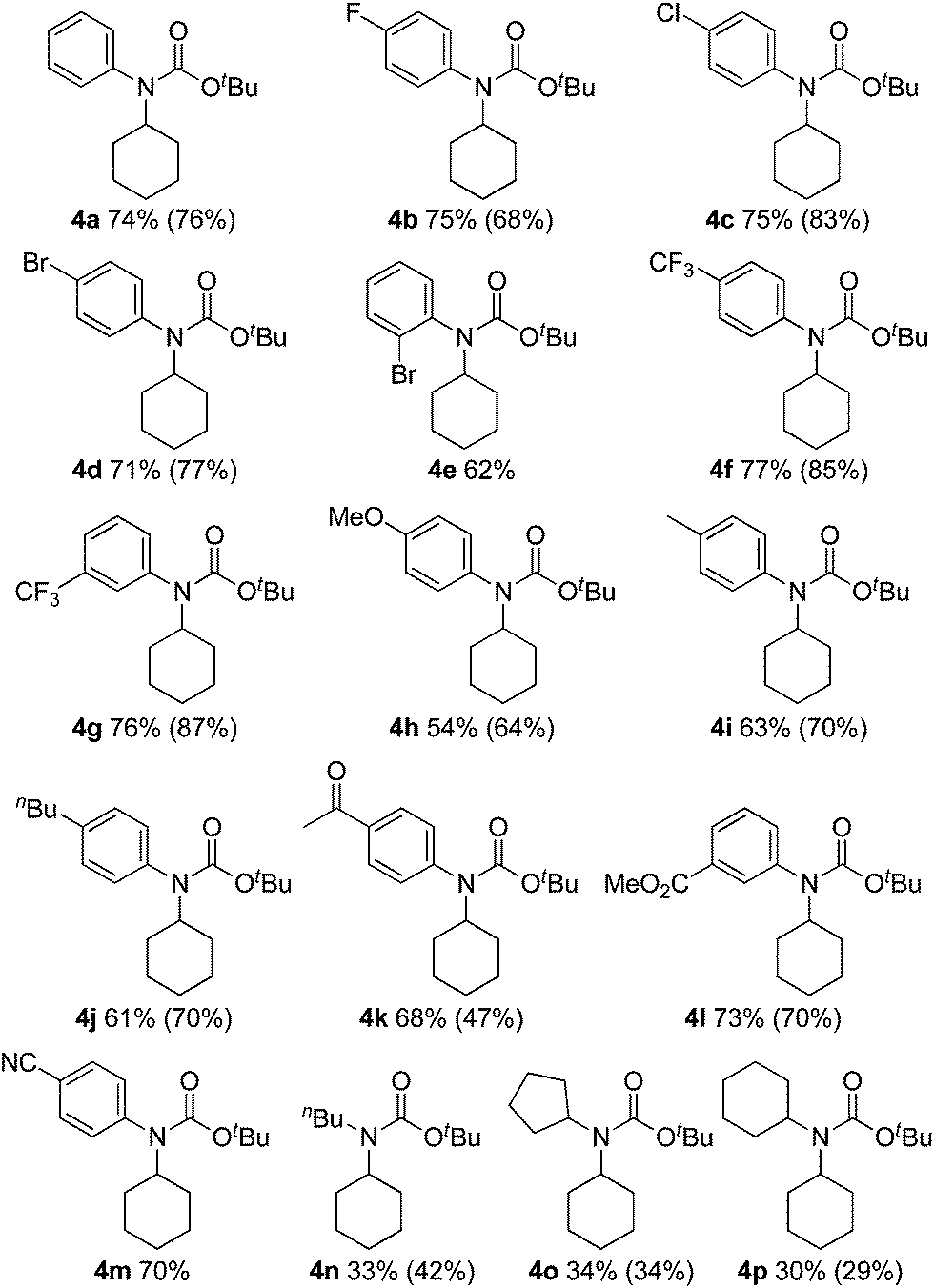
The substrate scope of isocyanates was investigated under the above optimal reaction conditions (Table 2). A variety of aryl and alkyl isocyanates were tested in cyclohexane (1a) or in a mixture of 1a/trifluorotoluene. Aryl isocyanates possessing a halogen atom, such as a fluorine, chlorine, or bromine atom, afforded the desired products 4b–e in excellent yields. Aryl isocyanates containing a trifluoromethyl group underwent carbamation of cyclohexane, affording the corresponding carbamates 4f and 4g in optimum yields. The carbamation reaction also proceeded from aryl isocyanates 4h–j containing an electron-donating group, such as a methoxy, methyl, or n-butyl group; however, the isolated yields of the products were slightly lower than those of aryl isocyanates with an electron-withdrawing group. Aryl isocyanates containing an acetyl, methoxycarbonyl, or cyano group were also effective, and the corresponding tertiary carbamates 4k–m were obtained in good yields. The carbamation reaction was next extended to more challenging substrates, alkyl isocyanates. Notably, reactions of 1a also occurred with alkyl isocyanates, such as n-butyl, cyclopentyl, and cyclohexylisocyanates, giving the corresponding N,N-dialkyl carbamates (4n–p).
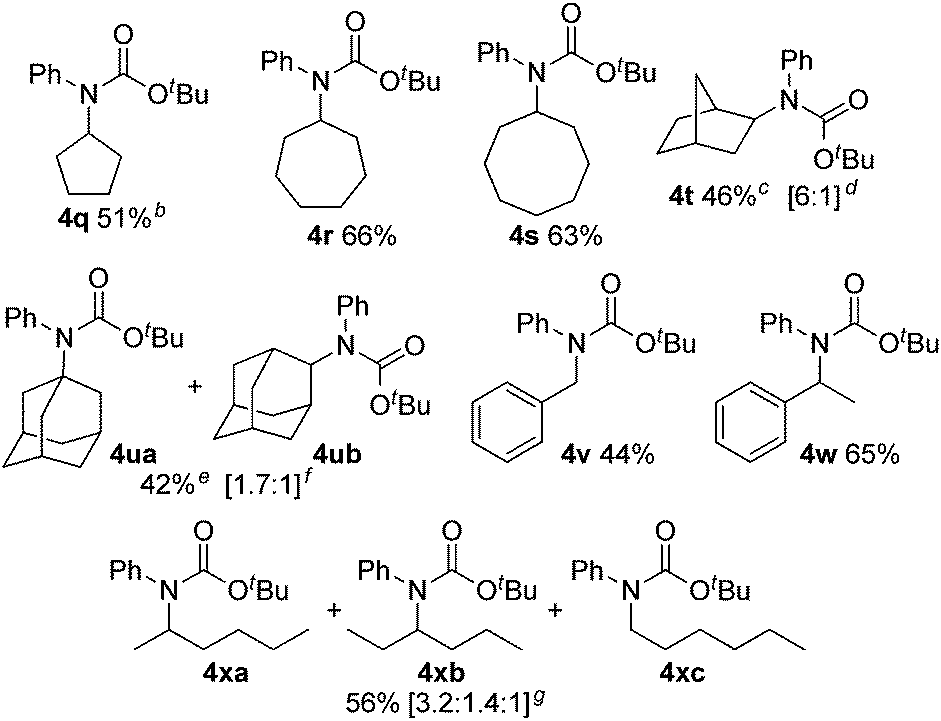
We next evaluated C(sp3)–H carbamation of other cyclic and linear unactivated alkanes (Table 3). C(sp3)–H carbamation of cycloalkanes such as cyclopentane, cycloheptane, and cyclooctane proceeded well, giving the corresponding tertiary carbamates 4q–s in good yields. C(sp3)–H carbamation of norbornane proceeded with single-site selectivity and gave a mixture of stereoisomers 4t in 46% yield (exo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :endo = 6:1). The reaction of adamantane with phenyl isocyanate (2a) afforded a regioisomeric mixture (4ua and 4ub) in 42% yield (4ua:4ub = 1.7:1). In the case of toluene, benzylic C(sp3)–H amidation proceeded and 4v was obtained in 44% yield. Ethylbenzene showed exclusive site selectivity, and furnished only a single carbamation product 4w in good yield. The reaction of 2a with a linear alkane (n-hexane), C(sp3)–H carbamation, proceeded nicely and afforded a mixture of carbamates 4xa, 4xb, and 4xc (3.2:1.4:1) in good yield. This is a highly valuable transformation of hydrocarbon feedstocks to tertiary carbamates.
:endo = 6:1). The reaction of adamantane with phenyl isocyanate (2a) afforded a regioisomeric mixture (4ua and 4ub) in 42% yield (4ua:4ub = 1.7:1). In the case of toluene, benzylic C(sp3)–H amidation proceeded and 4v was obtained in 44% yield. Ethylbenzene showed exclusive site selectivity, and furnished only a single carbamation product 4w in good yield. The reaction of 2a with a linear alkane (n-hexane), C(sp3)–H carbamation, proceeded nicely and afforded a mixture of carbamates 4xa, 4xb, and 4xc (3.2:1.4:1) in good yield. This is a highly valuable transformation of hydrocarbon feedstocks to tertiary carbamates.
|
|
|---|
| a Reaction conditions: 1 (5.00 mmol), 2a (0.500 mmol), 3 (1.25 mmol), [Cu(NCMe)4]BF4 (0.0250 mmol), neocuproine (0.0250 mmol), trifluorotoluene (1.0 mL), 100 °C, 24 h. b Cyclopentane (1.4 mL, 30 equiv.) was used. c Trifluorotoluene (1.5 mL) was used. d Ratio of exo- to endo-products. e Trifluorotoluene (2.0 mL) was used. f Ratio of 4ua to 4ub. g Ratio of 4xa to 4xb to 4xc. |
|

A proposed reaction mechanism is summarized in Scheme 2: (1) the copper(I) species-catalyzed7 or thermal homolytic cleavage of a peroxide generates tert-butoxy radical, which reacts with an isocyanate and sequential oxidation of the copper(I) species to give Cu(II)–amide species; (2) abstraction of a hydrogen atom from the C(sp3)–H bond of an alkane by tert-butoxy radical generates an alkyl radical with the release of tBuOH;8 (3) combination of the alkyl radical with Cu(II)–amide species produces a Cu(III) intermediate with alkyl and amide ligands;8a and (4) reductive elimination of the Cu(III) species affords a tertiary carbamate and regenerates the Cu(I) species.
 | ||
| Scheme 2 Proposed catalytic cycle for C(sp3)–H carbamation of alkanes. | ||
Next, kinetic isotopic effect studies with separate kinetic experiments were performed to gain insight into the rate-determining step for the amidation of cyclohexane (1a). Significant kinetic isotopic effects were observed (kH/kD = 2.9), suggesting that C–H bond cleavage was the rate-determining step of the reaction (Scheme 3, for details, see ESI‡).
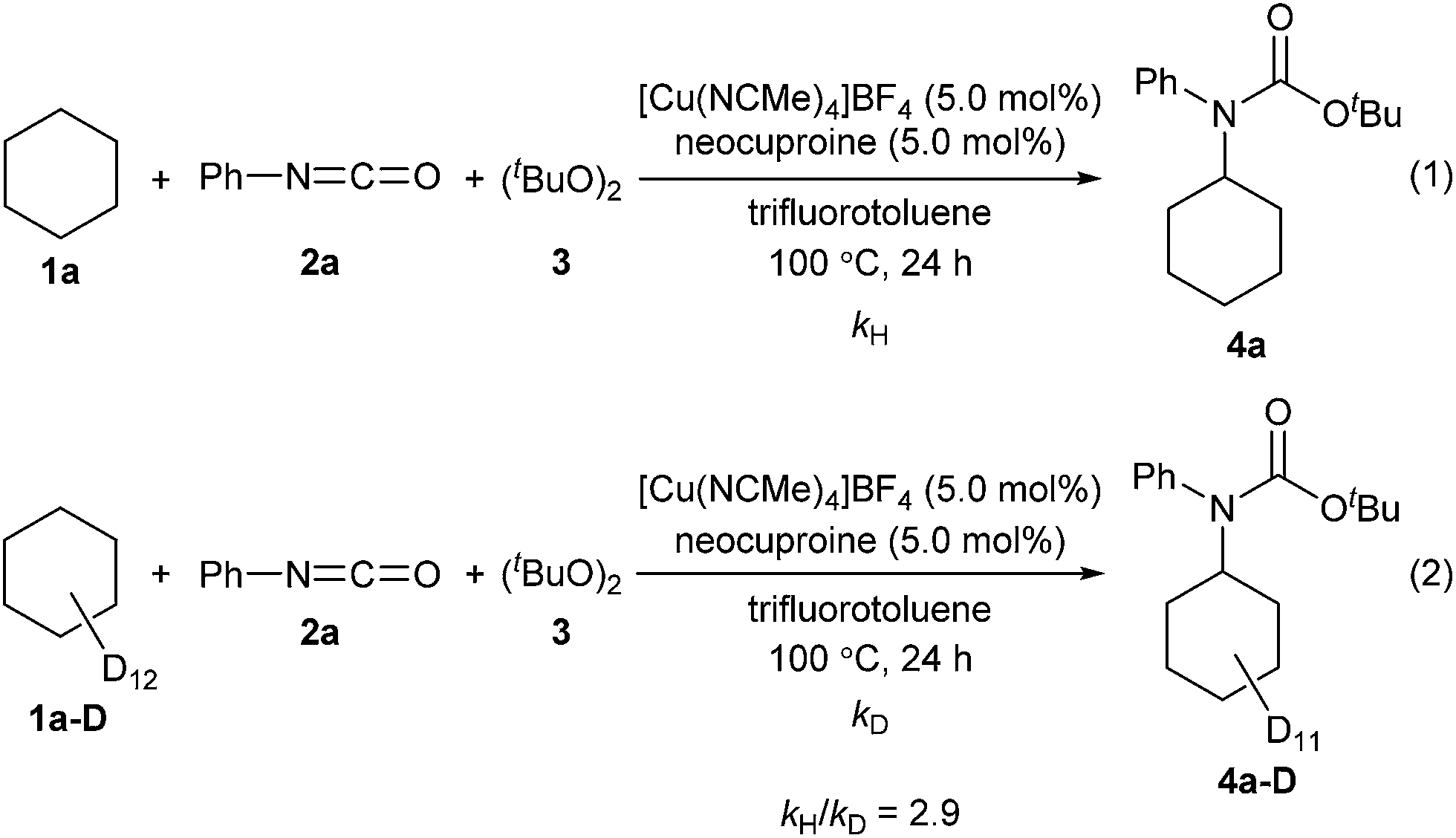 | ||
| Scheme 3 Kinetic isotopic effect studies. | ||
We also examined the reactivity difference between carbamate 5n and isocyanate 2n in the carbamation reaction (Scheme 4). Treatment of n-butyl carbamate 5n with cyclohexane (1a) under the optimized reaction conditions afforded 4n in only 11% yield. On the other hand, n-butyl isocyanate (2n) gave 4n in 42% yield under similar conditions. These results indicated that isocyanates are better carbamation reagents than secondary carbamates5d under the optimized conditions.
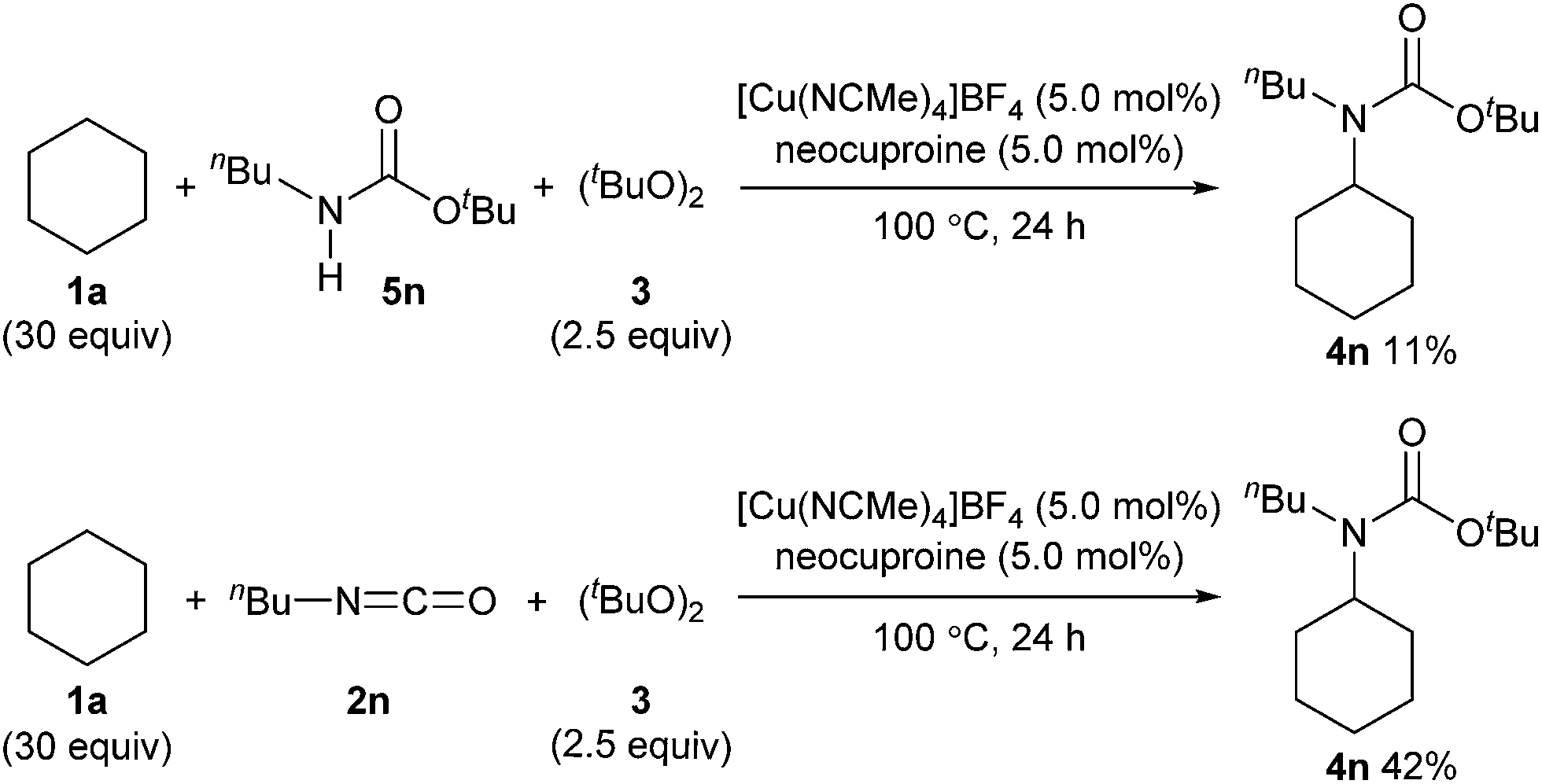 | ||
| Scheme 4 Evaluation of reactivity difference between secondary carbamates and isocyanates. | ||
To address the reactivity difference between isocyanates and secondary carbamates, and to gain further support for the proposed reaction mechanism in Scheme 2, we performed several reactions as follows (Scheme 5): (1) A reaction of phenyl isocyanate (2a) with cyclohexane (1a) under the optimized conditions without using a copper catalyst was performed. As a result, carbamate 5a was obtained in 76% yield, and tertiary carbamate 4a was not formed at all (Scheme 5(1)). This result showed that the copper catalyst was crucial for coupling between isocyanates and alkanes. (2) A reaction of phenyl isocyanate (2a) in the absence of 1a under the optimized conditions afforded carbamate 7 in 36% yield (Scheme 5(2)). A methyl radical was generated through β-methyl elimination of a tert-butoxy radical, and sequential coupling with an in situ generated copper(II)–carbamate species (6a) would lead to the formation of carbamate 7. (3) A reaction of n-butyl isocyanate (2n) with di-tert-butyl peroxide (3) in the presence of a stoichiometric amount of a copper complex was conducted in trifluorotoluene, and the course of the reaction was monitored by in situ-FT-IR (Scheme 5(3), see ESI‡ for details). As a result, an absorption band at 2266 cm−1 (assigned for the carbonyl group of isocyanate 2n) started to disappear after 1.4 h (induction period), and a new absorption band, possibly assigned to the carbonyl groups of copper complex 6n, appeared at 1725 cm−1. The maximum absorbance intensity at 1725 cm−1 was 0.063 (A.U.). (4) Under similar conditions, the progress of a reaction between n-butyl carbamate 5n and di-tert-butyl peroxide (3) and the formation of copper complex 6n was monitored by in situ-FT-IR (Scheme 5(4), see ESI‡ for details). An absorbance band for the carbonyl group of carbamate 5n was observed at 1720 cm−1, and an absorbance band at 1725 cm−1 corresponding to 6n started to appear after 3.9 h (induction period). The maximum absorbance intensity at 1725 cm−1 was 0.040 (A.U., the absorbance intensity of 6n must be less than 0.040 due to overlapping with an absorbance of 5n). The differences in the induction period and absorbance intensity of the bands at 1725 cm−1 for reactions (3) and (4) correlated well with the result that n-butyl isocyanate (2n) was more reactive compared with n-butyl carbamate 5n. In addition, the reaction of cyclohexane (1a) with phenylisocyanate (2a) was almost completely inhibited by 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO, 2 equivalents) under the optimized conditions. This result indicated that a radical pathway is involved as a key step in the catalytic cycle.
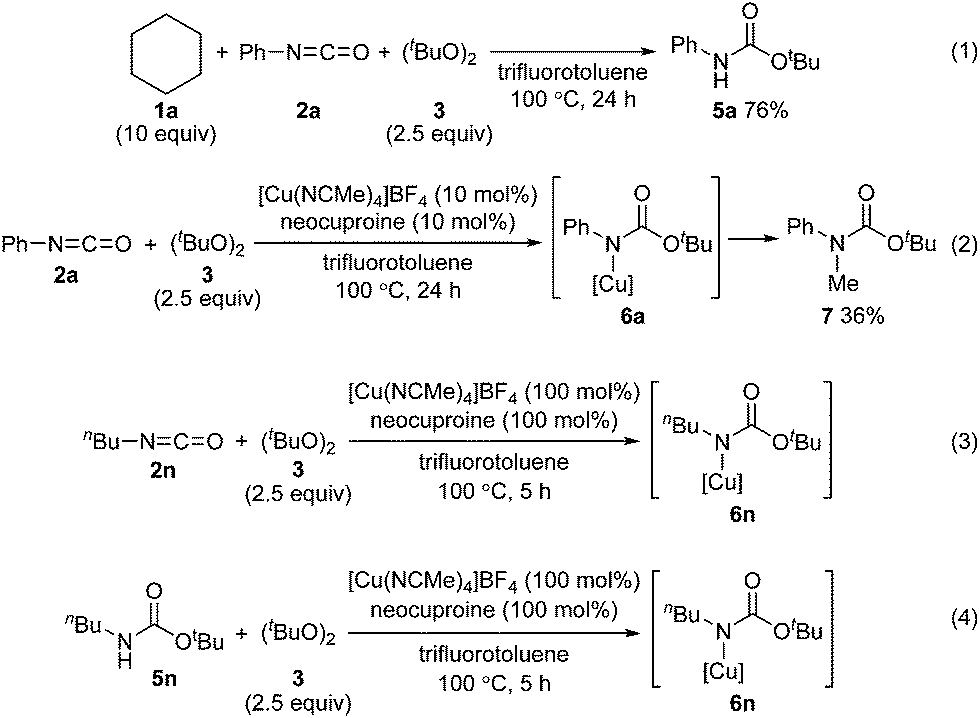 | ||
| Scheme 5 Several experiments performed to understand the reaction mechanism: (1) a reaction between cyclohexane (1a), isocyanate 2a, and peroxide 3 without using a copper catalyst; (2) a reaction between isocyanate 2a and peroxide 3 without addition of cyclohexane (1a); (3) in situ-FT-IR study of a reaction between [Cu(NCMe)4]BF4, isocyanate 2n, and peroxide 3; (4) in situ-FT-IR study of a reaction between [Cu(NCMe)4]BF4, carbamate 5n, and peroxide 3. | ||
C(sp3)–H carbamation could also be performed on a gram scale by treating cyclohexane (1a) and p-trifluoromethylphenyl isocyanate (2f) with a copper catalyst in trifluorotoluene solvent (Scheme 6, see also the ESI‡). As a result, 4f was obtained in 79% yield (1.35 g), which was comparable to the yield of 4f on a smaller scale (77% yield, 132 mg, Table 2).
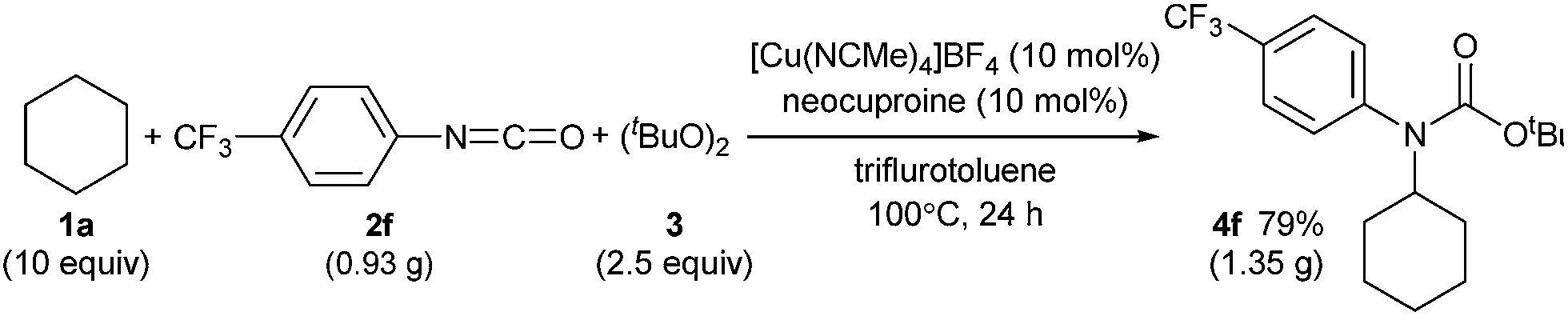 | ||
| Scheme 6 Gram scale synthesis of tert-butyl N-cyclohexyl-N-(4-trifluromethylphenyl)carbamate (4f). | ||
The developed method can be further extended to the direct synthesis of valuable amines from alkanes in combination with thermal cleavage of the tert-butoxycarbonyl group at a higher temperature (Scheme 5). The reaction of cyclohexane (1a) with phenyl isocyanate (2a) and (tBuO)2 (3) at 150 °C gave N-cyclohexylaniline (8) in 65% yield (Scheme 7).
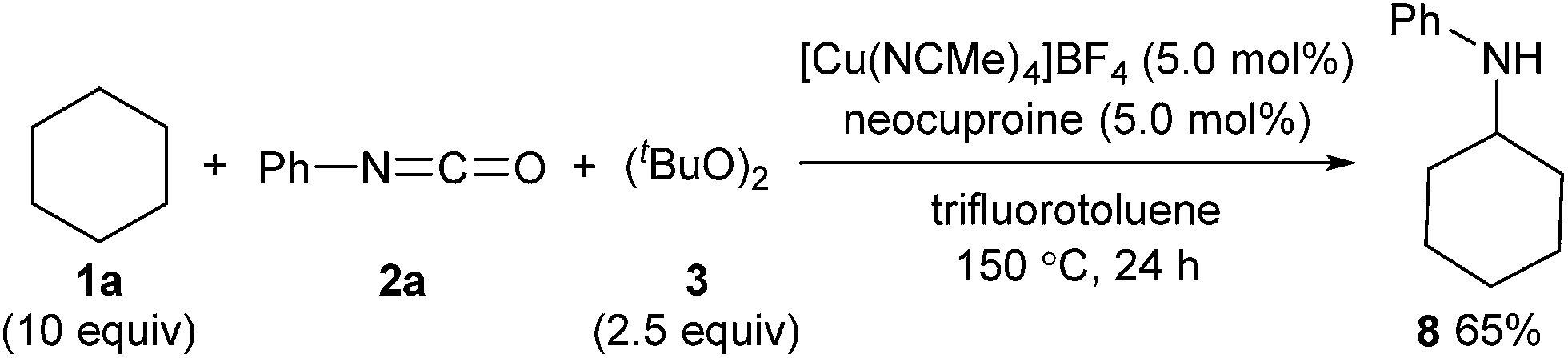 | ||
| Scheme 7 One-pot direct synthesis of a secondary amine from an alkane. | ||
Conclusions
In summary, we developed a copper-catalyzed, non-nitrene-based C(sp3)–H carbamation reaction. This new C(sp3)–H to C(sp3)–N bond transformation proceeded from unactivated alkanes and isocyanates. Although there are few examples of copper-catalyzed amidation or amination to afford secondary amides or amines, this novel protocol allowed us to obtain tertiary carbamates directly from hydrocarbon feedstocks. The reaction had a broad substrate scope, and the observed site selectivity was 3° > 2° > 1°. The reaction proceeded smoothly even on a gram scale, and a higher temperature directly produced the corresponding free amine without the addition of an acid. Kinetic studies suggested that radical mediated C(sp3)–H bond cleavage was the rate-limiting step. The copper-catalyzed C(sp3)–H carbamation reaction will be useful for rapid construction of synthetically useful and pharmaceutically valuable molecules bearing N-alkyl-N-aryl or N,N-dialkyl tertiary carbamate moieties.Acknowledgements
P.K.C. thanks JSPS for a research fellowship, and this work was supported in part by ERATO from JST (for M.K. and Y.K.) and the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Y.K.).References
- (a) W. S. Saari, W. Halczenko, W. C. Randall and V. J. Lotti, J. Med. Chem., 1983, 26, 1769 CrossRef CAS; (b) J. Charton, S. Girault-Mizzi, M.-A. Debreu-Fontaine, F. Foufelle, I. Hainault, J.-G. Bizot-Espiard, D.-H. Caignard and C. Sergheraert, Bioorg. Med. Chem., 2006, 14, 4490 CrossRef CAS PubMed; (c) K. Ohta, Y. Chiba, T. Ogawa and Y. Endo, Bioorg. Med. Chem. Lett., 2008, 18, 5050 CrossRef CAS PubMed; (d) C. D. Haffner, S. A. Thomson, Y. Guo, L. T. Schaller, S. Boggs, S. Dickerson, J. Gobel, D. Gillie and J. P. Condreay, Bioorg. Med. Chem. Lett., 2010, 20, 6983 CrossRef CAS PubMed; (e) V. Ehmke, C. Heindl, M. Rottmann, C. Freymond, W. B. Schweizer, R. Brun, A. Stich, T. Schirmeister and F. Diederich, ChemMedChem, 2011, 6, 273 CrossRef CAS PubMed; (f) P. Du, L. Xu, J. Huang, K. Yu, R. Zhao, B. Gao, H. Jiang, W. Zhao, X. Zhen and W. Fu, Chem. Biol. Drug Des., 2013, 82, 326 CrossRef CAS PubMed.
- (a) D. L. Boger, H. Zarrinmayeh, S. A. Munk, P. A. Kitos and O. Suntornwat, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 1431 CrossRef CAS; (b) S. A. Lawrence, Amines: Synthesis Properties and Applications, Cambridge University Press, Cambridge, 2004 Search PubMed; (c) C. Fischer, S. L. Zultanski, H. Zhou, J. L. Methot, S. Shah, H. Nuthhall, B. L. Hughes, N. Smotrov, A. Hill, A. A. Szewczak, C. M. Moxham, N. Bays, R. E. Middleton, B. Munoz and M. S. Shearman, Bioorg. Med. Chem. Lett., 2012, 22, 3140 CrossRef CAS PubMed; (d) C. Lu, Y. Guo, J. Li, M. Yao, Q. Liao, Z. Xie and X. Li, Bioorg. Med. Chem. Lett., 2012, 22, 7683 CrossRef CAS PubMed; (e) A. L. Wolfe, K. K. Duncan, N. K. Parelkar, S. J. Weir, G. A. Vielhauer and D. L. Boger, J. Med. Chem., 2012, 55, 5878 CrossRef CAS PubMed; (f) K. O. Yerdelen and H. I. Gul, Med. Chem. Res., 2013, 22, 4920 CrossRef CAS; (g) M. Guerrero, R. Poddutoori, M. Urbano, X. Peng, T. P. Spicer, P. S. Chase, P. S. Hodder, M.-T. Schaeffer, S. Brown, H. Rosen and E. Roberts, Bioorg. Med. Chem. Lett., 2013, 23, 6346 CrossRef CAS PubMed; (h) Y. Du, H. Ye, T. Gill, L. Wang, F. Guo, A. Cuconati, J.-T. Guo, T. M. Block and X. Xu, Bioorg. Med. Chem. Lett., 2013, 23, 2172 CrossRef CAS PubMed; (i) S. Butini, E. Gabellieri, M. Brindisi, S. Giovani, S. Maramai, G. Kshirsagar, S. Brogi, V. L. Pietra, M. Giustiniano, L. Marinelli, E. Novellino, G. Campiani, A. Cappelli and S. Gemma, Eur. J. Org. Chem., 2013, 70, 233 CAS.
- (a) X.-Q. Yu, J.-S. Huang, X.-G. Zhou and C.-M. Che, Org. Lett., 2000, 2, 2233 CrossRef CAS PubMed; (b) Z. Li, D. A. Capretto, R. Rahaman and C. He, Angew. Chem., Int. Ed., 2007, 46, 5184 CrossRef CAS PubMed; (c) C. Liang, F. Collet, F. Robert-Peillard, P. Muller, R. H. Dodd and P. Dauban, J. Am. Chem. Soc., 2008, 130, 343 CrossRef CAS PubMed; (d) K. Huard and H. Lebel, Chem.–Eur. J., 2008, 14, 6222 CrossRef CAS PubMed; (e) Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palmino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961 CrossRef CAS PubMed; (f) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061 RSC; (g) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (h) Y. Liu, X. Guan, E. L.-M. Wong, P. Liu, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2013, 135, 7194 CrossRef CAS PubMed; (i) V. Bagchi, P. Paraskevopoulou, P. Das, L. Chi, Q. Wang, A. Choudhury, J. S. Mathieson, L. Cronin, D. B. Pardue, T. R. Cundari, G. Motrikas, Y. Sanakis and P. Stavropoulos, J. Am. Chem. Soc., 2014, 136, 11362 CrossRef CAS PubMed.
- (a) J. J. Ritter and P. P. Minieri, J. Am. Chem. Soc., 1948, 70, 4045 CrossRef CAS; (b) Q. Michaudel, D. Thevent and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 2547 CrossRef CAS PubMed.
- (a) G. Pelletier and D. A. Powell, Org. Lett., 2006, 8, 6031 CrossRef CAS PubMed; (b) S. Wiese, Y. M. Badiei, R. T. Gephart, S. Mossin, M. S. Varonka, M. M. Melzer, K. Meyer, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2010, 49, 8850 CrossRef CAS PubMed; (c) R. T. Gephart III, D. L. Huang, M. J. B. Aguila, G. Schmidt, A. Shahu and T. H. Warren, Angew. Chem., Int. Ed., 2012, 51, 6488 CrossRef PubMed; (d) R. T. Gephart III and T. H. Warren, Organometallics, 2012, 31, 7728 CrossRef; (e) B. L. Tran, B. Li, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 2555 CrossRef CAS PubMed.
- (a) Y. Kohmura, K.-i. Kawasaki and T. Katsuki, Synlett, 1997, 1456 CrossRef CAS PubMed; (b) R. S. Srivastava, N. R. Traver and K. M. Nicholas, J. Am. Chem. Soc., 2007, 129, 15250 CrossRef CAS PubMed; (c) S. A. Reed and M. C. White, J. Am. Chem. Soc., 2008, 130, 3316 CrossRef CAS PubMed; (d) R. Fan, W. Li and L. Zhang, Org. Lett., 2009, 11, 1425 CrossRef CAS PubMed; (e) G. Deng, W. Chen and C.-J. Li, Adv. Synth. Catal., 2009, 351, 353 CrossRef CAS; (f) D. A. Powell and H. Fan, J. Org. Chem., 2010, 75, 2726 CrossRef CAS PubMed; (g) M. Ochiai, K. Miyamato, T. Kaneaki, S. Hayashi and W. Nakanishi, Science, 2011, 332, 448 CrossRef CAS PubMed; (h) J. A. Souto, D. Zian and K. Muniz, J. Am. Chem. Soc., 2012, 134, 7242 CrossRef CAS PubMed; (i) Z. Wang, J. Ni, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2014, 53, 3496 CrossRef CAS PubMed; (j) X. Wu, Y. Zhao, G. Zhang and H. Ge, Angew. Chem., Int. Ed., 2014, 53, 3706 CrossRef CAS PubMed.
- (a) J. K. Kochi, Tetrahedron, 1962, 18, 483 CrossRef CAS; (b) H. Paul, R. D. Small Jr and J. C. Scaiano, J. Am. Chem. Soc., 1978, 100, 4520 CrossRef CAS; (c) R. T. Gephart III, C. L. McMullin, N. G. Sapiezynski, E. S. Jang, M. J. B. Aguila, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2012, 134, 17350 CrossRef PubMed.
- (a) M. B. Andrus, A. K. Argade, X. Chen and M. G. Pamment, Tetrahedron Lett., 1995, 36, 2945 CrossRef CAS; (b) M. Finn, R. Friedline, N. K. Suleman, C. J. Wohl and J. M. Tanko, J. Am. Chem. Soc., 2004, 126, 7578 CrossRef CAS PubMed; (c) G. Deng and C.-J. Li, Tetrahedron Lett., 2008, 49, 5601 CrossRef CAS PubMed; (d) R. Xia, H.-Y. Niu, G.-R. Qu and H.-M. Guo, Org. Lett., 2012, 14, 5546 CrossRef CAS PubMed; (e) Z.-Q. Zhu, T.-T. Wang, P. Bai and Z.-Z. Huang, Org. Biomol. Chem., 2014, 12, 5839 RSC.
Footnotes |
| † Dedicated to Professor Iwao Ojima on his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and 1H and 13C NMR charts. See DOI: 10.1039/c5sc00238a |
| This journal is © The Royal Society of Chemistry 2015 |