
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrite reduction by copper through ligand-mediated proton and electron transfer†
Cameron M.
Moore
and
Nathaniel K.
Szymczak
*
Department of Chemistry, University of Michigan, 930 N. University Ave., Ann Arbor, MI 48109, USA. E-mail: nszym@umich.edu
First published on 14th April 2015
Abstract
Nitrite reduction by a copper complex featuring a proton-responsive tripodal ligand is demonstrated. Gaseous nitric oxide was confirmed as the sole NOX by-product in quantitative yield. DFT calculations predict that nitrite reduction occurs via a proton and electron transfer process mediated by the ligand. The reported mechanism parallels nitrite reduction by copper nitrite reductase.
Introduction
Nitrite reductases (NiRs) are enzymes found in prokaryotic organisms which catalyze the one-electron (e−) reduction of nitrite to nitric oxide (NO).1 Copper nitrite reductases (CuNiRs) are homotrimeric enzymes with each monomer containing two copper centers: a T1 site for electron transfer, and a catalytically-active T2 site.2 Although X-ray crystallographic studies have provided structural snapshots of intermediates along the reduction pathway,3 the precise mechanism by which CuNiR catalyzes nitrite reduction has been disputed.1c In one proposed pathway, nitrite coordinates to the reduced T2 center, then following two proton (H+) transfer events, water is released and a copper–nitrosyl species is generated (described as either Cu(I)–NO+ or Cu(II)–NO); Fig. 1A.3c,4 In support of this mechanism, a crystal structure of CuNiR with NO bound to the reduced T2 site was reported with an unusual side-on binding mode of NO,5 which suggests that NO coordination to copper is at least possible under reducing conditions.6 However, an oxidized Cu(NO) unit (i.e.Fig. 1A) would be capable of nitrosylating nearby amino acid residues,7 and thus is unlikely to be formed under catalytic conditions. An alternative pathway for nitrite reduction catalyzed by CuNiR has been described wherein nitrite first coordinates to the oxidized T2 center followed by H+ transfer from a nearby aspartic acid residue to protonate the coordinated nitrite.8 In this case, protonation of nitrite triggers e− transfer from the T1 center to the T2 center, with release of NO to form a copper-hydroxide (Fig. 1B). This mechanism is consistent with isolated crystal structures of CuNiR with nitrite bound to the oxidized T2 center,3c steady-state kinetics and pulsed radiolysis experiments,9 and computational modeling.10 Although the intimate pathway of nitrite reduction may be disputed, the network of hydrogen bonds (H-bonds) provided by nearby amino acid residues is widely accepted to play a key role in positioning substrate and facilitating e− transfer.1c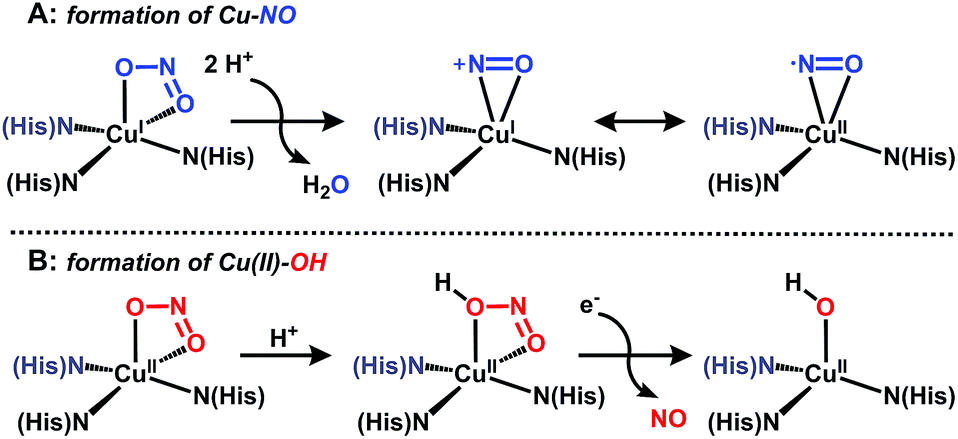 | ||
| Fig. 1 Postulated intermediates for nitrite reduction by CuNiR. | ||
With the goal of clarifying the fundamental pathways of nitrite reduction, the reactivity of synthetic copper complexes toward nitrite has been extensively studied,1c although limited examples have been reported that demonstrate secondary sphere interactions with a copper nitrite complex.11 Prior reports have largely focused on the preparation of copper(I) nitrite adducts and subsequent reactivity with exogenous H+ sources to release NO. One critical distinction between synthetic copper(I) nitrite adducts and CuNiR is the observed mode of nitrite coordination: there are no reported synthetic copper(I) complexes supported by biologically-relevant ligands that feature the κO-nitrite coordination observed in CuNiR, and instead exclusively feature κN-coordination.12
While select systems have been shown to produce NO from a copper(I) nitrite complex,1c,13 the mechanism by which these reactions proceed are often not fully resolved, thus precluding direct mechanistic comparisons to CuNiR. One reason for limited mechanistic insight has been the isolation of terminal copper(II) complexes which do not contain the inorganic products of nitrite reduction (i.e. H2O or NO).13b,13d,13f Although copper(II)–nitrosyls have been implicated in many synthetic nitrite reduction pathways, their isolation has remained elusive,7b,14,15 thus calling into question their role in synthetic and biological copper nitrite reduction schemes.
A nitrite reduction pathway that circumvents the formation of a highly reactive copper–nitrosyl species is likely operative in CuNiR (Fig. 1B). In this Edge Article, we demonstrate that nitrite reduction by a copper complex supported by a proton-responsive ligand proceeds through a parallel pathway. A mechanism is described whereby a H+/e− transfer pathway to nitrite releases NO, which bypasses the formation of an unstable copper(II)–nitrosyl species, and provides a synthetic mechanistic analogue for nitrite reduction in CuNiR.
Results and discussion
Recently we described copper complexes supported by H3thpa, a tripodal ligand featuring pendent hydroxyl groups capable of engaging in H-bonding interactions with metal bound substrates.16 We previously showed that the H-bonding manifold presented by H3thpa allows access to a unique copper(I) fluoride complex (CuF(H3thpa), 1) that is best described as containing a ‘captured’ fluoride anion in the secondary coordination sphere.16b We envisioned that anions capped by SiR3+ units would react with 1via metathesis to provide new copper complexes in which we could interrogate H-bonding and H+/e− transfer reactivity toward reducible substrates. Specifically, we sought to exploit this methodology to examine the reactivity of 1 with nitrite in an effort to experimentally distinguish between two mechanistic pathways of nitrite reduction. Given the capability of the H3thpa ligand scaffold to deliver H+ and subsequently provide H-bond donors and acceptors, we hypothesized that the inorganic products of nitrite reduction would be captured within a hydrogen bonding network surrounding the copper center.In order to assess the ability of 1 to engage in metathesis reactivity with silyl-anions, we first examined the substitution of the fluoride anion in 1 for chloride. When 1 was treated with an equivalent of Ph3SiCl at room temperature, the previously described CuCl(H3thpa)16a complex and Ph3SiF were generated quantitatively; both of which were confirmed by 1H and 19F NMR spectroscopy (Fig. S1 and S2†). Based on the clean reactivity observed with Ph3SiCl, we synthesized a reagent capable of transferring nitrite to 1 by preparing Ph3Si(ONO) via salt metathesis of Ph3SiCl and AgNO2 in benzene solvent.17
The reaction of 1 with Ph3Si(ONO) occurs immediately in dichloromethane solvent: when Ph3Si(ONO) is added to a yellow solution of 1, a rapid color change to green occurs. 19F NMR spectra of the resulting solution confirm the quantitative formation of Ph3SiF, indicating metathesis of fluoride. The color change is indicative of oxidation from copper(I) to copper(II), a supposition confirmed by EPR and UV-vis spectra collected of reaction solutions.18 Solid state IR spectra of the isolated green material from this reaction do not show bands associated with nitrite or a metal–nitrosyl species, however a new ligand C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1658 cm−1 was visualized (consistent with a change in protonation state of the ligand), along with broadened −OH bands. Based on the above findings, we hypothesized that the terminal copper-containing product in this reaction was a copper(II)–aquo complex (Cu(OH2)Hthpa, 2, Fig. 2).
O stretch at 1658 cm−1 was visualized (consistent with a change in protonation state of the ligand), along with broadened −OH bands. Based on the above findings, we hypothesized that the terminal copper-containing product in this reaction was a copper(II)–aquo complex (Cu(OH2)Hthpa, 2, Fig. 2).
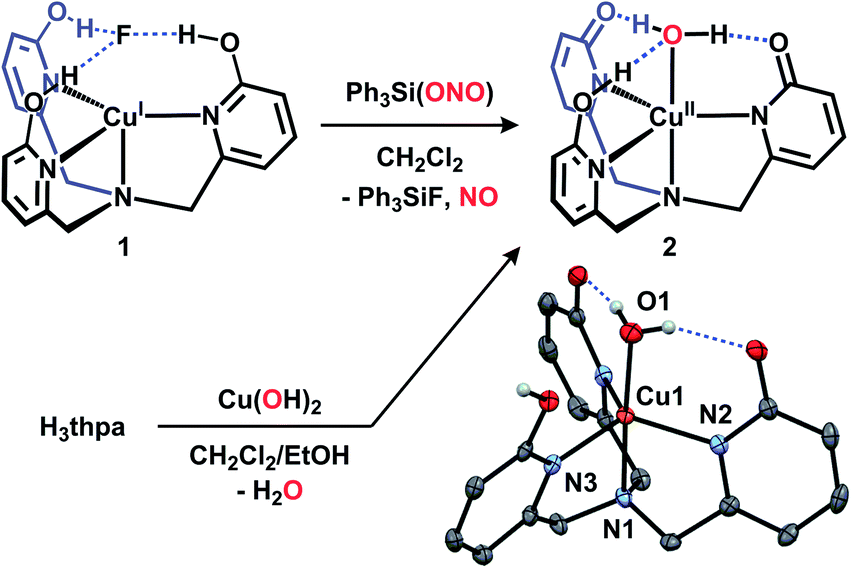 | ||
| Fig. 2 Syntheses and crystal structure of 2 (thermal ellipsoids shown at 50% probability, H-atoms not involved in H-bonding and solvent omitted for clarity). | ||
We sought an alternative preparation of 2 to confirm its formation during the reaction of 1 with Ph3Si(ONO). Authentic samples of complex 2 were prepared by allowing equimolar amounts of the ligand, H3thpa, and Cu(OH)2 to react in dichloromethane/ethanol solution. The solution characterization data (UV-vis and EPR) for 2 prepared in this manner were identical to those from the reaction of 1 with Ph3Si(ONO).18 The crystal structure of 2 contains two independent molecules along with an ethanol solvate which is engaged in H-bonding interactions with only one of the molecules. The solid state structure of one of the independent molecules of 2 is presented in Fig. 2 and reveals a trigonal bipyramidal coordination geometry at copper, consistent with the solution state coordination geometry as determined by EPR and UV-vis spectroscopy.16a,18 The Cu–OH2 is coordinated at a distance of 1.956(2) Å from copper and features H-bonds (O⋯O separations 2.567(3) and 2.664(3) Å) to the asymmetric Hthpa ligand. The asymmetry of the ligand environment is confirmed by examination of the ligand's C–O bond lengths (1.299(3) Å and 1.284(3) Å for the deprotonated ‘arms’ and 1.325(3) Å for the protonated ‘arm’) which reflect the anionic nature of the two ligand ‘arms’. The protonated ‘arm’ (N3) is engaged in intermolecular H-bonding interactions with another molecule of 2, as opposed to the Cu-bound O atom. This network of intermolecular H-bonding, which forms a 1-D chain in the extended structure, is presumably a manifestation of crystal packing and unlikely to persist in solution under dilute conditions.19
In addition to the formation of 2 from 1 and Ph3Si(ONO), NO was also confirmed as a reaction product by gas-phase IR spectroscopy, as well as by trapping experiments with CoTPP (TPP = tetraphenylporphyrin) and quantification using UV-vis spectroscopy.20 Headspace analysis of reaction mixtures revealed two broad bands at 1904 and 1844 cm−1 in the IR spectrum, consistent with NO, as well as bands associated with dichloromethane solvent vapors (Fig. S12†). To further support the assignment of gaseous NO as a by-product, we prepared the 15N isotopologue, Ph3Si(O15NO), and allowed it to react with 1. Headspace analysis of the reaction mixture by IR spectroscopy showed two broad bands at 1868 and 1817 cm−1, consistent with 15NO, further substantiating NO formation during the reaction of 1 and Ph3Si(ONO). The formation of NO was quantitative, as revealed by CoTPP trapping experiments.
Control reactions confirmed that the quantitative generation of NO was unique to 1. For instance, to examine whether a direct reaction of the silyl-reagent with a metal fluoride induced NO extrusion, we performed a control experiment where the known copper(I) fluoride CuF(PPh3)3 (3)21 was allowed to react with Ph3Si(ONO).22 Headspace analysis of this reaction mixture by IR spectroscopy revealed negligible bands associated with NO (Fig. S12†). Trapping experiments with CoTPP did not reveal any significant formation of NO above the background during the reaction of 3 and Ph3Si(ONO).
The quantitative yield of NO from complex 1 cannot be attributed solely to a disproportionation reaction of nitrite. Under sufficiently acidic conditions, nitrite disproportionates to form NO, along with other NOx species.23 To examine a possible acid-promoted nitrite disproportionation pathway that produces NO, we probed the reactivity of Ph3Si(ONO) with CuF(H3thpa)BF4 (4).16b Complex 4 serves as an ideal platform to test the ability of the ligand framework to deliver H+ equivalents to nitrite since (1) e− transfer is not possible and (2) the –OH groups within the putative Cu(H3thpa)2+ dication are expected to be more acidic than in the analogous Cu(H3thpa)+ monocation.24 Although NO was detected by IR spectroscopy in the headspace of reaction samples containing 4 and Ph3Si(ONO), quantification using CoTPP revealed formation of NO in only ca. 10% yield above the background decomposition.18 Taken together, these results confirm the necessity of the H3thpa ligand framework on copper(I) to mediate nitrite reduction in the absence of any exogenous H+ source. The quantitative formation of NO from 1 and Ph3Si(ONO) confirms that 1 serves to deliver H+and e−, as opposed to initiating acid-mediated disproportionation.
The generation of a copper(II)–(OHx) species from copper(I) and nitrite is reminiscent of a proposed pathway for biological nitrite reduction in CuNiR (Fig. 1B). We sought to investigate key intermediates along the nitrite reduction pathway in our system to provide insight into the reaction sequence that may be applied to CuNiR. However, no intermediates were detected by 1H NMR spectroscopy during the reaction of 1 with Ph3Si(ONO) at or below −50 °C, which precluded mechanistic analysis in solution.18 Accordingly, we examined the reaction profile using density functional theory (DFT) calculations.18 The initial nitrite binding step was first interrogated for a series of possible Cu(H3thpa)–nitrite adducts. Three nearly isoenergetic (<0.5 kcal mol−1 difference) structures were computationally identified that feature an interaction of nitrite with the putative Cu(H3thpa)+ cation (Fig. S16†).25 These included an ‘encounter’ complex (I, Fig. 3A) where, reminiscent of the F− binding in 1, nitrite is positioned in the second coordination sphere by H-bonding interactions. Additionally, two other isomers were located containing two nitrite coordination modes; an η1-κO isomer (I′) and an η1-κN isomer (I′′, Fig. S16†).
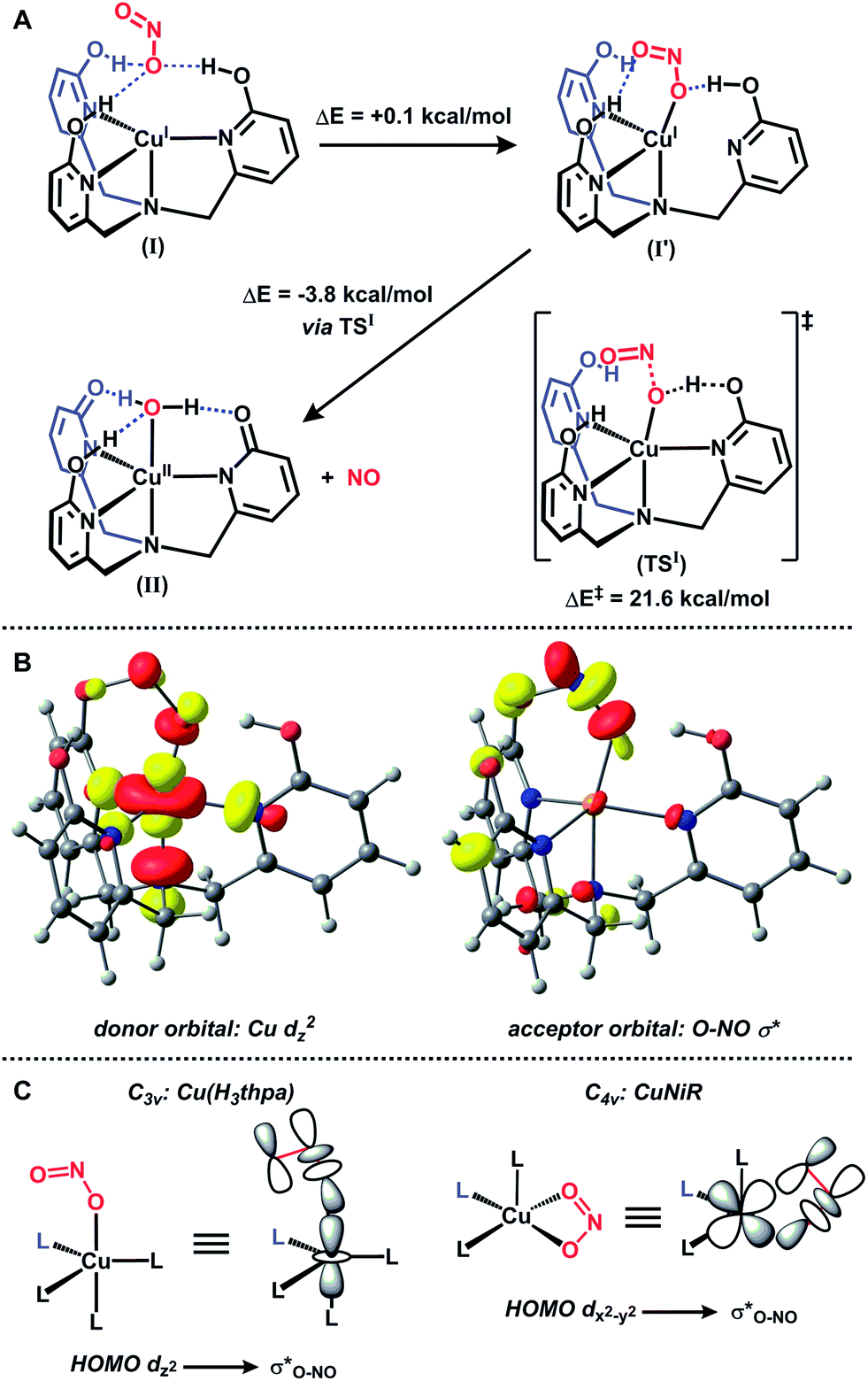 | ||
| Fig. 3 DFT-calculated pathway for nitrite reduction (a), donor and acceptor molecular orbitals along the N–O bond cleavage reaction coordinate (b, isovalue = 0.05) and comparison of nitrite binding mode and donor/acceptor orbitals for Cu(H3thpa)+ (c, left) and CuNiR (c, right). | ||
The nitrite reduction sequence subsequent to the initial binding event was evaluated. In analogy to Fig. 1A, nitrite reduction via water elimination from the η1-κN isomer (I′′) to provide a copper–nitrosyl (III, Scheme S2†) was found to be an endothermic process by 17.9 kcal mol−1, presumably due to the formation of a high energy copper(II)–nitrosyl (Fig. S17†). In contrast, and in analogy to Fig. 1B, reduction via NO elimination from the η1-κO isomer (I′) was found to be an exothermic process by 3.8 kcal mol−1 to generate the experimentally observed copper(II)–aquo (II) species (Fig. 3A).
The transition state for the critical N–O bond breaking step (TSI) was located computationally and is 21.6 kcal mol−1 above the starting ‘encounter’ complex I. In the calculated transition state TSI, H+ transfer from the H3thpa scaffold to the coordinated nitrite is accompanied by elongation of the O–NO bond. There is substantial Mulliken spin density on the Cu–O fragment (0.565) in the transition state. This value is intermediate between the Mulliken spin density of the starting species I (0.0) and the Mulliken spin density on the Cu–O fragment in the product II (0.929). The spin density on the Cu–O fragment in the transition state (along with spin density of the opposite sign on NO, Fig. S19†) indicates charge transfer from copper to nitrite concomitant with H+ transfer from H3thpa to nitrite. This H+/e− transfer, accompanied by NO ejection, is followed by an additional H+ transfer from the H2thpa ligand to copper-bound hydroxide to form the final copper(II)–aquo product II.
To understand the impact of the binding mode of nitrite in our system as compared to CuNiR we analyzed the frontier molecular orbitals of structures along the O–NO bond cleavage reaction coordinate. The empty σ*O–NO orbital overlaps with the filled Cu dz2 orbital (Fig. 3B). This is in contrast to CuNiR, where the highest occupied molecular orbital (HOMO) is primarily dx2−y2 character and subsequently nitrite coordinates in a bidentate manner to maximize orbital overlap between dx2−y2 and σ*O–NO for electron transfer.10 For Cu(H3thpa)+, the HOMO is dz2 and favors an alternative mode of nitrite binding. The binding of nitrite as a monodentate ligand κO maximizes overlap between dz2 and σ*O–NO for electron transfer (Fig. 3C). The dz2 ground state predicted for II, subsequent to electron transfer and NO release, is in agreement with the experimental EPR data for 2.16a,b,18
The difference in mode of nitrite binding between our synthetic system and CuNiR has ramifications on the calculated barrier height for O–NO bond cleavage. In CuNiR, bidentate coordination of nitrite to copper maximizes back-bonding interactions and significantly lowers the calculated barrier to O–NO bond cleavage (16 kcal mol−1).10 When a monodentate coordination was examined in silico, the extent of back-bonding between copper and nitrite was significantly lower and consequently, a higher barrier to O–NO bond cleavage was observed (26 kcal mol−1).10 The barrier in the present system (21 kcal mol−1) is intermediate of these regimes in CuNiR. The H-bonding interactions in TSI between the distal oxygen of the nitrite anion and the −OH groups of the H3thpa ligand serves to lower the σ*O–NO orbital energy, similar in fashion to bidentate coordination to copper. In contrast to energetic consequences on orbital energies imposed by geometric constraints, the hydrogen bonding interactions in H3thpa provide an alternative means by which to lower the σ*O–NO orbital energy, and this approach may well be exploited as a general strategy to minimize energetic costs associated with substrate reduction. Such strategies have been thoroughly described for biological systems.26
Conclusions
In conclusion, we have demonstrated nitrite reduction by a copper complex featuring a proton-responsive tripodal ligand. The formation of NO was confirmed by gas-phase IR spectroscopy, isotope labelling, and trapping experiments. DFT calculations predict that nitrite binds in an η1-κO fashion to the copper center prior to reduction via a H+/e transfer. This reaction, facilitated by the secondary sphere environment, parallels the crucial role of H-bonding residues near the active site of CuNiR, which serve to position nitrite and facilitate electron transfer. This is, to our knowledge, the first synthetic copper system to reduce nitrite in such a fashion.27 Ongoing efforts on this system are focused at utilizing the H3thpa scaffold to facilitate multiple H+/e− transfer events to coordinated substrates.Acknowledgements
This work was supported by the University of Michigan Department of Chemistry, NSF-GRFP (CMM), UM Rackham Graduate School (CMM), NSF grant CHE-0840456 for X-ray instrumentation and through computational resources and services provided by Advanced Research Computing at UM. NKS is a Dow Corning Assistant Professor and Alfred P. Sloan Research Fellow. We thank Dr Jeff W. Kampf for X-ray assistance and Prof. Nicolai Lehnert and Prof. Abhishek Dey for helpful discussions.Notes and references
- (a) B. A. Averill, Chem. Rev., 1996, 96, 2951 CrossRef CAS PubMed; (b) W. G. Zumft, Microbiol. Mol. Biol. Rev., 1997, 61, 533 CAS; (c) A. C. Merkle and N. Lehnert, Dalton Trans., 2012, 3355 RSC and references therein.
- S. Suzuki, K. Kataoka, K. Yamaguchi, T. Inoue and Y. Kai, Coord. Chem. Rev., 1999, 190–192, 245 CrossRef CAS.
- (a) M. E. P. Murphy, S. Turley and E. T. Adman, J. Biol. Chem., 1997, 272, 28455 CrossRef CAS PubMed; (b) M. J. Ellis, M. Prudencio, F. E. Dodd, R. W. Strange, G. Sawers, R. R. Eady and S. S. Hasnain, J. Mol. Biol., 2002, 316, 51 CrossRef CAS PubMed; (c) S. V. Antonyuk, R. W. Strange, G. Sawers, R. R. Eady and S. S. Hasnain, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12041 CrossRef CAS PubMed.
- B. A. Averill, Angew. Chem., Int. Ed., 1994, 33, 2057 CrossRef PubMed.
- E. I. Tocheva, F. I. Rosell, A. G. Mauk and M. E. P. Murphy, Science, 2004, 304, 867 CrossRef CAS PubMed.
- Note that the NO adduct of reduced CuNiR can also be generated in solution, see: O. M. Usov, Y. Sun, V. M. Grigoryants, J. P. Shapleigh and C. P. Scholes, J. Am. Chem. Soc., 2006, 128, 13102 CrossRef CAS PubMed.
- (a) C. L. Hulse, J. M. Tiedje and B. A. Averill, J. Am. Chem. Soc., 1989, 111, 2322 CrossRef CAS; (b) M. Sarma and B. Mondal, Inorg. Chem., 2011, 50, 3206 CrossRef CAS PubMed.
- K. Kataoka, H. Furusawa, K. Takagi, K. Yamaguchi and S. Suzuki, J. Biochem., 2000, 127, 345 CrossRef CAS.
- (a) Y. Zhao, D. A. Lukoyanov, Y. V. Toropov, K. Wu, J. P. Shapleigh and C. P. Scholes, Biochemistry, 2002, 41, 7464 CrossRef CAS PubMed; (b) S. Suzuki, K. Kataoka and K. Yamaguchi, Acc. Chem. Res., 2000, 33, 728 CrossRef CAS PubMed; (c) K. Kobayashi, S. Tagawa, Deligeer and S. Suzuki, J. Biochem., 1999, 126, 408 CrossRef CAS.
- S. Ghosh, A. Dey, Y. Sun, C. P. Scholes and E. I. Solomon, J. Am. Chem. Soc., 2009, 131, 277 CrossRef CAS PubMed.
- For select Cu(II) examples, see: (a) F. Hueso-Ureña, A. L. Peñas-Chamorro, M. N. Moreno-Carretero, M. Quirós-Olozábal and J. M. Salas-Peregrín, Polyhedron, 1999, 18, 351 CrossRef; (b) M. Harata, K. Jitsukawa, H. Masuda and H. Einaga, Bull. Chem. Soc. Jpn., 1998, 71, 637 CrossRef CAS.
- Note that copper(I)–nitrite adducts featuring κO-coordination have been observed using phosphine ligands, see: J. A. Halfen and W. B. Tolman, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 215 CrossRef , along with ref. 13d and 13e.
- For selected examples, see: (a) J. A. Halfen, S. Mahapatra, M. M. Olmstead and W. B. Tolman, J. Am. Chem. Soc., 1994, 116, 2173 CrossRef CAS; (b) J. A. Halfen, S. Mahapatra, E. C. Wilkinson, A. Gengenbach, V. G. Young Jr, L. Que and W. B. Tolman, J. Am. Chem. Soc., 1996, 118, 763 CrossRef CAS; (c) J. A. Halfen and W. B. Tolman, J. Am. Chem. Soc., 1994, 116, 5475 CrossRef CAS; (d) W.-J. Chuang, I. J. Lin, H.-Y. Chen, Y.-L. Chang and S. C. N. Hsu, Inorg. Chem., 2010, 49, 5377 CrossRef CAS PubMed; (e) S. C. N. Hsu, Y.-L. Chang, W.-J. Chuang, H.-Y. Chen, I. J. Lin, M. Y. Chiang, C.-L. Kao and H.-Y. Chen, Inorg. Chem., 2012, 51, 9297 CrossRef CAS PubMed; (f) M. Kujime and H. Fujii, Angew. Chem., Int. Ed., 2006, 45, 1089 CrossRef CAS PubMed; (g) P. P. Paul and K. D. Karlin, J. Am. Chem. Soc., 1991, 113, 6331 CrossRef CAS.
- (a) M. Sarma, A. Kalita, P. Kumar, A. Singh and B. Mondal, J. Am. Chem. Soc., 2010, 132, 7846 CrossRef CAS PubMed; (b) M. Sarma, A. Singh, G. S. Gupta, G. Das and B. Mondal, Inorg. Chim. Acta, 2009, 363, 63 CrossRef PubMed.
- A. M. Wright, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 14336 CrossRef CAS PubMed.
- (a) C. M. Moore, D. A. Quist, J. W. Kampf and N. K. Szymczak, Inorg. Chem., 2014, 53, 3278 CrossRef CAS PubMed; (b) C. M. Moore and N. K. Szymczak, Chem. Commun., 2015, 51, 5490 RSC.
- For a similar synthesis to generate SiiPr3(ONO), see: M. Weidenbruch and F. Sabeti, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1976, 31, 1212 Search PubMed.
- See the Electronic ESI† for more details.
- DFT optimizations of 2 using a CH2Cl2 dielectric predict that all OH groups engage in intramolecular H-bonding interactions. Under dilute conditions where intermolecular interactions are less favored, we presume this is an accurate depiction of the solution structure and have therefore represented 2 in accordance with the computational results.
- M. Kumar, N. A. Dixon, A. C. Merkle, M. Zeller, N. Lehnert and E. T. Papish, Inorg. Chem., 2012, 51, 7004 Search PubMed.
- F. H. Jardine, L. Rule and A. G. Vohra, J. Chem. Soc. A, 1970, 238 Search PubMed.
- CuF(PPh3)3 was selected as a viable copper(I) fluoride given its reported synthesis and characterization, in contrast to CuF(tpa), which is not known. See ref. 16b.
- M. S. Rayson, J. C. Mackie, E. M. Kennedy and B. Z. Dlugogorski, Inorg. Chem., 2012, 51, 2178 Search PubMed.
- Note that CuCl(H3thpa) (see ref. 16a) can be deprotonated with weak bases such as sodium acetate, suggesting an upper limit for the first pKa of CuX(H3thpa) complexes is ∼4. Please see the ESI† for more details.
- Note that the presence of nearly isoenergetic coordination isomers is in accordance with previous studies on copper–nitrite adducts, see: N. Lehnert, U. Cornelissen, F. Neese, T. Ono, Y. Noguchi, K. Okamoto and K. Fujisawa, Inorg. Chem., 2007, 46, 3916 Search PubMed.
- (a) G. A. Jeffery and W. Saenger, Hydrogen Bonding in Biological Structures, Springer-Verlag, Berlin, 1991 Search PubMed; (b) K. M. Lancaster, Struct. Bonding, 2012, 142, 119 Search PubMed.
- Note that a similar mechanism for iron-mediated nitrite reduction was recently reported, see: E. M. Matson, Y. J. Park and A. R. Fout, J. Am. Chem. Soc., 2014, 136, 17398 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and experimental procedures, spectral data and computational details. CCDC 1051416 and 1056984. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00720h |
| This journal is © The Royal Society of Chemistry 2015 |