
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic asymmetric chlorinative dearomatization of naphthols†
Qin
Yin
,
Shou-Guo
Wang
,
Xiao-Wei
Liang
,
De-Wei
Gao
,
Jun
Zheng
and
Shu-Li
You
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China. E-mail: slyou@sioc.ac.cn; Fax: +86-21-5492-5087
First published on 27th April 2015
Abstract
An organocatalytic asymmetric chlorinative dearomatization of naphthols was realized for the first time, providing chiral naphthalenones with a Cl-containing all-substituted stereocenter in excellent yields and enantioselectivity (up to 97% yield and 96% ee). The reaction features mild reaction conditions, good tolerance of diverse functional groups and simple reaction operation.
Phenol and its derivatives are readily accessible chemical feedstocks and are widely utilized in chemical synthesis.1 Among the versatile transformations, the catalytic asymmetric dearomatization (CADA) reaction2 of phenol derivatives offers a facile and straightforward route to access chiral cyclic enones with one quaternary carbon stereogenic center. Therefore, development of the CADA reaction of phenol derivatives has received increasing attention recently.3 Strategies for the direct catalytic asymmetric dearomatization of phenols, including hypervalent iodine4 or transition metal-catalyzed5 oxidation, transition metal-catalyzed allylic alkylation6 or arylation,7 and chiral phosphoric acid catalyzed amination,8 have been elegantly unveiled.9 Very recently, Toste and co-workers reported a highly enantioselective dearomative fluorination of phenols by chiral anion phase-transfer catalysis.10 Inspired by these pioneering works, we envisaged that the asymmetric chlorinative dearomatization of phenols via homogeneous catalysis might be possible, providing interesting products with a C–Cl bond-containing chiral center.11 However, compared with electrophilic fluorination reagents such as Selectfluor, electrophilic chlorination reagents such as N-chlorosuccinimide (NCS) and DCDMH (1,3-dichloro-5,5-dimethylhydantoin) have much higher electrophilic reactivity, which may cause a significant amount of background reaction or undesired reactions such as electrophilic aromatic substitution at the ortho or para-position (the problem of regioselectivity). In addition, the construction of a Cl-containing all-substituted stereocenter with high enantioselectivity via dearomatization of phenols remains underexplored. To test our hypothesis, commercially available cinchonine derivatives such as (DHQD)2PHAL were chosen as chiral catalysts since they are privileged catalysts for the asymmetric halofunctionalization of alkenes.12,13 After extensive preliminary investigation of substituted phenols, we found that naphthols are suitable substrates for the chlorinative dearomatization process.14 Herein, we report such a highly enantioselective dearomative chlorination of naphthols under catalysis by (DHQD)2PHAL, providing an efficient synthesis of chiral naphthalenones with an α-Cl-containing all-substituted stereocenter (Scheme 1).15
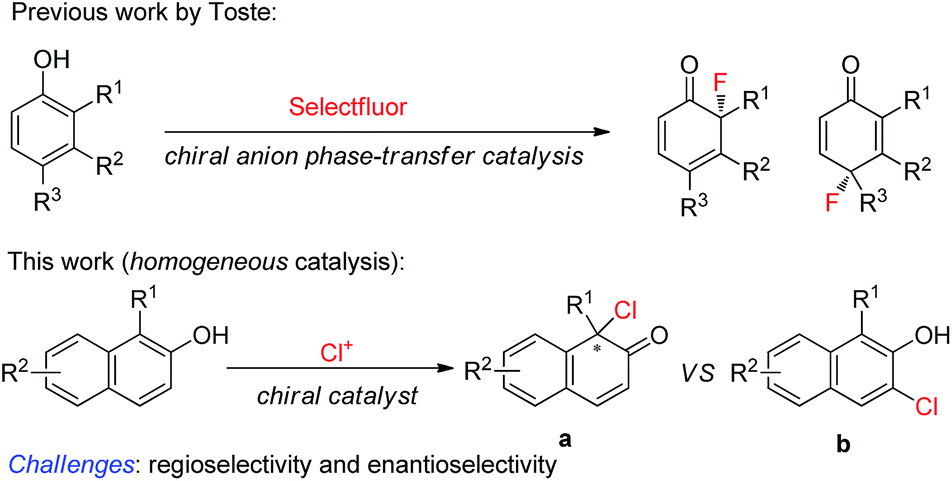 | ||
| Scheme 1 Asymmetric chlorination of naphthol derivatives via homogeneous catalysis. | ||
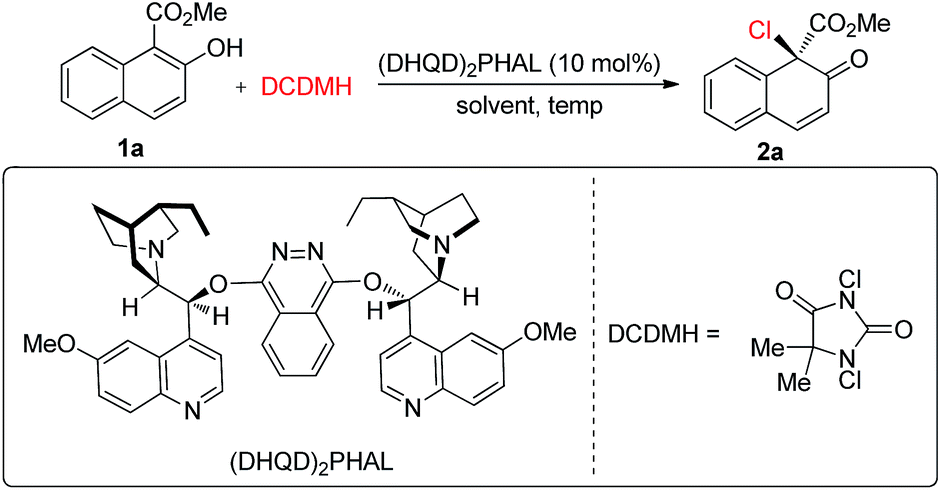
We commenced our studies by testing the reactions between commercially available substrate 1a and 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) in the presence of 10 mol% (DHQD)2PHAL.16 Firstly, various solvents were surveyed at room temperature. The dearomative product 2a could be obtained in 94% yield in toluene with encouraging enantioselectivity observed (52% ee, entry 1). Further screening of chlorine-containing solvents revealed that CCl4 could give comparable results, affording 2a in 54% ee (entries 2–5). To our surprise, the screening of other solvents revealed that CS2 gave the best results and 2a could be produced in 90% yield with 62% ee with a prolonged reaction time (entries 6–8). However, the enantioselectivity of 2a was only slightly elevated from 62% ee to 64% ee when the reaction was carried out at −30 °C in CS2 (entry 9). Gratifyingly, a significant increase in enantioselectivity could be achieved when the reaction was performed in toluene at a decreased temperature. 2a was obtained in 95% yield with 92% ee by carrying out the reaction at −78 °C (entry 11). Notably, by changing the catalyst from (DHQD)2PHAL to (DHQ)2PHAL, 2a with 90% ee, with the opposite configuration, could be obtained in an almost quantitative yield (entry 12). A decrease in the catalyst loading from 10 mol% to 2 mol% led to a prolonged reaction time, however, the yield and enantioselectivity of 2a remained at an excellent level (98% yield, 90% ee, entry 13) (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Solvent | Temp (°C) | Time (h) | Yieldb (%) | Eec (%) |
| a Reactions were performed with 1a (0.1 mmol), DCDMH (0.12 mmol) and 10 mol% of (DHQD)2PHAL at rt in an open flask. b Isolated yield. c Determined by HPLC analysis. d 10 mol% of (DHQ)2PHAL was utilized. e 2 mol% of (DHQD)2PHAL was utilized. | |||||
| 1 | Toluene | Rt | 1 | 94 | 52 |
| 2 | DCM | Rt | 1 | 92 | 36 |
| 3 | DCE | Rt | 1 | 93 | 25 |
| 4 | CHCl3 | Rt | 1 | 92 | 34 |
| 5 | CCl4 | Rt | 1 | 92 | 54 |
| 6 | Hexane | Rt | 1 | 89 | 32 |
| 7 | THF | Rt | 1 | 88 | 12 |
| 8 | CS2 | Rt | 4 | 90 | 62 |
| 9 | CS2 | −30 | 24 | 91 | 64 |
| 10 | Toluene | −30 | 8 | 94 | 75 |
| 11 | Toluene | −78 | 10 | 95 | 92 |
| 12d | Toluene | −78 | 10 | 94 | −90 |
| 13e | Toluene | −78 | 31 | 98 | 90 |
Under the optimized reaction conditions, 2-naphthols with different substituents were synthesized to test the generality of this asymmetric chlorination process (Scheme 2). Firstly, the substituent effect of the ester group (Me, Et, allyl) was evaluated and, in all cases, excellent yields were achieved. With the increase in steric hindrance from a methyl, to an ethyl, to an allyl group, the enantioselectivity of the corresponding products 2a–2c showed a decreasing trend. However, the levels were still excellent (2a–2c, 91–95% yields, 86–92% ee). The substituent effect on the core of 2-hydroxy-1-naphthoate was next investigated. Electron-withdrawing groups such as 6-Br and 6-CN, or aryl groups such as Ph, 3,5-(Me)2C6H3 and 4-F-C6H4 were well tolerated. The corresponding products were all obtained in excellent yields and enantioselectivity (2d–2h, 80–88% yields, 93–96% ee). Electron-donating groups such as 6-Me and 6-phenethyl were also well tolerated and the corresponding products 2i and 2j were obtained in 87% yield, 93% ee and 90% yield, 94% ee, respectively. To our delight, the unsaturated double bond or triple bond in the substrates did not interfere with the reactivity or enantiocontrol of the reaction. For instance, reactions with substrate 1k with a styryl group and substrate 1l with a phenylethynyl group could proceed smoothly to give the corresponding products 2k and 2l in 88% yield, 94% ee and 91% yield, 93% ee, respectively. Furthermore, substrate 1m with both a triple bond and a hydroxyl group was also well tolerated, and product 2m was obtained in 80% yield with 78% ee. Various substituents at other positions of 2-hydroxy-1-naphthoate were also surveyed. Substrate 1n with 4-Br, substrate 1o with 7-Br and substrate 1p with 7-MeO could all be successfully converted to their corresponding products in excellent yields and enantioselectivity (2n–2p, 85–95% yields, 90–94% ee). In addition, product 2q with a 3-Br substituent was obtained in 88% yield with 73% ee. Apart from 2-hydroxy-1-naphthoates, 2-naphthols with an electron-donating group at the 1-position were also suitable substrates. For instance, substrate 1r with 1,3-dimethyl groups could be smoothly transformed to the corresponding product 2r (91% yield, 82% ee). To be noted, in the presence of (DHQ)2PHAL, substrates 1r and 1s with 1-Me and 3-Ph, respectively, could also work well in this reaction to yield 2r (94% yield, 86% ee) and 2s (89% yield, 82% ee) respectively. The absolute configuration of the product was determined, by X-ray analysis of enantiopure 2d, as R (see the ESI† for details).
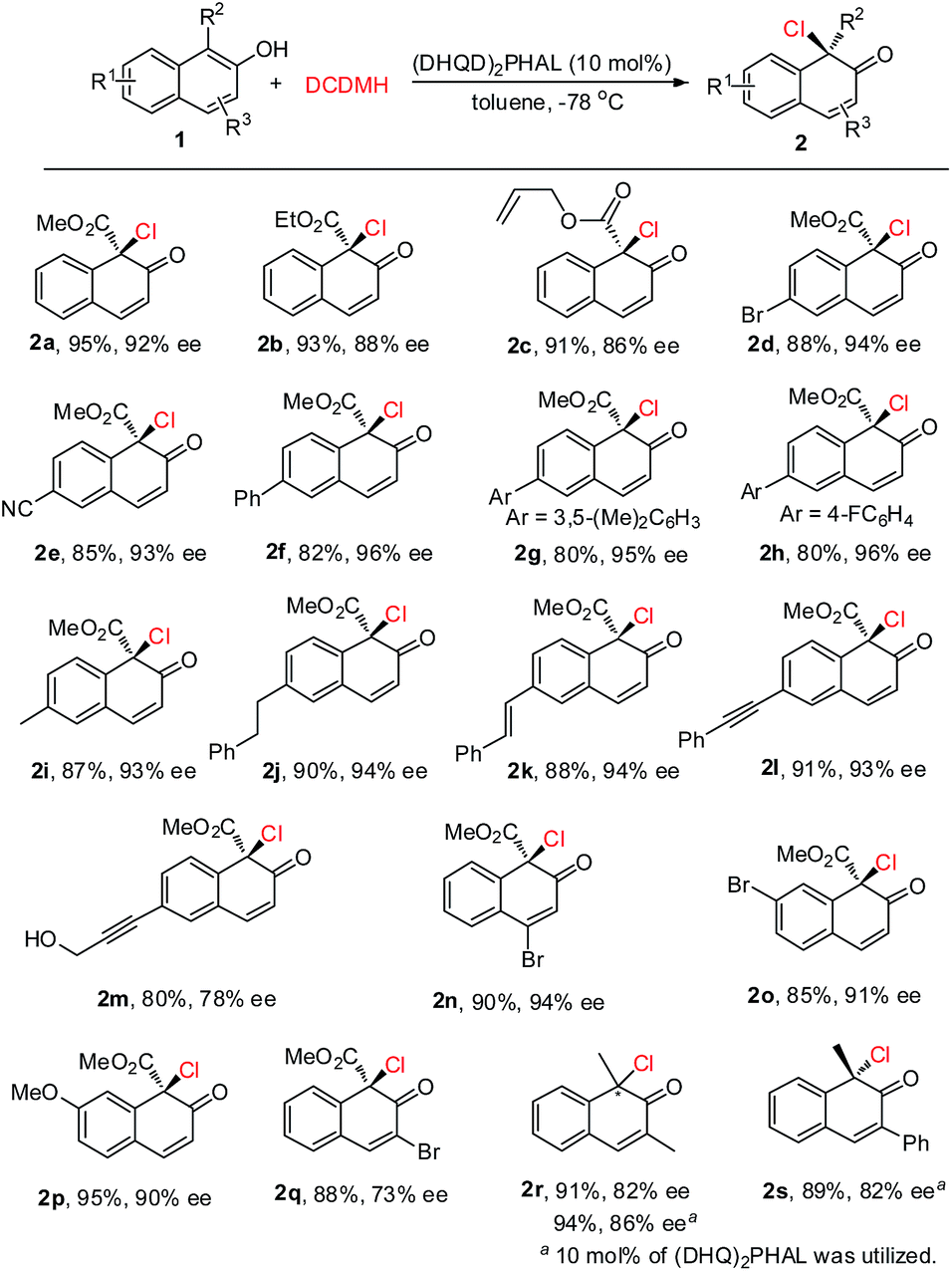 | ||
| Scheme 2 Evaluation of substrate scope. | ||
Besides 2-naphthols, methyl 1-hydroxy-2-naphthoate, 1t, was also well tolerated in this dearomative chlorination reaction. Under slightly optimized conditions (in the presence of 10 mol% of (DHQD)2PYR in CHCl3/CCl4 at −70 °C), product 2t was obtained in 94% yield with 90% ee (Scheme 3), and its structure was confirmed by X-ray analysis. To our knowledge, highly enantioselective intermolecular dearomatization of 1-naphthol derivatives has not been reported yet.4b
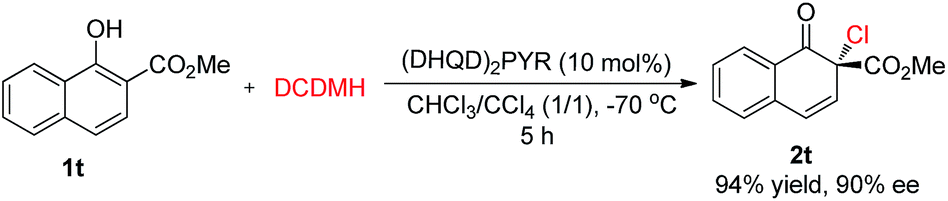 | ||
| Scheme 3 Asymmetric chlorinative dearomatization reaction of a 1-naphthol derivative. | ||
We also tested the asymmetric bromination of 1a with 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) under the standard reaction conditions (eqn (1), Scheme 4). The desired brominative product 2u was obtained in 96% yield with 9% ee. The almost racemic result was possibly due to the very strong background reaction. To our surprise, a further attempt using 2-hydroxy-1-naphthoic acid (1v) as the substrate in the presence of (DHQD)2PHAL provided the achiral decarboxylative compound 2v in quantitative yield (eqn (2), Scheme 4).
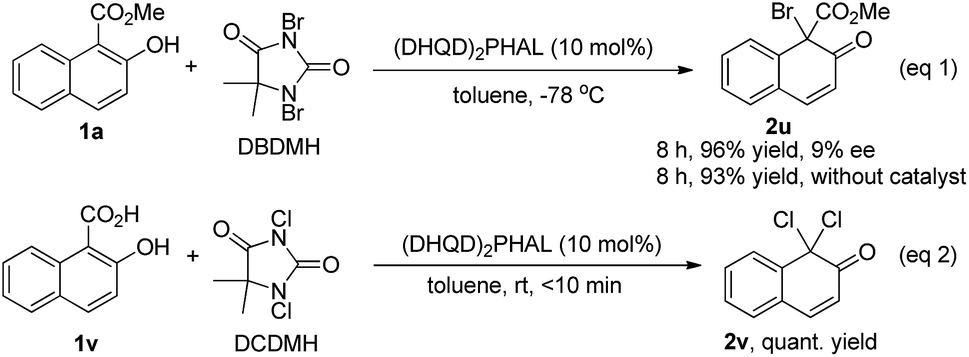 | ||
| Scheme 4 Bromination of 1a with DBDMH (eqn 1) and reaction of 2-hydroxy-1-naphthoic acid (1v) with DCDMH (eqn 2). | ||
To evaluate the practicality of this dearomative strategy, gram-scale reactions of 1a and 1t were performed. As displayed in Scheme 5, the corresponding products 2a and 2t could be obtained in excellent yields without a notable reduction in the enantioselectivity (91% ee and 87% ee, respectively).
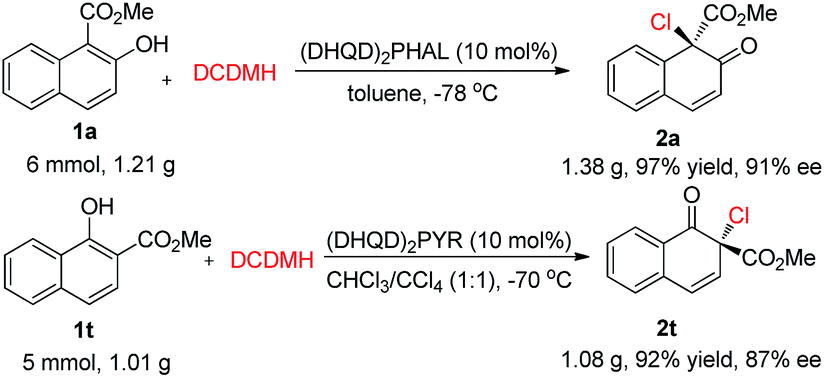 | ||
| Scheme 5 Gram-scale reactions. | ||
To further show the synthetic utility of this newly developed protocol, several transformations of the products were carried out (Scheme 6). With different workup procedures, 2a could be converted to the chiral allylic alcohol 3a or epoxide 3b in moderate yields with excellent diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) via the reduction of carbonyl by Dibal-H (eqn (1) and (2), Scheme 6). When 2a was subjected to oxidative bromination conditions, 2q could be achieved in 74% yield with 89% ee, serving as a complementary route to access 2q with high enantioselectivity (eqn (3), Scheme 6). In addition, 2t could be converted to highly functionalized compounds through stereoselective halogenation of the double bond. For instance, the dibromination product 3c could be obtained as a single diastereoisomer under bromination conditions, without reduction in the enantioselectivity (85% yield, 88% ee, eqn (4), Scheme 6). Furthermore, multi-functionalized chlorohydrin 3d was obtained under electrophilic chlorination reaction conditions in 70% yield with good stereochemical integrity and high diastereoselectivity (dr = 11:1, 86% ee for the major diastereoisomer, eqn (5), Scheme 6).17
:1) via the reduction of carbonyl by Dibal-H (eqn (1) and (2), Scheme 6). When 2a was subjected to oxidative bromination conditions, 2q could be achieved in 74% yield with 89% ee, serving as a complementary route to access 2q with high enantioselectivity (eqn (3), Scheme 6). In addition, 2t could be converted to highly functionalized compounds through stereoselective halogenation of the double bond. For instance, the dibromination product 3c could be obtained as a single diastereoisomer under bromination conditions, without reduction in the enantioselectivity (85% yield, 88% ee, eqn (4), Scheme 6). Furthermore, multi-functionalized chlorohydrin 3d was obtained under electrophilic chlorination reaction conditions in 70% yield with good stereochemical integrity and high diastereoselectivity (dr = 11:1, 86% ee for the major diastereoisomer, eqn (5), Scheme 6).17
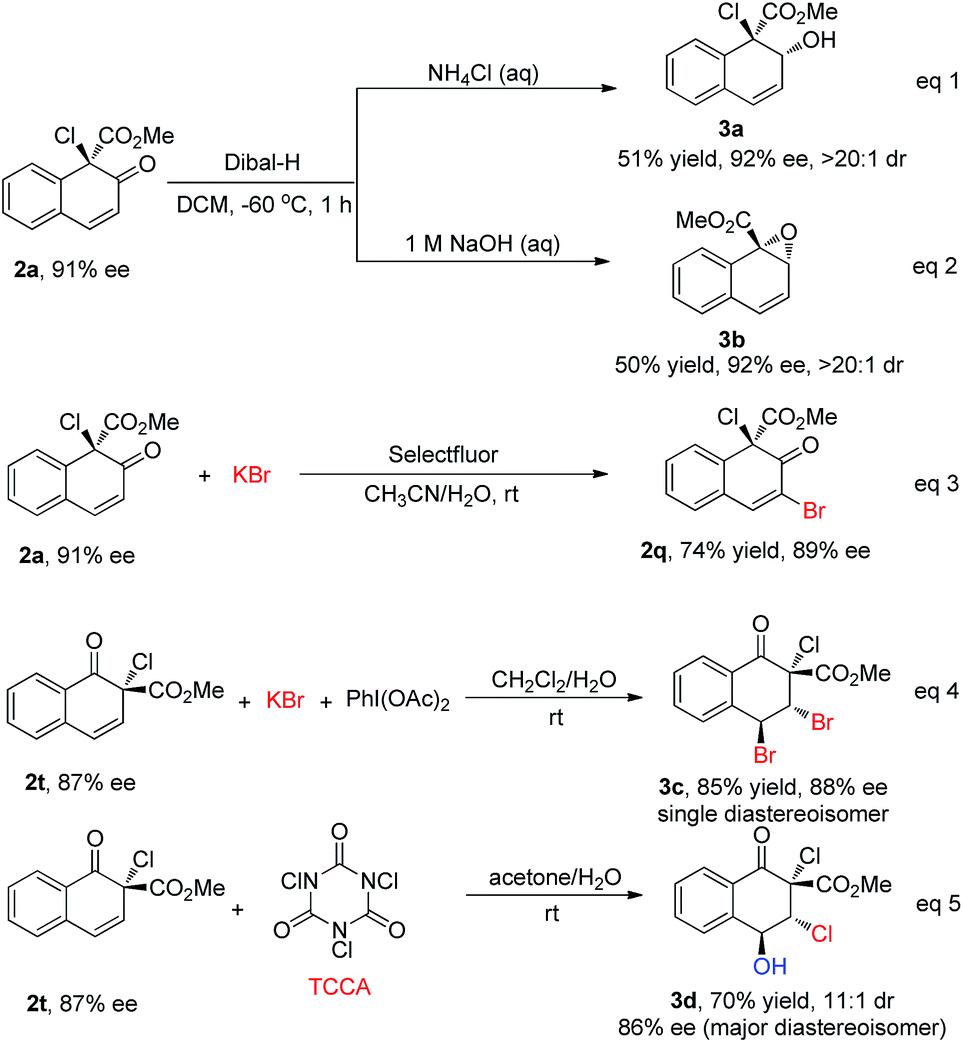 | ||
| Scheme 6 Transformations of products. | ||
As for the working model of this reaction, inspired by the pioneering studies by Nicolaou,13c Hennecke13k and Tang,13l we speculate that the phthalazine nitrogen in the catalyst interacts with the hydroxyl group of naphthol via a hydrogen bond to increase the nucleophilic property of the 1-position (Fig. 1). In addition, the intramolecular hydrogen bond of 1a itself also makes a contribution to a relatively rigid chiral environment. On the other hand, the tertiary amine nitrogen in quinuclidine acts as a Lewis base to activate the chloronium species to provide a bifunctional catalytic model, which is in line with Borhan's research.13a
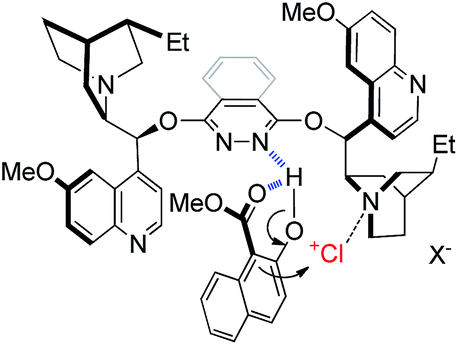 | ||
| Fig. 1 Proposed working model. | ||
To investigate this proposal, several control experiments were carried out, as shown in Scheme 7. Firstly, when substrate 1u, of which the hydroxyl was protected by a methyl group, was subjected to the chlorination conditions, no reaction occurred (eqn (1), Scheme 7). When the protecting group was changed to TMS, the reaction of 1v proceeded very slowly to give the desired product 2a in only 45% yield with 20% ee (eqn (2), Scheme 7). When a homogeneous toluene solution of the potassium salt of 1a, prepared in situ by treating 1a with 1.05 equiv of KOMe and 18-crown-6, was subjected to the standard conditions, product 2a was obtained in 99% yield, however in an almost racemic form (eqn (3), Scheme 7). The control experiment (eqn (4), Scheme 7) revealed that the addition of methanol and 18-crown-6 did not have any effect on the yield or enantioselectivity of 2a. All these experiments suggested that the hydroxyl group in the substrate is relevant not only to the reactivity but also to the enantiocontrol, possibly playing a role as a hydrogen bond donor. Furthermore, the addition of benzoic acid dramatically decreased the reaction rate as well as the enantioselectivity of 2a from 92% ee to 64% ee (eqn (5), Scheme 7). The possible protonation of the quinuclidine nitrogen atom by the acid decreased the catalytic efficiency of the catalyst. Despite the fact that some promising experimental evidence was obtained, the working model is postulated and needs further studies.
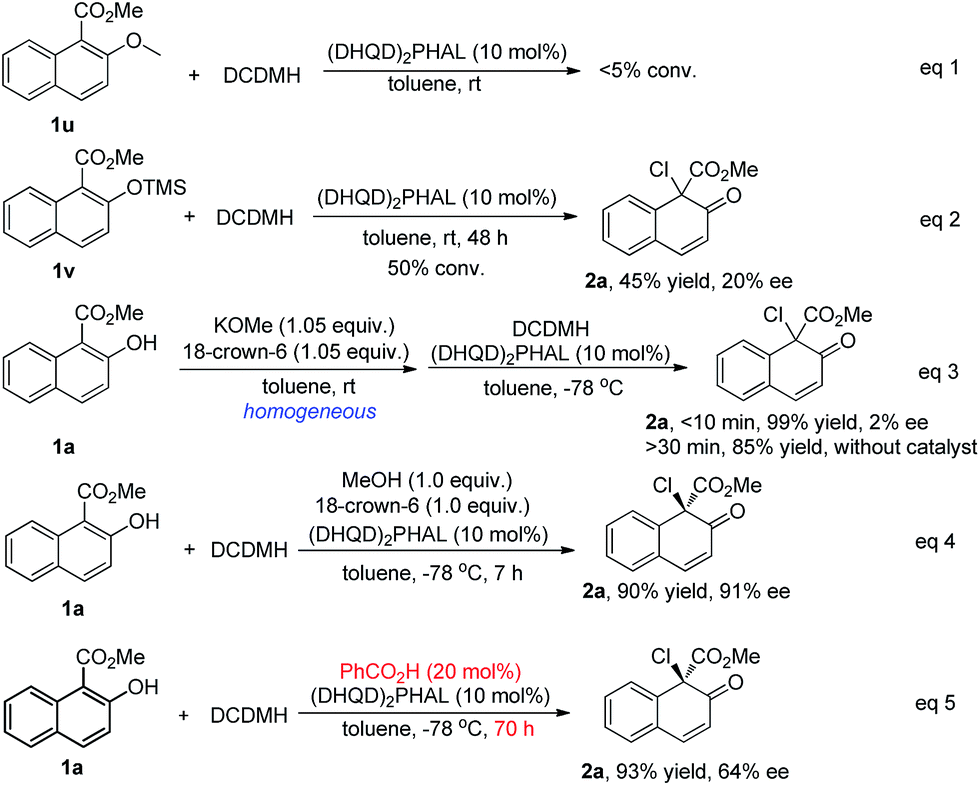 | ||
| Scheme 7 Control experiments. | ||
In summary, we have realized for the first time the organocatalytic asymmetric chlorinative dearomatization of naphthols, providing chiral naphthalenones with a Cl-containing all-substituted stereocenter in excellent yields and enantioselectivity. The reaction features mild reaction conditions, good tolerance of diverse functional groups and simple reaction operation. Notably, highly enantioselective intermolecular dearomative chlorination of 1-naphthol derivative was also realized. In addition, the gram-scale reactions and practical transformations of the products reveal the potential synthetic utility of this method.
Acknowledgements
We thank the National Basic Research Program of China (973 Program 2015CB856600), the National Natural Science Foundation of China (21332009, 21421091, 21361140373), and the Chinese Academy of Sciences for generous financial support.Notes and references
- M. Weber, M. Weber and M. Kleine-Boymann, “Phenol” in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2004, pp. 503–519 Search PubMed.
- For reviews, see: (a) S. P. Roche and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 4068 CrossRef CAS PubMed; (b) L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2011, 66, 2235 CrossRef PubMed; (c) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed.
- For selected examples of stepwise asymmetric dearomatization of phenols, see: (a) R. Imbos, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2002, 124, 184 CrossRef CAS PubMed; (b) Y. Hayashi, H. Gotoh, T. Tamura, H. Yamaguchi, R. Masui and M. Shoji, J. Am. Chem. Soc., 2005, 127, 16028 CrossRef CAS PubMed; (c) Q. Liu and T. Rovis, J. Am. Chem. Soc., 2006, 128, 2552 CrossRef CAS PubMed; (d) N. T. Vo, R. D. M. Pace, F. O'Hara and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 404 CrossRef CAS PubMed; (e) Q. Gu, Z.-Q. Rong, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2010, 132, 4056 CrossRef CAS PubMed; (f) Q. Gu and S.-L. You, Chem. Sci., 2011, 2, 1519 RSC; (g) Q. Gu and S.-L. You, Org. Lett., 2011, 13, 5192 CrossRef CAS PubMed; (h) R. Leon, A. Jawalekar, T. Redert and M. J. Gaunt, Chem. Sci., 2011, 2, 1487 RSC; (i) M.-Q. Jia and S.-L. You, Chem. Commun., 2012, 48, 6363 RSC; (j) M.-Q. Jia, C. Liu and S.-L. You, J. Org. Chem., 2012, 77, 10996 CrossRef CAS PubMed; (k) D. M. Rubush, M. A. Morges, B. J. Rose, D. H. Thamm and T. Rovis, J. Am. Chem. Soc., 2012, 134, 13554 CrossRef CAS PubMed; (l) S. Takizawa, T. M. Nguyen, A. Grossmann, D. Enders and H. Sasai, Angew. Chem., Int. Ed., 2012, 51, 5423 CrossRef CAS PubMed; (m) W. Wu, X. Li, H. Huang, X. Yuan, J. Lu, K. Zhu and J. Ye, Angew. Chem., Int. Ed., 2013, 52, 1743 CrossRef CAS PubMed; (n) M. T. Corbett and J. S. Johnson, Chem. Sci., 2013, 4, 2828 RSC; (o) Z.-T. He, B. Tian, Y. Fukui, X. Tong, P. Tian and G.-Q. Lin, Angew. Chem., Int. Ed., 2013, 52, 5314 CrossRef CAS PubMed.
- For selected examples, see: (a) T. Dohi, A. Maruyama, N. Takenaga, K. Senami, Y. Minamitsuji, H. Fujioka, S. B. Caemmerer and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 3787 CrossRef CAS PubMed; (b) S. Quideau, G. Lyvinec, M. Marguerit, K. Bathany, A. Ozanne-Beaudenon, T. Buffeteau, D. Cavagnat and A. Chénedé, Angew. Chem., Int. Ed., 2009, 48, 4605 CrossRef CAS PubMed; (c) M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2010, 49, 2175 CrossRef CAS PubMed; (d) M. Uyanik, T. Yasui and K. Ishihara, Tetrahedron, 2010, 66, 5841 CrossRef CAS PubMed; (e) T. Dohi, N. Takenaga, T. Nakae, Y. Toyoda, M. Yamasaki, M. Shiro, H. Fujioka, A. Maruyama and Y. Kita, J. Am. Chem. Soc., 2013, 135, 4558 CrossRef CAS PubMed.
- (a) S. Dong, J. Zhu and J. A. Porco Jr, J. Am. Chem. Soc., 2008, 130, 2738 CrossRef CAS PubMed; (b) A. Rudolph, P. H. Bos, A. Meetsma, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2011, 50, 5834 CrossRef CAS PubMed; (c) T. Oguma and T. Katsuki, J. Am. Chem. Soc., 2012, 134, 20017 CrossRef CAS PubMed; (d) T. Oguma and T. Katsuki, Chem. Commun., 2014, 50, 5053 RSC.
- (a) T. Nemoto, Y. Ishige, M. Yoshida, Y. Kohno, M. Kanematsu and Y. Hamada, Org. Lett., 2010, 12, 5020 CrossRef CAS PubMed; (b) Q.-F. Wu, W.-B. Liu, C.-X. Zhuo, Z.-Q. Rong, K.-Y. Ye and S.-L. You, Angew. Chem., Int. Ed., 2011, 50, 4455 CrossRef CAS PubMed; (c) C.-X. Zhuo and S.-L. You, Angew. Chem., Int. Ed., 2013, 52, 10056 CrossRef CAS PubMed.
- (a) S. Rousseaux, J. García-Fortanet, M. A. Del Aguila Sanchez and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 9282 CrossRef CAS PubMed; (b) R.-Q. Xu, Q. Gu, W.-T. Wu, Z.-A. Zhao and S.-L. You, J. Am. Chem. Soc., 2014, 136, 15469 CrossRef CAS PubMed.
- S.-G. Wang, Q. Yin, C.-X. Zhuo and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 647 CAS.
- During the preparation of this manuscript, two examples of Lewis acid catalyzed asymmetric dearomatization of naphthols were reported, see: (a) J. Nan, J. Liu, H. Zheng, Z. Zuo, L. Hou, H. Hu, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2015, 54, 2356 CrossRef CAS PubMed; (b) D. Yang, L. Wang, F. Han, D. Li, D. Zhao and R. Wang, Angew. Chem., Int. Ed., 2015, 54, 2185 CrossRef CAS PubMed.
- R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2013, 135, 1268 CrossRef CAS PubMed.
- L. Benati, D. Nanni, C. Sangiorgi and P. Spagnolo, J. Org. Chem., 1999, 64, 7836 CrossRef CAS.
- For selected reviews on asymmetric halogenation of olefins, see: (a) G. Chen and S. Ma, Angew. Chem., Int. Ed., 2010, 49, 8306 CrossRef CAS PubMed; (b) A. Castellanos and S. P. Fletcher, Chem.–Eur. J., 2011, 17, 5766 CrossRef CAS PubMed; (c) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938 CrossRef CAS PubMed; (d) U. Hennecke, Chem.–Asian J., 2012, 7, 456 CrossRef CAS PubMed; (e) K. Murai and H. Fujioka, Heterocycles, 2013, 87, 763 CrossRef CAS PubMed; (f) C. K. Tan and Y.-Y. Yeung, Chem. Commun., 2013, 49, 7985 RSC; (g) Y. A. Cheng, W. Z. Yu and Y.-Y. Yeung, Org. Biomol. Chem., 2014, 12, 2333 RSC; (h) S. Zheng, C. M. Schienebeck, W. Zhang, H.-Y. Wang and W. Tang, Asian J. Org. Chem., 2014, 3, 366 CrossRef CAS PubMed.
- For selected examples of asymmetric halogenation of olefins catalyzed by cinchonine derivatives, see: (a) D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298 CrossRef CAS PubMed; (b) W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664 CrossRef CAS PubMed; (c) K. C. Nicolaou, N. L. Simmons, Y. Ying, P. M. Heretsch and J. S. Chen, J. Am. Chem. Soc., 2011, 133, 8134 CrossRef CAS PubMed; (d) O. Lozano, G. Blessley, T. Martinez del Campo, A. L. Thompson, G. T. Giuffredi, M. Bettati, M. Walker, R. Borman and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 8105 CrossRef CAS PubMed; (e) A. Jaganathan, A. Garzan, D. C. Whitehead, R. J. Staples and B. Borhan, Angew. Chem., Int. Ed., 2011, 50, 2593 CrossRef CAS PubMed; (f) Z.-M. Chen, Q.-W. Zhang, Z.-H. Chen, H. Li, Y.-Q. Tu, F.-M. Zhang and J.-M. Tian, J. Am. Chem. Soc., 2011, 133, 8818 CrossRef CAS PubMed; (g) H. Li, F.-M. Zhang, Y.-Q. Tu, Q.-W. Zhang, Z.-M. Chen, Z.-H. Chen and J. Li, Chem. Sci., 2011, 2, 1839 RSC; (h) K. Ikeuchi, S. Ido, S. Yoshimura, T. Asakawa, M. Inai, Y. Hamashima and T. Kan, Org. Lett., 2012, 14, 6016 CrossRef CAS PubMed; (i) A. Jaganathan, R. J. Staples and B. Borhan, J. Am. Chem. Soc., 2013, 135, 14806 CrossRef CAS PubMed; (j) W. Zhang, N. Liu, C. M. Schienebeck, K. Decloux, S. Zheng, J. B. Werness and W. Tang, Chem.–Eur. J., 2012, 18, 7296 CrossRef CAS PubMed; (k) M. Wilking, C. Mück-Lichtenfeld, C. G. Daniliuc and U. Hennecke, J. Am. Chem. Soc., 2013, 135, 8133 CrossRef CAS PubMed; (l) W. Zhang, N. Liu, C. M. Schienebeck, X. Zhou, I. I. Izhar, I. A. Guzei and W. Tang, Chem. Sci., 2013, 4, 2652 RSC; (m) Q. Yin and S.-L. You, Org. Lett., 2013, 15, 4266 CrossRef CAS PubMed; (n) Q. Cai, Q. Yin and S.-L. You, Asian J. Org. Chem., 2014, 3, 408 CrossRef CAS PubMed; (o) Q. Yin and S.-L. You, Org. Lett., 2014, 16, 1810 CrossRef CAS PubMed; (p) Q. Yin and S.-L. You, Org. Lett., 2014, 16, 2426 CrossRef CAS PubMed; (q) M. Wilking, C. G. Daniliuc and U. Hennecke, Synlett, 2014, 1701 Search PubMed.
- The dearomatized products of phenol derivatives could be observed by NMR study, but unstable to be isolated.
- For examples of catalytic asymmetric chlorination of carbonyl compounds, see: K. Shibatomi and A. Narayama, Asian J. Org. Chem., 2013, 2, 812 CrossRef CAS PubMed , and references therein.
- N-Chlorosuccinimide was also tested as an electrophilic chlorination reagent, however, the reaction was slow and took several days for complete conversion.
- The relative configurations of products 3c and 3d were determined by NOESY NMR analysis, see the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analysis data for the new compounds. CCDC 1048128 ((R)-2t) and 1048302 ((R)-2d). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00494b |
| This journal is © The Royal Society of Chemistry 2015 |