DOI:
10.1039/C5SC00268K
(Edge Article)
Chem. Sci., 2015,
6, 2501-2505
Identifying lead hits in catalyst discovery by screening and deconvoluting complex mixtures of catalyst components†
Received
23rd January 2015
, Accepted 16th February 2015
First published on 16th February 2015
Abstract
A reaction-economic combinatorial strategy is described for lead hit identification in catalyst discovery efforts directed towards a specific transformation. Complex mixtures of rationally chosen precatalysts and ligands are screened against various reaction parameters to identify lead conditions in a small number of reactions. Iterative deconvolution of the resulting hits identifies which components contribute to the lead in situ generated catalyst. Application of this strategy rapidly uncovered a new mild in situ generated catalyst for the dehydrative Friedel–Crafts reaction as well as conditions for selective monoarylation in catalytic ortho-C–H arylation of unsubstituted N-(quinolin-8-yl)benzamide.
Introduction
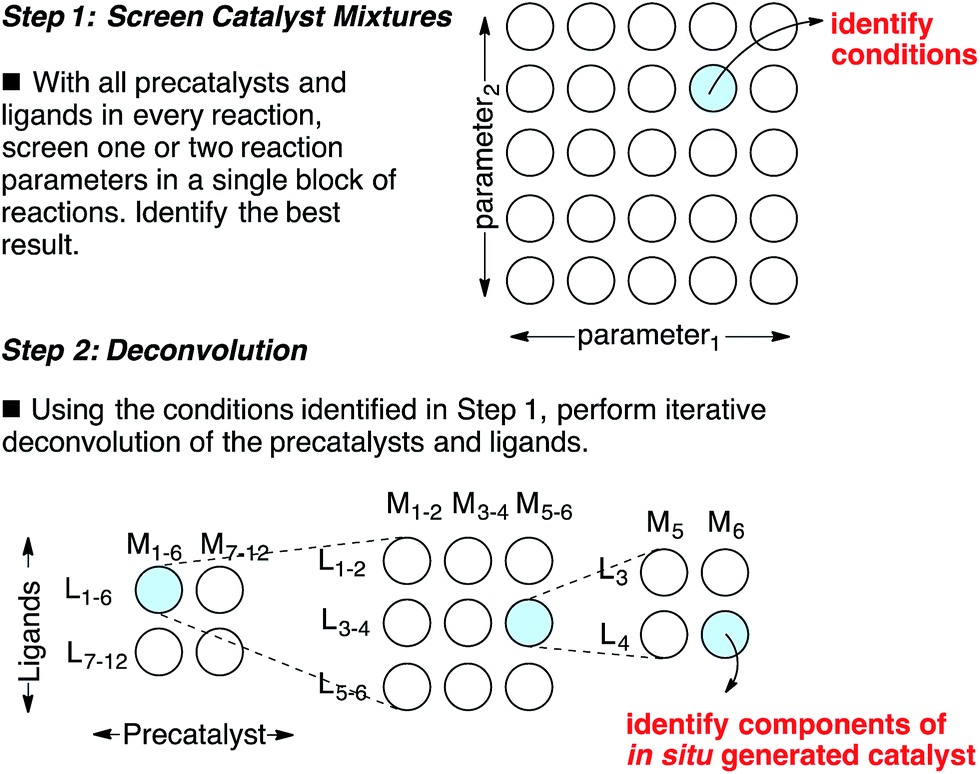
Catalyst discovery is a complex multidimensional problem that often requires extensive experimentation to obtain a lead result. In a representative catalyst discovery scenario, precatalyst, ligand, solvent, acid/base additive or temperature may all be critical reaction parameters, and incorrect choice of any one of these may result in failure to observe an initial “hit” for the desired product. In response to this reality, many creative screening approaches have been developed for catalyst discovery, each with different purposes, strengths and weaknesses.1–4 Of those screening approaches that are directed towards discovering a specific new catalytic transformation or a new class of catalysts for an existing transformation, few are highly general and several require laborious synthesis of labeled starting materials. Though advances in analytical technology are decreasing the barriers to general reaction development approaches using high throughput experimentation,5 the large number of reactions necessary to thoroughly explore the intersection of just three or four reaction parameters might still deter chemists who do not have access to high throughput reactionware and instrumentation. Complementary approaches that can reduce the number of reactions required to obtain a lead result for a specific transformation and that do not require high throughput instrumentation are appealing. Towards this goal, we have devised an approach for the identification of lead results in catalyst discovery based on the assumption that precatalysts and ligands can be screened as mixtures and later deconvoluted.6 By employing a complex mixture of all precatalysts and ligands in every reaction, one or two additional reaction parameters (e.g. solvent, acid/base, etc.) are screened to identify a promising result. In this way, up to four reaction parameters can be surveyed at once in a single small block of reactions, effectively ‘front-loading’ the problem of catalyst discovery (Scheme 1, Step 1). The precatalysts and ligands that contribute most to catalysis are identified by iterative deconvolution of the mixture in a manner similar to that described by Breit for mixtures of self-assembling ligands (Scheme 1, Step 2).4 As the aim of this approach is to accelerate catalyst discovery, the primary goal is simply to detect product formation. Thus, the fact that different precatalyst/ligand combinations exhibit different binding constants only leads to false negatives if a component critical to catalysis is completely inhibited. Risks of encountering such catalyst poisoning scenarios can be rationally minimized by tuning the ratio of precatalysts to ligands during screening. Given these considerations, we anticipated that this screening approach could be useful for reactions that involve significant molecular recognition between catalyst and substrate. Herein, we describe the successful application of this strategy to developing new catalysts for two completely different catalytic reactions. In the first example, a Friedel–Crafts reaction is used to discover a new powerful boron catalyst that arises by covalent assembly from a complex mixture of boronic acids and bidentate O-ligands. In a second example, application of the strategy has uncovered a catalytic system for selective mono ortho-C–H arylation of N-(quinolin-8-yl)benzamide in the absence of blocking groups at the ortho or meta positions. Our collective results indicate that a rationally guided screening approach using complex mixtures of catalyst components, while not necessarily an analytical solution to a catalyst discovery problem, is a useful complementary strategy to rapidly identifying lead “hits” in a small number of reactions without the use of high throughput equipment.
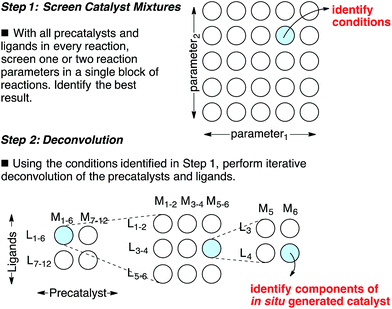 |
| | Scheme 1 A combinatorial strategy for identifying lead “hits” in catalyst discovery by screening complex mixtures of catalyst components. | |
Results and discussion
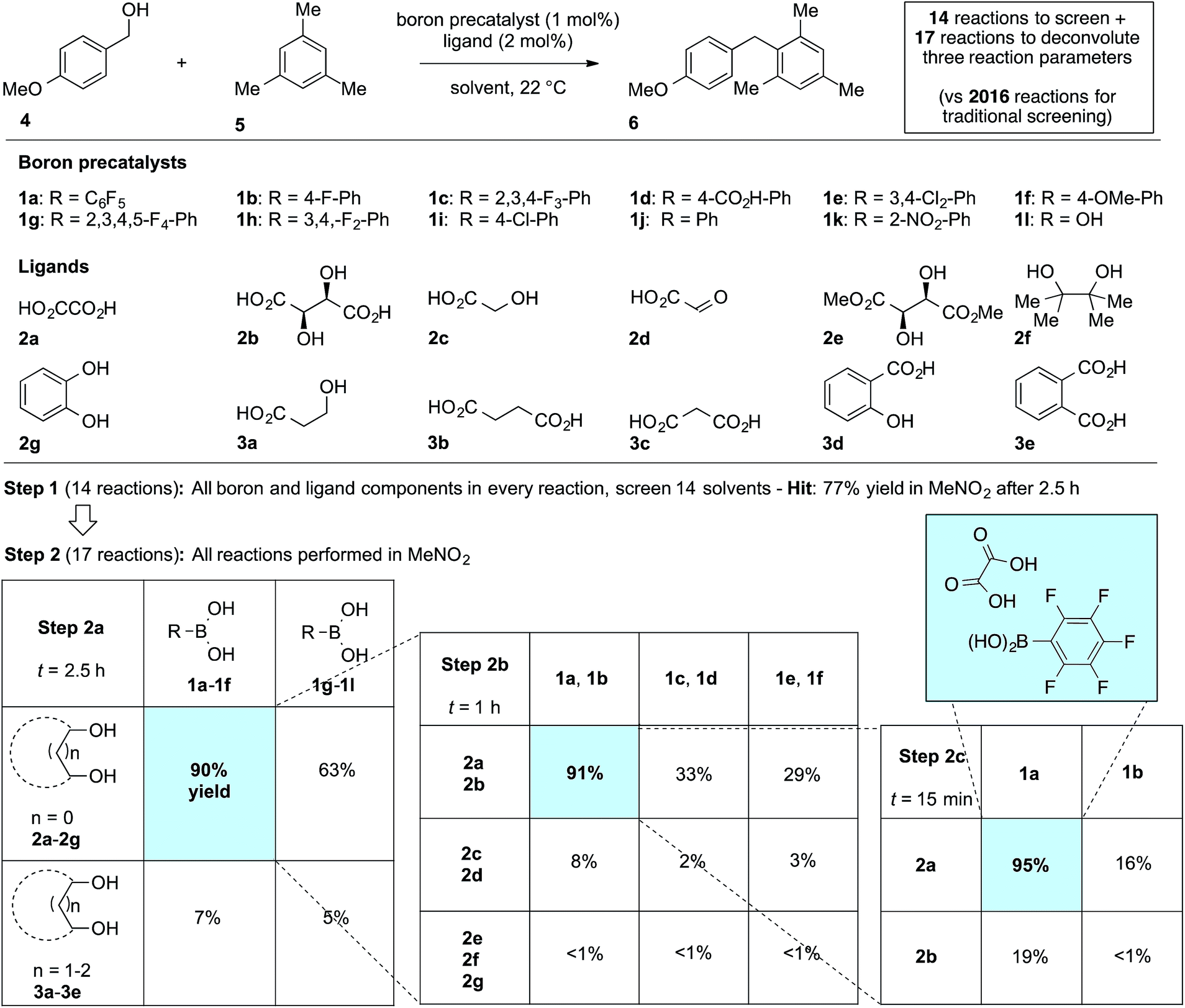
We first applied the catalyst discovery strategy to the selection of an in situ generated boron catalyst from a complex mixture of boronic acids and bidentate O-ligands.7,8 The dehydrative Friedel–Crafts reaction9 between p-methoxybenzyl alcohol (4) and mesitylene (5) to give diarylmethane (6) was chosen as a target reaction since it can be triggered by strong Brønsted or Lewis acids but is not enabled by boronic acids10 or carboxylic acids. Twelve boron precatalysts were chosen for screening, including eleven electronically diverse boronic acids and boric acid. Twelve bidentate O-ligands were chosen with the goal of maximizing their structural diversity,11 including diols, catechols, hydroxyacids and diacids. Screening a mixture of all boron precatalysts (1 mol% each) and all O-ligands (2 mol% each) against 14 different solvents at 22 °C resulted in 77% yield of 6 in MeNO2, 26% in MeCN, 3% in CH2Cl2 or DCE and <1% in the ten other solvents after 2.5 h (Fig. 1, Step 1). Thus, a lead result was identified in a three-dimensional screen requiring just 14 reactions, though the identities of the most active components were not yet known. Control experiments showed negligible reactivity when boronic acids alone or O-ligands alone were employed as catalysts. To facilitate the deconvolution process in MeNO2, the boron compounds were arbitrarily divided into two groups (1a–1f and 1g–1l). The O-ligands were also divided into two groups (2a–2g and 3a–3e). The four possible combinations of groups were screened with 1 mol% of each boron precatalyst and 2 mol% of each O-ligand present in the reaction at the same time (Fig. 1, Step 2a). The best result was found to come from the mixture of boron precatalysts 1a–1f and O-ligands 2a–2g, which went to completion after 2.5 h. The winning boron precatalysts were broken up into three arbitrary groups, 1a–1b, 1c–1d and 1e–1f. Likewise, the winning O-ligands were divided into three groups, 2a–2b, 2c–2d and 2e–2g. The most rapid of the nine reactions was that containing a mixture of 1a–1b and 2a–2b, which went to completion after 1 h (Fig. 1, Step 2b). The remaining four combinations were tested individually. Impressively, mixing 1 mol% of 1a with 2 mol% of 2a led to 95% yield of 6 after 15 min at room temperature (Fig. 1, Step 2c). As expected, the time for reaction completion decreased with each deconvolution step as the optimal catalyst is present in higher concentration. Only 31 reactions were required to screen and deconvolute three reaction parameters, with eventual success foreshadowed in Step 1 by a block of just 14 reactions. In contrast, screening the same three reaction parameters one at a time would require 2016 reactions, with no indication of success at the outset. Indeed, traditional linear screening arrives at the same result (see ESI†). Remarkably, the catalytic effect of 1a + 2a is faster than benchmark boron catalysts BF3·THF and B(C6F5)3·H2O,12 which gave 80% and 20% yield, respectively, with the remainder being starting materials under identical conditions and time. In contrast, addition of 1 mol% 1a or 2 mol% 2a in isolation led to <5% product after 4 h in both cases. Though the covalent assembly of boronic acids with oxalic acid to give dioxaborolanediones is known,13,14 catalysis by the combination of those components or by the resulting covalent adduct has not been reported.15
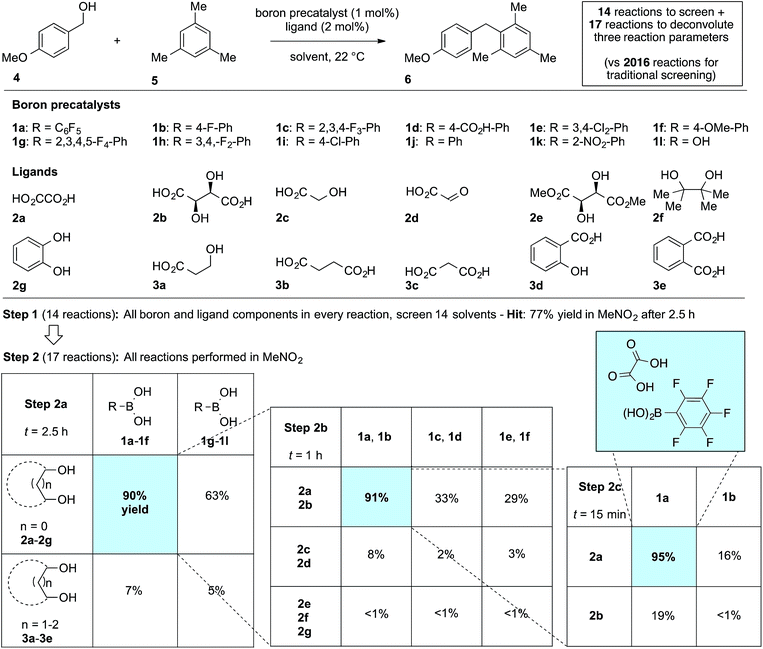 |
| | Fig. 1 Combinatorial discovery and deconvolution of in situ-generated boron catalysts for the dehydrative Friedel–Crafts reaction. Conditions: 1.0 equiv. 4 (0.2 M), 3.0 equiv. 5. Yield determined by 1H NMR in CDCl3 with DMSO (1 equiv.) as internal standard. | |
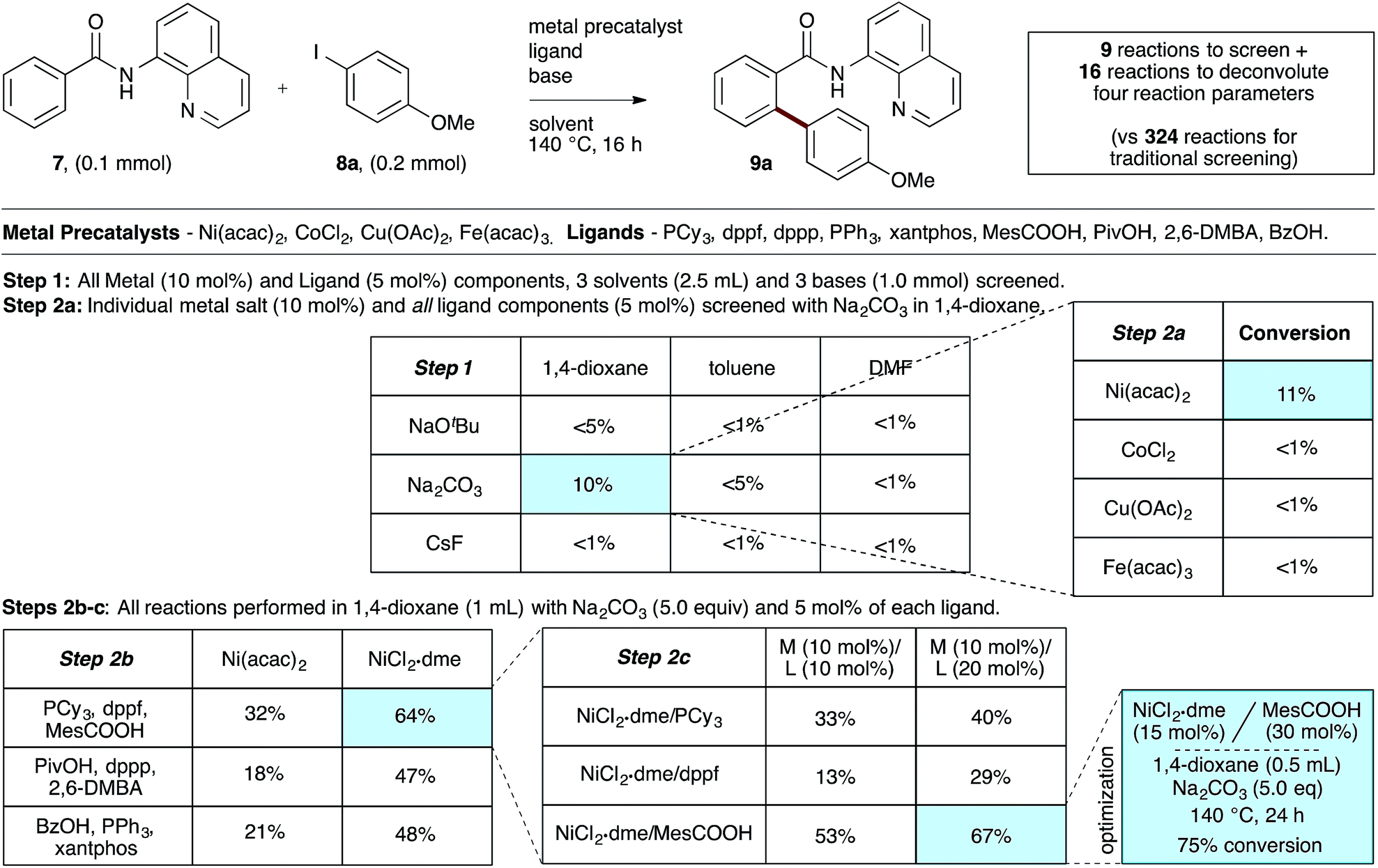
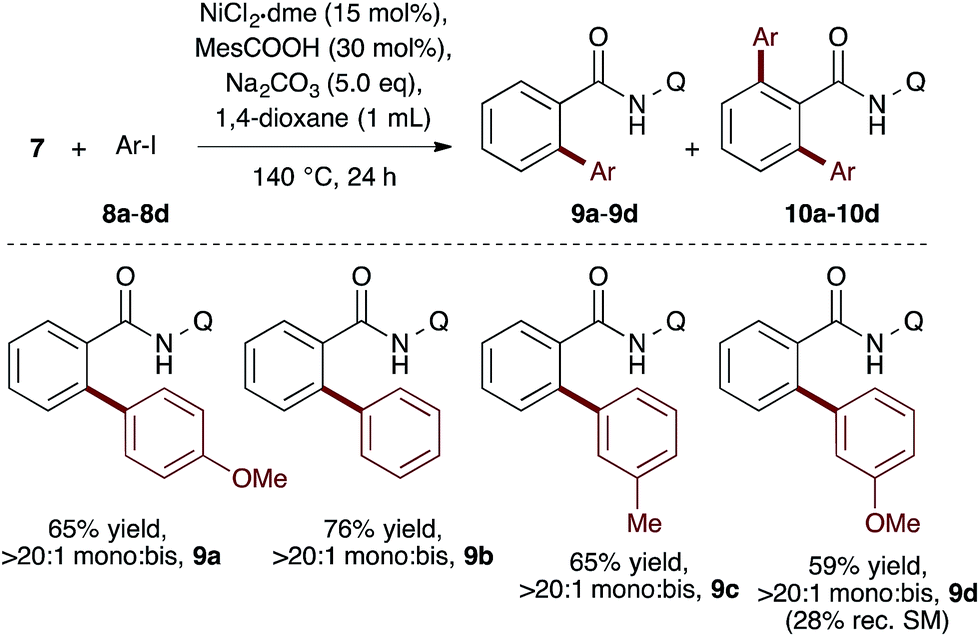
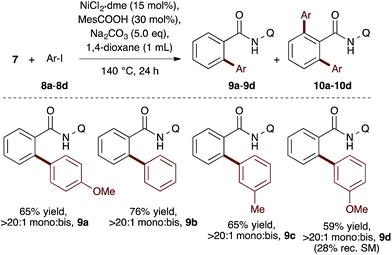
To evaluate the catalyst discovery strategy in a more complex system, we elected to explore transition metal catalyzed C–H activation, a reaction class we hypothesized would be highly applicable to such a combinatorial approach owing to the significant molecular recognition between catalyst and substrate. Bidentate chelation assistance for directed C–H activation has found increasing application in the past decade owing to the desirability of functionalizing ‘inert’ C–H bonds.16 Since the seminal report of Daugulis,17 the 8-aminoquinoline motif has proven effective as a directing group for C–H activation reactions in combination with Pd, Ru, Cu, Ni, Co and Fe catalyst systems.18 8-Aminoquinoline directed o-arylation of benzamides with aryl halides was described by Daugulis and coworkers under Pd catalysis17 and by Chatani and co-workers under Ru catalysis.19 In both cases, a blocking group at the ortho or meta position of the benzamide is required to avoid undesired products of bisarylation. As these were the sole reports of such a reaction, we felt the search for alternative catalytic systems represented an attractive challenge on which to refine our screening approach. After surveying the literature for metals and ligands employed in directed C–H activation reactions, four metal precatalysts and nine labile ligands were selected as potentially viable co-catalysts for such a transformation (Fig. 2). The coupling of unsubstituted benzamide 7 with 4-iodoanisole (8a) was chosen as the target reaction. With a mixture of 10 mol% of each metal precatalyst and 5 mol% of each ligand present in all reactions, three solvents and three bases were screened at 140 °C in nine total reactions (Step 1). Only the combination of Na2CO3 in 1,4-dioxane gave moderate (10%) conversion to the desired o-arylation product 9a. Thus, a lead result was identified in a four-dimensional screen in just nine reactions. Deconvolution of the metal salts revealed that Ni(acac)2 was the active catalyst precursor, whilst Fe(acac)3, CoCl2 and Cu(OAc)2 all exhibited no catalytic activity (Step 2a). Maintaining Na2CO3 as base and 1,4-dioxane as solvent, the nine ligands were arbitrarily divided into groups of three, and evaluated in combination with Ni(acac)2 and NiCl2·dme. A significant enhancement of reaction efficiency was observed in the reactions employing PCy3, MesCOOH and dppf as ligand (Step 2b). Continued deconvolution established NiCl2·dme and MesCOOH as an efficient combination (Step 2c, 67% conversion). Further tweaking of the reaction conditions enabled the conversion to be increased to 75%. Surprisingly, the ratio of 9a to bisarylation product 10a was >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by 1H NMR. Application of these reaction conditions with various aryl iodides allowed for the desired o-arylation products to be isolated in 59–76% yields, all with >20:1 selectivity for monoarylation (Table 1).
:1 by 1H NMR. Application of these reaction conditions with various aryl iodides allowed for the desired o-arylation products to be isolated in 59–76% yields, all with >20:1 selectivity for monoarylation (Table 1).
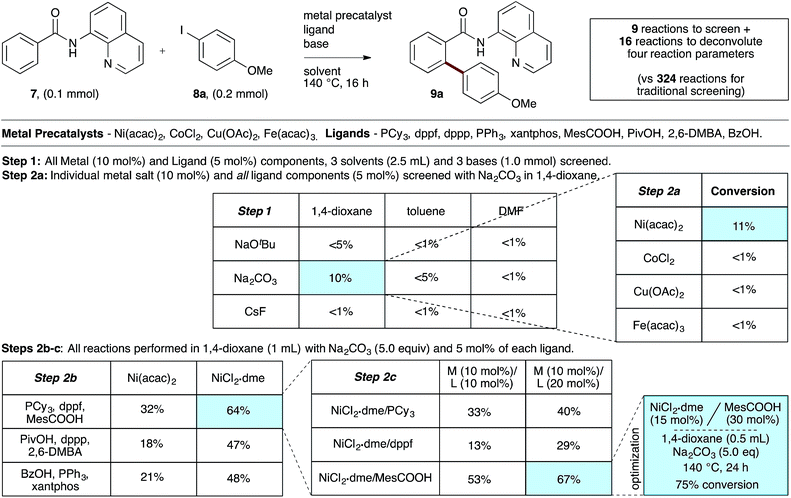 |
| | Fig. 2 Combinatorial discovery and deconvolution of in situ-generated catalysts for C–H monoarylation of unsubstituted benzamide 7. Conversion determined by 1H NMR of the crude mixtures in CDCl3. 2,6-DMBA = 2,6-dimethoxybenzoic acid. | |
Table 1 Reaction scope of monoarylationa
|
Isolated yield after column chromatography. Q = 8-quinoline.
|
|
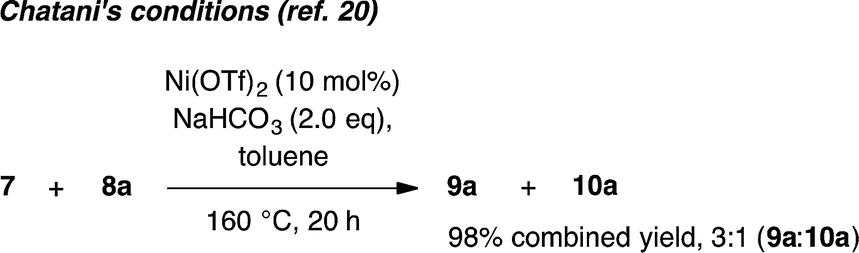
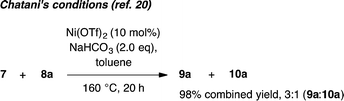
Whilst completing the deconvolution of this reaction system, Chatani and coworkers disclosed a closely related Ni-catalyzed ortho-arylation system.20,21 Their system proceeds under Ni(OTf)2 catalyzed ligand-free conditions at 160 °C and, like existing Pd17 and Ru19 catalyzed methods, is typically not useful for unsubstituted phenyl benzamides due to the significant formation of bisarylation adducts. Indeed, reaction of 7 with 8a under Chatani's conditions gave complete conversion to a 3:1 mixture of mono- and bisarylation products in our hands (eqn (1)). In contrast, the system described herein exhibits a significant ligand effect,22 proceeds at 140 °C and furnishes >20:1 selectivity for monoarylation products 9a–9d. Four parallel screens requiring only 25 total reactions identified a selective set of conditions for directed monoarylation of 7. Using an inexpensive Ni(II) salt and 2,4,6-trimethylbenzoic acid, hitherto unreported biaryl products have been accessed in synthetically useful yields.
| |  | (1) |
Conclusion
In conclusion, we have described an approach for catalyst discovery using complex mixtures of potential catalyst components. Our “proof of concept” attempts to apply this strategy have uncovered a mild and novel boron catalyst for the dehydrative Friedel–Crafts reaction and new conditions for Ni-catalyzed directed C–H arylation of benzamide that are highly selective for monoarylation in the absence of blocking groups. Initial hits for both cases were uncovered in 14 reactions or less and required a small number of additional deconvolution reactions to identify key catalyst components. The number of steps in the deconvolution process can be tailored to the preference of the user by adjusting the number of reactions used per deconvolution step. Though the approach's generality and its ability to handle larger sets of catalyst components remains to be seen, screening approaches employing rationally chosen complex mixtures should be useful for chemists lacking the resources for high throughput experimentation, particularly for reactions involving significant molecular recognition between substrate and catalyst. We anticipate this approach will also increase the chances of uncovering unexpected cooperative effects between catalytic components that would not otherwise be assayed in the same reaction vessel.
Acknowledgements
This work was supported in part by a LabEx CSC “Chemistry of Complex Systems” grant and the Marie Curie Actions FP7-PEOPLE Program (CIG-2012-326112). E.W. thanks the French government for a MRT fellowship. E.R. thanks the Leverhulme Trust for a fellowship. We thank Dr Malik Hellal for valuable discussions.
Notes and references
- For a review, see: K. D. Collins, T. Gensch and F. Glorius, Nat. Chem., 2014, 6, 859–871 CrossRef CAS PubMed.
-
(a) C. Hinderling and P. Chen, Int. J. Mass Spectrom., 2000, 195–196, 377–383 CrossRef;
(b) P. J. Fagan, E. Hauptman, R. Shapiro and A. Casalnuovo, J. Am. Chem. Soc., 2000, 122, 5043–5051 CrossRef CAS;
(c) J. W. Szewczyk, R. L. Zuckerman, R. G. Bergman and J. A. Ellman, Angew. Chem., Int. Ed., 2001, 40, 216–219 CrossRef CAS;
(d) T. R. Boussie, G. M. Diamond, C. Goh, K. A. Hall, A. M. LaPointe, M. Leclerc, C. Lund, V. Murphy, J. A. W. Shoemaker, U. Tracht, H. Turner, J. Zhang, T. Uno, R. K. Rosen and J. C. Stevens, J. Am. Chem. Soc., 2003, 125, 4306–4317 CrossRef CAS PubMed;
(e) M. W. Kanan, M. M. Rozenman, K. Sakurai, T. M. Snyder and D. R. Liu, Nature, 2004, 431, 545–549 CrossRef CAS PubMed;
(f) A. B. Beeler, S. Su, C. A. Singleton and J. A. Porco, J. Am. Chem. Soc., 2007, 129, 1413–1419 CrossRef CAS PubMed;
(g) M. M. Rozeman, M. W. Kanan and D. R. Liu, J. Am. Chem. Soc., 2007, 129, 14933–14938 CrossRef PubMed;
(h) O. Trapp, S. K. Weber, S. Bauch and W. Hofstadt, Angew. Chem., Int. Ed., 2007, 46, 7307–7310 CrossRef CAS PubMed;
(i) A. Teichert and A. Pfaltz, Angew. Chem., Int. Ed., 2008, 47, 3360–3362 CrossRef CAS PubMed;
(j) C. A. Müller and A. Pfaltz, Angew. Chem., Int. Ed., 2008, 47, 3363–3366 CrossRef PubMed;
(k) C. Markert, P. Rösel and A. Pfaltz, J. Am. Chem. Soc., 2008, 130, 3234–3235 CrossRef CAS PubMed;
(l) H. Kinoshita, O. J. Ingham, W. W. Ong, A. B. Beeler and J. A. Porco Jr, J. Am. Chem. Soc., 2010, 132, 6412–6418 CrossRef CAS PubMed;
(m) T. J. Montavon, J. Li, J. R. Cabrera-Pardo, M. Mrksich and S. A. Kozmin, Nat. Chem., 2012, 4, 45–51 CrossRef CAS PubMed;
(n) J. Quinton, S. Kolodych, M. Chaumonet, V. Bevilacqua, M.-C. Nevers, H. Volland, S. Gabillet, P. Thuéry, C. Créminon and F. Taran, Angew. Chem., Int. Ed., 2012, 51, 6144–6148 CrossRef CAS PubMed.
-
(a) D. W. Robbins and J. F. Hartwig, Science, 2011, 333, 1423–1427 CrossRef CAS PubMed;
(b) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed.
- J. Wieland and B. Breit, Nat. Chem., 2010, 2, 832–837 CrossRef CAS PubMed.
-
(a) J. R. Schmink, A. Bellomo and S. Berritt, Aldrichimica Acta, 2013, 46, 71–80 CAS;
(b) A. Bellomo, N. Celebi-Olcum, X. Bu, N. Rivera, R. T. Ruck, C. J. Welch, K. N. Houk and S. D. Dreher, Angew. Chem., Int. Ed., 2012, 51, 6912–6915 CrossRef CAS PubMed;
(c) A. B. Santanilla, E. L. Regalado, T. Pereira, M. Shevlin, K. Bateman, L.-C. Campeau, J. Schneeweis, S. Berritt, Z.-C. Shi, P. Nantermet, Y. Liu, R. Helmy, C. J. Welch, P. Vachal, I. W. Davies, T. Cernak and S. D. Dreher, Science, 2015, 347, 49–53 CrossRef PubMed.
- For a pioneering example of screening mixtures of two ligands, see: M. T. Reetz, T. Sell, A. Meiswinkel and G. Mehler, Angew. Chem., Int. Ed., 2003, 42, 790–793 CrossRef CAS PubMed.
-
Boronic Acids – Preparation and Applications in Organic Synthesis Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2nd edn, 2011 Search PubMed.
- For recent reviews on boron catalysis, see:
(a) J. N. Payette and H. Yamamoto, in ref. 6, pp. 551–590;
(b) E. Dimitrijevic and M. S. Taylor, ACS Catal., 2013, 3, 945–962 CrossRef CAS.
- For recent reviews on the dehydrative Friedel-Crafts reaction, see:
(a) M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 6 Search PubMed;
(b) E. Emer, R. Sinisi, M. G. Capdevila, D. Petruzziello, F. De Vincentiis and P. G. Cozzi, Eur. J. Org. Chem., 2011, 647–666 CrossRef CAS.
- Boronic acid-catalyzed activation of allylic and benzylic alcohols requires much high temperatures and catalyst loadings, see:
(a) J. A. McCubbin, H. Hosseini and O. V. Krokhin, J. Org. Chem., 2010, 75, 959–962 CrossRef CAS PubMed;
(b) J. A. McCubbin and O. V. Krokhin, Tetrahedron Lett., 2010, 51, 2447–2449 CrossRef CAS PubMed;
(c) H. Zheng, S. Ghanbari, S. Nakamura and D. G. Hall, Angew. Chem., Int. Ed., 2012, 51, 6187–6190 CrossRef CAS PubMed.
- K. D. Shimizu, B. M. Cole, C. A. Krueger, K. W. Kuntz, M. L. Snapper and A. H. Hoveyda, Angew. Chem., Int. Ed., 1997, 36, 1704–1707 CrossRef CAS.
- M. Hellal, F. C. Falk, E. Wolf, M. Dryzhakov and J. Moran, Org. Biomol. Chem., 2014, 12, 5990–5994 CAS.
-
(a) K. Kustin and R. Pizer, J. Am. Chem. Soc., 1969, 91, 317–322 CrossRef CAS;
(b) S. Friedman, B. Pace and R. Pizer, J. Am. Chem. Soc., 1974, 96, 5381–5384 CrossRef CAS;
(c) S. Friedman and R. Pizer, J. Am. Chem. Soc., 1975, 97, 6059–6062 CrossRef CAS;
(d) L. Babcock and R. Pizer, Inorg. Chem., 1980, 19, 56–61 CrossRef CAS;
(e) R. Pizer and P. J. Ricatto, Inorg. Chem., 1994, 33, 2402–2406 CrossRef CAS.
-
(a) P. I. Paetzold, W. Sheibitz and E. Scholl, Z. Naturforsch., B: Chem. Sci., 1971, 26, 646–649 CAS;
(b) P. Paetzold, S. Neyses and L. Géret, Z. Anorg. Allg. Chem., 1995, 621, 732–736 CrossRef CAS;
(c) A. S. Voisin, A. Bouillon, J.-C. Lancelot, A. Lesnard, H. Oulyadi and S. Rault, Tetrahedron Lett., 2006, 47, 2165–2169 CrossRef CAS PubMed;
(d) L. F. Li, H. S. Lee, H. Li, X. Q. Yang and X. J. Huang, Electrochem. Commun., 2009, 11, 2296–2299 CrossRef CAS PubMed.
- Tri(pentafluorophenyl)boroxine was ruled out as an in situ generated catalyst as it rapidly hydrolysed under the reaction conditions.
- G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed.
-
(a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed;
(b) E. T. Nadres, G. I. F. Santos, D. Shabashov and O. Daugulis, J. Org. Chem., 2013, 78, 9689–9714 CrossRef CAS PubMed.
- M. Corbet and F. De Campo, Angew. Chem., Int. Ed., 2013, 52, 9896–9898 CrossRef CAS PubMed.
- Y. Aihara and N. Chatani, Chem. Sci., 2013, 4, 664–670 RSC.
- A. Yokota, Y. Aihara and N. Chatani, J. Org. Chem., 2014, 79, 11922–11932 CrossRef CAS PubMed.
- For related Ni-catalyzed alkylation of 8-aminoquinoline benzamides, see:
(a) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 136, 898–901 CrossRef PubMed;
(b) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2013, 136, 1789–1792 CrossRef PubMed.
- In the absence of carboxylate ligand only 52% conversion of 9a was observed instead of 75%. In contrast, Chatani and coworkers found that carboxylate ligands suppressed reactivity in substrates possessing a blocking group (see Table 1, entry 13 in ref. 20).
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data. See DOI: 10.1039/c5sc00268k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a