
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReaction-activated palladium catalyst for dehydrogenation of substituted cyclohexanones to phenols and H2 without oxidants and hydrogen acceptors†
Jingwu
Zhang
,
Qiangqiang
Jiang
,
Dejun
Yang
,
Xiaomei
Zhao
,
Yanli
Dong
and
Renhua
Liu
*
School of Pharmacy, East China University of Science and Technology, Meilong Road 130, Shanghai 200237, China. E-mail: liurh@ecust.edu.cn; Fax: +86-21-64250627; Tel: +86-21-64250627
First published on 19th May 2015
Abstract
It is widely believed that the dehydrogenation of organic compounds is a thermodynamically unfavorable process, and thus requires stoichiometric oxidants such as dioxygen and metal oxides or sacrificial hydrogen acceptors to remove the hydrogen from the reaction mixture to drive the equilibrium towards the products. Here we report a previously unappreciated combination of common commercial Pd/C and H2 which dehydrogenates a wide range of substituted cyclohexanones and 2-cyclohexenones to their corresponding phenols with high isolated yields, with H2 as the only byproduct. The reaction requires no oxidants or hydrogen acceptors because instead of removing the generated hydrogen with oxidants or hydrogen acceptors, we demonstrated it can be used as a cocatalyst to help power the reaction. This method for phenol synthesis manifests a high atom economy, and is inherently devoid of the complications normally associated with oxidative dehydrogenations.
Introduction
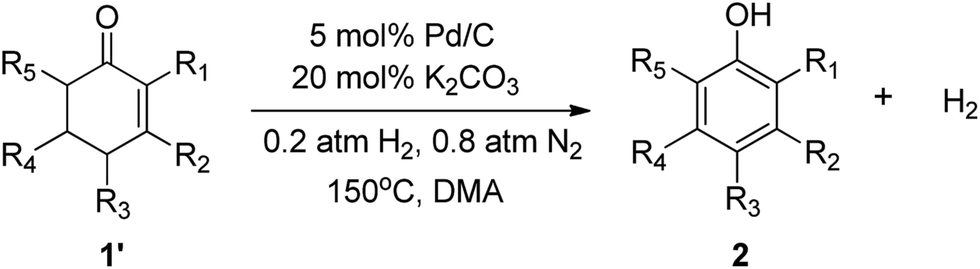
Phenols are common precursors and key structural motifs of industrial chemicals ranging from fine (e.g. agrochemicals, dyes, pigments, and pharmaceuticals) to bulk chemicals (polymers).1 The synthesis of substituted phenols typically relies on the introduction of chemical functional groups on to the aromatic ring with patterns of nucleophilic and electrophilic aromatic substitution, and metal-catalyzed cross-coupling reactions.2 However, the nucleophilic reactions of aromatic substitution are largely hampered due to the electron-donating properties of the hydroxyl group. As such, the strong electronic directing effect of the hydroxyl group result in the limited availability of electrophilic reactions of aromatic substitution for the preparation of ortho- and para-substituted derivatives.3 Although metal-catalyzed cross-coupling reactions are useful approaches for phenols, these methods generally require an expensive metal catalyst,4 and differential prefunctionalization of the arene coupling partners with a halide and an electropositive group.5 Thus achieving a versatile method for the facile synthesis of substituted phenols, especially for meta-substituted phenols, remains a key challenge. Recently, the oxidative dehydrogenation of substituted cyclohexanones,6 which is designed to be a complementary route for the convenient synthesis of meta-substituted phenols, is emerging as a versatile strategy to achieve this goal. However, most of reported methods for the synthesis of phenols via oxidative dehydrogenation of cyclohexanones have met with a limited degree of success in regards to atom economy. The existing reactions of this type are typically carried out with stoichiometric oxidizing reagents or hydrogen acceptors, e.g. DDQ,7 and/or harsh reaction conditions and limited substrate scope (≥200 °C high reaction temperature, and are of limited use for the refinement of more complex substrates) or requiring a stepwise procedure (bromination and dehydrobromination). Very recently, Stahl and his co-workers made significant progress in the oxidative dehydrogenation of cyclohexanones to phenols.6a,8 They developed a palladium(II) catalyst (incorporating a sophisticated ligand) for the dehydrogenation. High conversions and product yields were obtained with this process, and the use of dioxygen as the oxidant was advantageous from a green viewpoint (Scheme 1a and b). However, oxidative dehydrogenations for the synthesis of phenols are frequently hindered by over-oxidation because the phenol products are inherently susceptible to oxidation, thus giving rise to byproducts.9 Moreover, the substrates possessing a chelating function, a nitrogen atom or a sulfur substituent, and/or containing other easily oxidizable functionalities are not compatible with palladium(II) catalyzed oxidation methods and are therefore of limited use in such oxidative dehydrogenations. Herein we report an oxidant- and hydrogen-acceptor-free dehydrogenation of a wide range of substituted cyclohexanones and 2-cyclohexenones to their corresponding phenols with H2 release. The reaction is catalyzed by a commercial Pd/C in combination with H2. The incorporation of H2 into the palladium catalyst does not hold back the dehydrogenation reaction and in many instances it also leads to a great increase in the catalyst activity for dehydrogenation. The method is characterized by requiring no oxidants and hydrogen acceptors, and therefore circumvents the problems of over-oxidation and the compatibility with easily oxidizable functionalities. Likewise, the method provides a promising approach to green hydrogen production from organic compounds, which represents an additional benefit.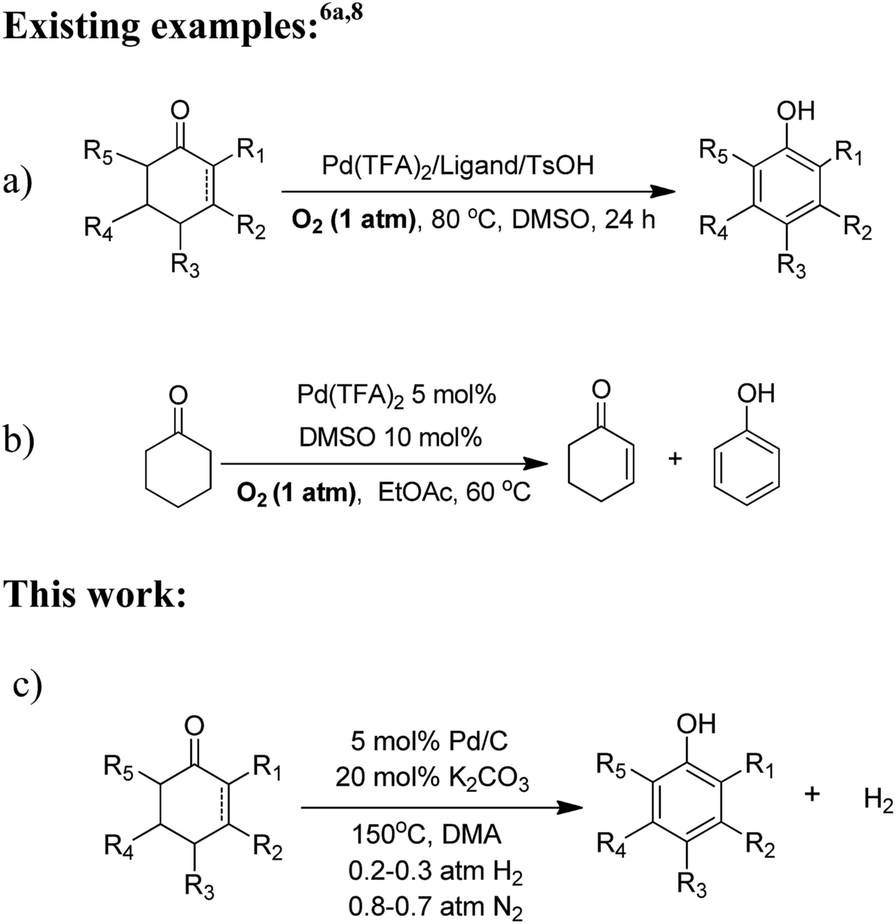 | ||
| Scheme 1 Dehydrogenation of substituted cyclohexanones to phenols. The reactions of both (a) and (b) proceeded with Pd(II) complex catalysts requiring molecular oxygen as the oxidant. (c) The palladium(0)-catalyzed reaction for dehydrogenation of cyclohexanones to phenols under 1 atm of the gas mixture atmosphere (70–80 vol% N2 and 30–20 vol% H2) which requires no oxidants and hydrogen acceptors, and H2 is the only side product. | ||
Results and discussion
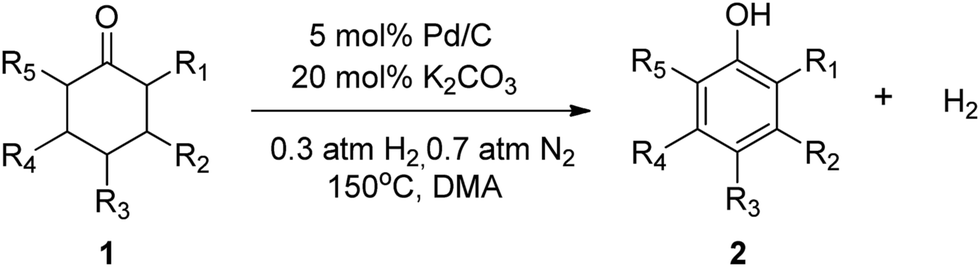
Recently, interest in exploring oxidant- and hydrogen acceptor-free dehydrogenation synthesis has been burgeoning for both the economic and environmental benefits. Many milder liquid-phase catalyst systems for the acceptorless dehydrogenation of alcohols to carbonyl compounds and alcohol dehydrogenation coupling reactions have been reported.10 It was intriguing to see if an effective catalyst could be developed for oxidant- and acceptor-free dehydrogenation of substituted cyclohexanones and 2-cyclohexenones to phenols with the release of H2. On the basis of our recently developed palladium(0) catalyzed dehydrogenation of amines to imines,11 we anticipated that palladium(0) could also catalytically dehydrogenate cyclohexanones and 2-cyclohexenones to phenols with the release of H2. Herein we report our results towards achieving this goal and give a full account of the influence of the reactions.To begin our study we investigated the reactivity of cyclohexanone for dehydrogenation with 5 mol% of Pd/C and N,N-dimethyl-acetamide (DMA) as the solvent under 1 atm of N2 atmosphere (a N2 balloon) at 130 °C for 24 h; the results showed 89% conversion and 82% selectivity (Table 1, entry 1). These encouraging results inspired us to further optimize the reaction conditions. A variety of additives were screened for their ability to promote the dehydrogenation reaction. The acidic additives tested were unable to efficiently yield the desired product although some of the reactions obtained a high conversion (Table 1, entries 20–22). On the contrary, most alkaline additives provided excellent product yields. The effects of different solvents were also evaluated. Strong polar aprotic solvents were generally effective for the conversion of the substrates to the desired phenols. In contrast, weak polar aprotic solvents, e.g. toluene, were unfavorable for the reaction. These observations could be rationalized in terms of the alkaline additives having relatively low solubility in weak polar aprotic solvents. Although water is conducive in making the added bases soluble, the addition of water to the reaction system is deleterious for the dehydrogenation.
|
|
|||
|---|---|---|---|
| Entry | Conditions | Conv. (%) | Select. (%) |
| a Basic reaction conditions: cyclohexanone (1 mmol), solvent (2 mL), 24 h, 1 atm gas atmosphere (gas balloon). The percentage is mol%. Conversion and selectivity were based on gas chromatography (GC) with area normalization. b The water content of the solvent was the volume fraction. c 94% of cyclohexanol was detected. d With palladium acetate instead of Pd/C. No phenol product was detected. | |||
| 1 | DMA/5% Pd/130 °C/N2 | 89 | 82 |
| 2 | DMF/5% Pd/130 °C/N2 | 85 | 70 |
| 3 | DMI/5% Pd/130 °C/N2 | 65 | 82 |
| 4 | DMP/5% Pd/130 °C/N2 | 67 | 84 |
| 5 | Toluene/5% Pd/100 °C/N2 | 0 | — |
| 6 | DMA/5% Pd/100 °C/N2 | 20 | 93 |
| 7 | DMA/1% Pd/130 °C/N2 | 25 | 84 |
| 8 | DMA/2.5% Pd/130 °C/N2 | 80 | 85 |
| 9 | DMA/10% Pd/130 °C/N2 | 99 | 88 |
| 10 | DMA/5% Pd/150 °C/N2 | 99 | 85 |
| 11 | DMA/5% Pd/160 °C/N2 | 99 | 81 |
| 12 | DMA/5% Pd/50% Li2CO3/150 °C/N2 | 88 | 82 |
| 13 | DMA/5% Pd/50% Na2CO3/150 °C/N2 | 99 | 76 |
| 14 | DMA/5% Pd/50% K2CO3/150 °C/N2 | 99 | 98 |
| 15 | DMA/5% Pd/50% Cs2CO3/150 °C/N2 | 82 | 78 |
| 16 | DMA/5% Pd/50% NaOH/150 °C/N2 | 65 | 62 |
| 17 | DMA/5% Pd/50% KOH/150 °C/N2 | 57 | 49 |
| 18 | DMA/5% Pd/50% CH3ONa/150 °C/N2 | 99 | 91 |
| 19 | DMA/5% Pd/50% NaOEt/150 °C/N2 | 99 | 92 |
| 20 | DMA/5% Pd/50% CF3SO3H/150 °C/N2 | 90 | 15 |
| 21 | DMA/5% Pd/50% TsOH/150 °C/N2 | 99 | 5 |
| 22 | DMA/5% Pd/50% CH3COOH/150 °C/N2 | 99 | 12 |
| 23 | DMA/2.5% Pd/10% K2CO3/150 °C/N2 | 94 | 85 |
| 24 | DMA/5% Pd/20% K2CO3/150 °C/N2 | 99 | 98 |
| 25 | DMA/5% Pd/30% K2CO3/150 °C/N2 | 99 | 98 |
| 26b | DMA/5% Pd/10% H2O/20% K2CO3/150 °C/N2 | 55 | 91 |
| 27 | DMA/5% Pd/20% K2CO3/150 °C/O2 | 99 | 0 |
| 28 | DMA/5% Pd/20% K2CO3/150 °C/Ar2 | 99 | 97 |
| 29 | DMA/5% Pd/20% K2CO3/150 °C/CO2 | 99 | 92 |
| 30c | DMA/5% Pd/20% K2CO3/150 °C/H2 | 99 | 5 |
| 31d | DMA/5% Pd/20% K2CO3/150 °C/N2 | 32 | — |
Several interesting phenomena observed during the gas atmosphere condition screening are worth noting. First, the dehydrogenation of cyclohexanone requires a long time to completely convert the substrate. At first we thought this arises because the dehydrogenation lacked oxidants and hydrogen acceptors. It is widely believed that the dehydrogenation of organic compounds with the loss of H2 is typically unfavorable in thermodynamics and thus requires an oxidant or a hydrogen acceptor to react with the generated hydrogen to provide an external driving force. Control experiments were therefore carried out using O2 in place of N2. Indeed, the incorporation of O2 into the reaction system increases the rates of the dehydrogenation, but the molecular oxygen also dramatically reduces the selectivity of the reaction. The reaction under 1 atm of O2 (a pure O2 balloon) is totally ineffective in selectivity for the desired product: only miscellaneous oxidation products were detected (Table 1, entry 27). Thus a variety of gas atmospheres was screened for their effects on the reaction. The results shown in Table 1 showed that inert gases, e.g. Ar, N2 and CO2, enabled the reaction to achieve high selectivity and conversion. When the reaction was carried out under 1 atm of H2 atmosphere (a H2 balloon), it provided 94% of the hydrogenation product, cyclohexanol, and 5% of desired phenol product (Table 1, entry 30). The reaction can still generate the desired dehydrogenation product, phenol, under the hydrogenation conditions of 1 atm of pure H2. This was unexpected. To further verify the influence of gas atmospheres on the dehydrogenation reaction for other substrates, we selected another substrate, 3-isobutyl-5-phenyl-cyclohexanone, to carry out control experiments under a variety of gas atmospheres (N2, O2, and a gas mixture of N2 and H2). Although the reaction under 1 atm of pure O2 atmosphere showed a relatively fast rate of dehydrogenation (the starting material was completely consumed within 20 h), the yield of the target product was not more than 20% (Fig. 1, curve ○ (green)). This arises because the target product, 3-isobutyl-5-phenyl-phenol, was further oxidized to by-products during the reaction. When the reaction was carried out under 1 atm of the gas mixture atmosphere (70 vol% of N2 and 30 vol% of H2), we only detected the dehydrogenation product, 3-isobutyl-5-phenyl-phenol and did not detect the hydrogenation product, 3-isobutyl-5-phenyl-cyclohexanol in the reaction mixture, while the starting material was completely converted. We did observe 7% of hydrogenation product, 3-isobutyl-5-phenyl-cyclohexanol, during the initial stages of the reaction, but the hydrogenation product was also completely dehydrogenated to the desired phenol gradually in the following reaction. More surprisingly, such a gas mixture atmosphere did not cause any decrease in dehydrogenation reaction rate, conversion and selectivity, but on the contrary did cause a large increase compared with the reaction under 1 atm of N2 atmosphere (Fig. 1, curve ■ (black) vs. curve ▲ (red)). The observation contradicts the generally accepted point of view that removing the hydrogen from the reaction mixture is beneficial for dehydrogenation reactions. To find out how the Pd/C in combination with H2 works, we treated the Pd/C with 1 atm of H2 at room temperature for 1 h and then used the treated Pd/C to dehydrogenate 3-isobutyl-5-phenyl-cyclohexanone under 1 atm of N2 atmosphere and at 150 °C. There were two key findings: (i) a small amount (4%) of hydrogenation product, corresponding to cyclohexanol, was generated during the initial stages of the reaction (in contrast, such a hydrogenation product was not observed in the reaction with the untreated Pd/C under 1 atm of N2 atmosphere and at 150 °C), (ii) the Pd/C treated with H2 exhibits a higher catalytic reactivity for the dehydrogenation (under N2 atmosphere) compared with the untreated Pd/C (under N2 atmosphere) (Fig. 1, curve ● (blue) vs. curve ▲ (red)). The Pd/C treated with H2 is able to convert 3-isobutyl-5-phenyl-cyclohexanone to the corresponding cyclohexanol without reducing reagents (H2). This result shows that the HPd(II)H active species are most likely being generated during the treatment of Pd/C with H2 because HPd(II)H can reduce cyclohexanones to cyclohexanols, and the HPd(II)H active species also act as the veritable active catalyst for the dehydrogenation. In contrast, the rate of the reaction with untreated Pd/C under 1 atm of N2 gradually increased during the initial stages of the reaction (Fig. 1, curve ▲ (red)), which suggests the catalyst is gradually activated by the generated H2. Thus we presumed that removing the generated H2 from the reaction system would suppress the dehydrogenation reaction. To verify this, we performed the reaction under 10 atm of highly pure N2 in an autoclave at 150 °C because the nitrogen pressure increase is equivalent to reducing the partial pressure of the hydrogen generated in the reaction, which is equivalent to removing the H2 from the reaction system. The results showed that the reaction implemented under 10 atm of N2 and at 150 °C for 27 h and 36 h provided yields of 0% and 25% of the target product respectively, compared with a product yield of 20% and 48% respectively but with only 1 atm of N2 atmosphere with the same reaction conditions (Fig. 2, curve c). These observations clearly showed removing the hydrogen from the reaction system (or reducing the partial pressure of hydrogen in the system) slowed down the reaction and the hydrogen created in the reaction can activate the Pd/C catalyst for the dehydrogenation process. This phenomenon manifests a catalyst that can be activated by its reaction products, which is controlled by the reaction, and so we refer to these catalysts as “reaction-activated catalysts”.
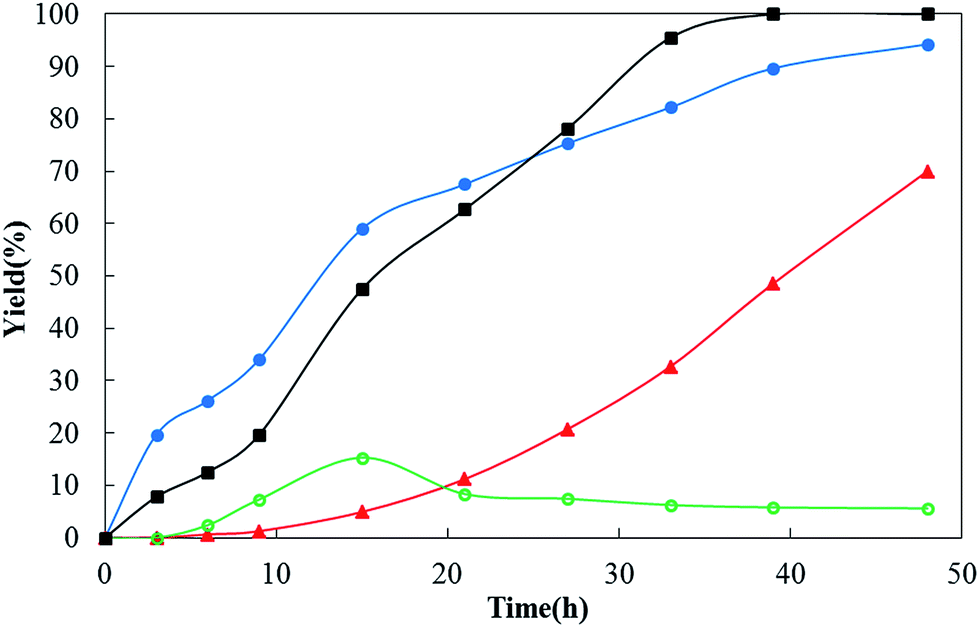 | ||
| Fig. 1 Reaction time course of the dehydrogenation of 3-isobutyl-5-phenyl-cyclohexanone to 3-isobutyl-5-phenyl-phenol under variable gas atmosphere conditions. The reaction conditions are as follows: 3-isobutyl-5-phenyl-cyclohexanone (2 mmol), DMA (4 mL), Pd/C (0.1 mmol Pd), K2CO3 (0.4 mmol), 150 °C. The yields were determined by GC analysis using n-dodecane as the internal standard. Curve ■ (black): the reaction was performed under 1 atm of the gas mixture atmosphere (70 vol% N2 and 30 vol% H2). Curve ● (blue): the Pd/C catalyst was pretreated with 1 atm H2 at room temperature for 1 h, and the dehydrogenation reaction with the pretreated Pd/C was performed under 1 atm N2 atmosphere. Curve ▲ (red): the reaction was performed under 1 atm N2 atmosphere. Curve ○ (green): the reaction was performed under 1 atm O2 atmosphere. | ||
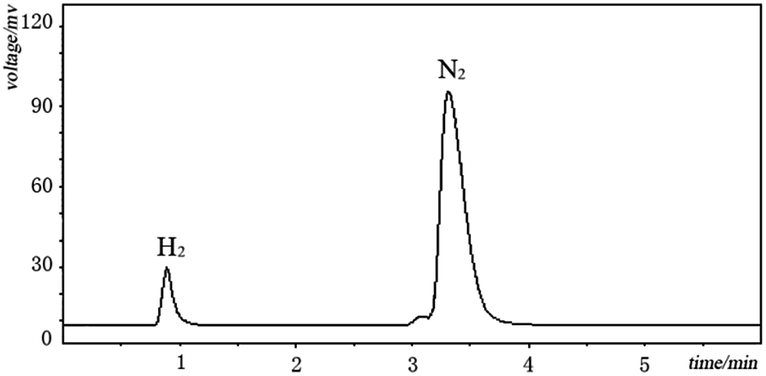 | ||
| Fig. 2 GC H2 detection for the dehydrogenation of cyclohexanone. The reactions were under N2 atmosphere. The gas sample was sampled from the reaction gas atmosphere (reaction conditions: 1 mmol cyclohexanone, 5% mol Pd/C, 150 °C, 1.5 h, 2 mL DMA under 1 atm N2 atmosphere). The H2 and N2 were confirmed by comparing with standard H2 and N2 GC spectra. | ||
The activation effect of the hydrogen generated in the reaction on the Pd/C catalyst might be less obvious in some cases because the resulting partial pressure of hydrogen still may not be high enough. Thus for some specific reactions, it is still required to add a certain extra amount of hydrogen gas to the catalytic dehydrogenation systems. The effect of H2 on the dehydrogenation depends on the amount of H2 used: too high partial pressure of hydrogen could lead to more hydrogenation product generation, which is unfavorable for the dehydrogenation reaction; a proper amount of H2 could help power the reaction; some dehydrogenation reactions do not need extra H2 added because the generated hydrogen gas is sufficient for the catalyst activation.
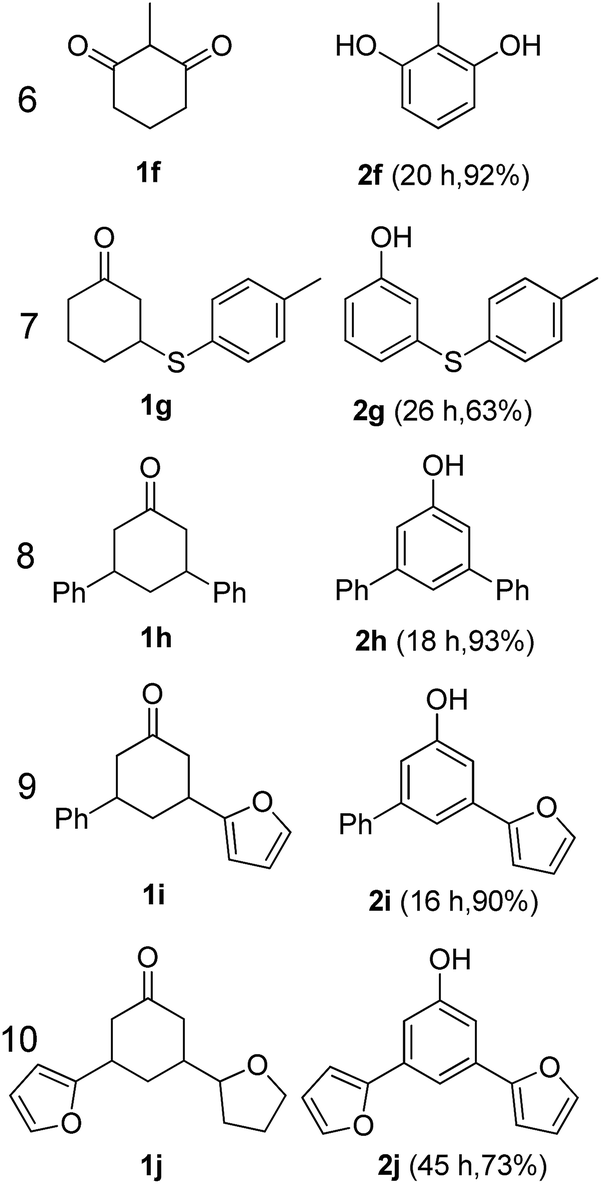
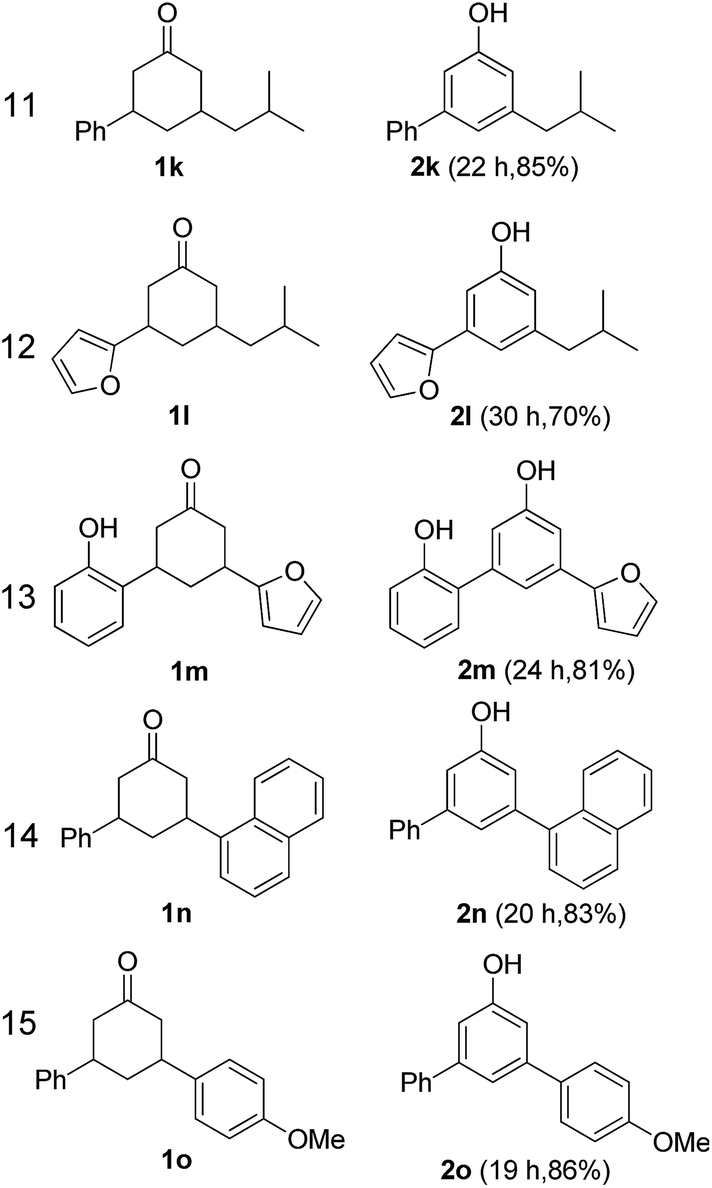
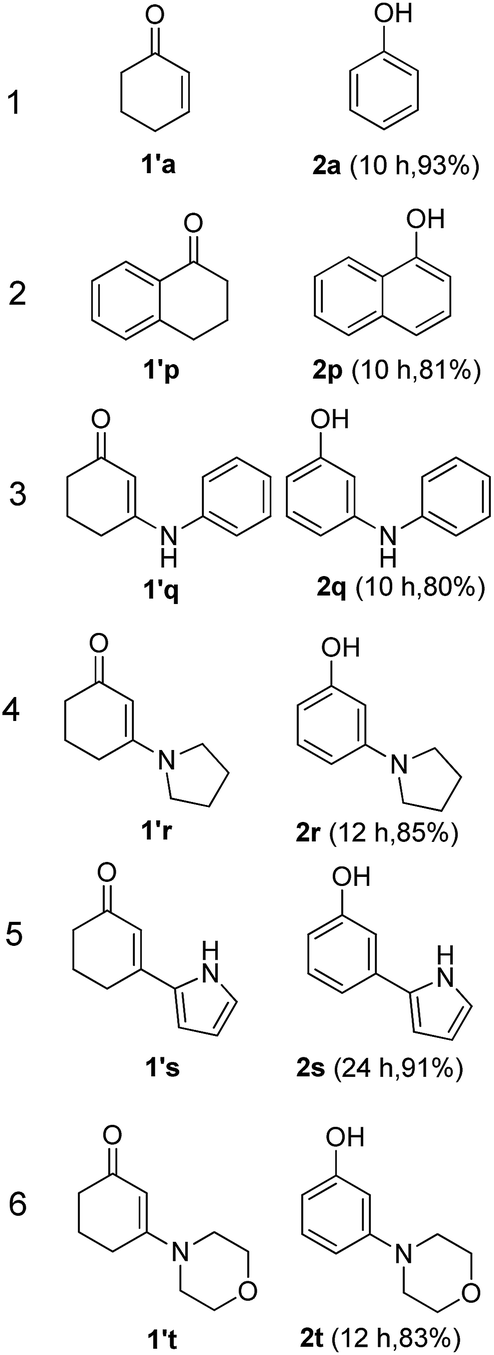
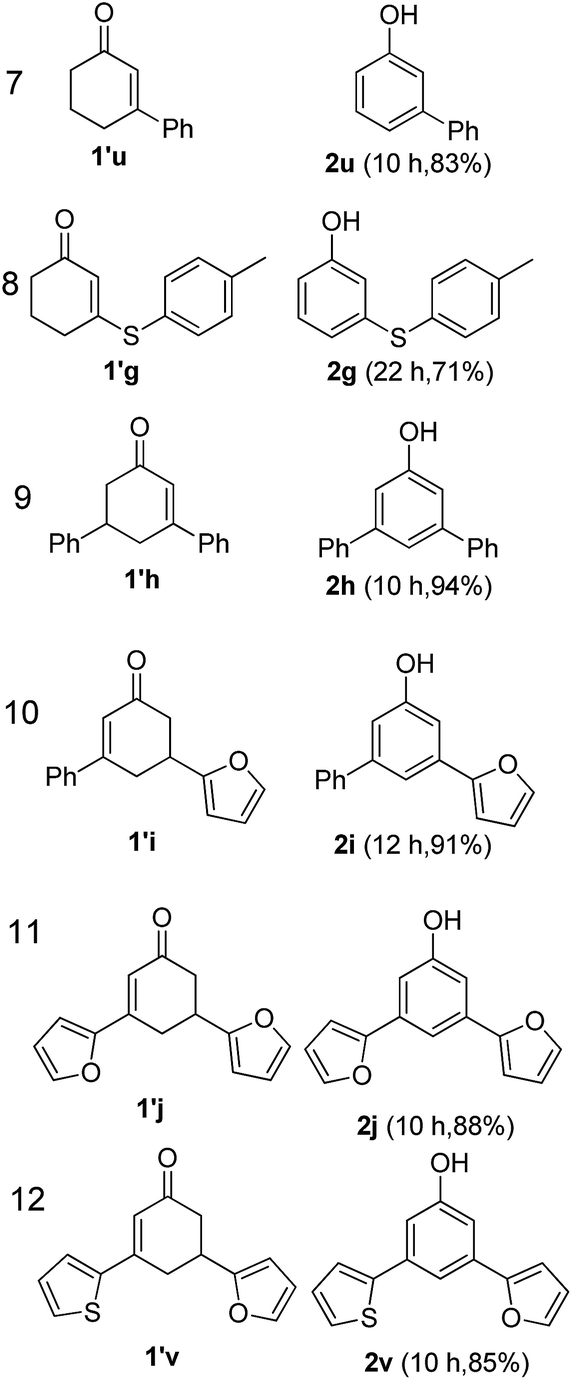
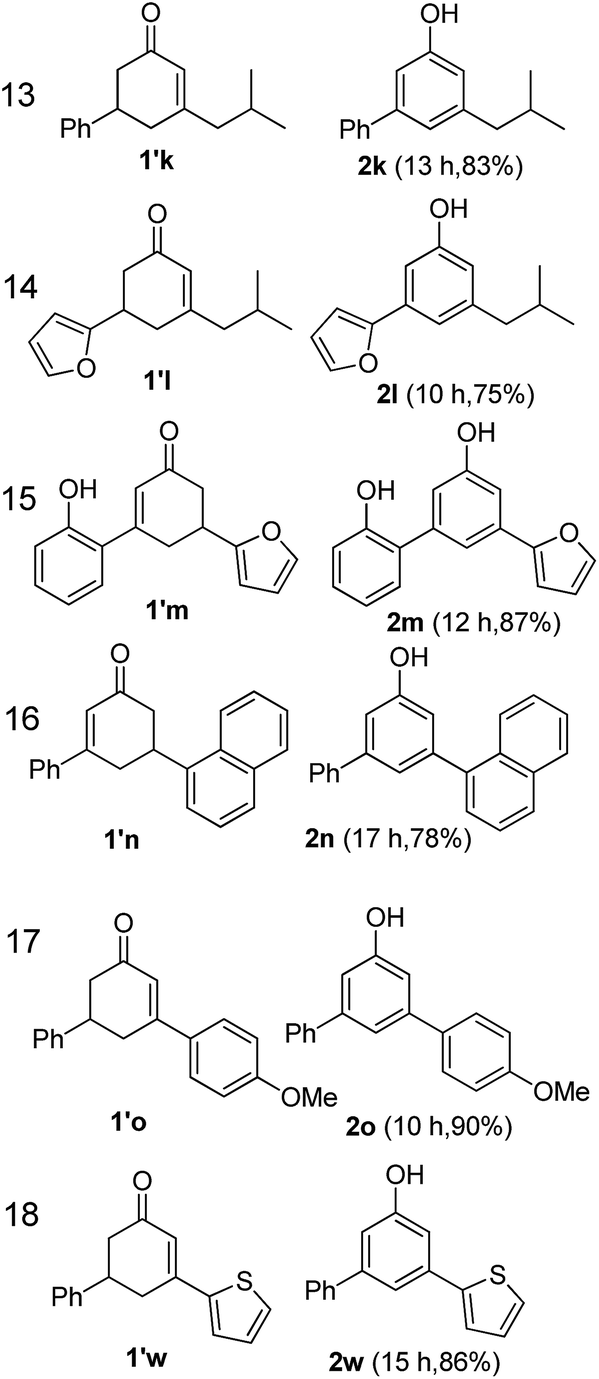
The hydrogen-containing gas atmosphere reaction conditions were applied to the dehydrogenation of a variety of substituted cyclohexanones and 2-cyclohexenones. A selection of pertinent examples are displayed in Tables 2 and 3. As can be seen, a broad spectrum of substituted cyclohexanones and cyclohexenones were dehydrogenated to their corresponding phenols with high isolated yields. The outcome of the dehydrogenation was not significantly affected by varying the position of the same substituent, e.g. a methyl group on the cyclohexanones. A number of 3-substituted cyclohexanones, which were readily synthesized with known methods, were efficiently dehydrogenated to their corresponding phenols with the present dehydrogenation method. This may be of great interest in the synthesis of meta-fragment substituted phenol derivatives. It is noteworthy that sulfur-, nitrogen- and oxygen-containing compounds (typically: Table 2, entries 7, 10, 13 and Table 3, entries 3–6, 8, 15, 18) were also very smoothly converted into their corresponding phenols in high yields with the catalytic system. However, these heteroatom-containing ketones are usually regarded as difficult substrates in most transition metal-catalyzed oxidative protocols due to their strong coordinating abilities. Of particular interest is the fact that under the oxidant- and acceptor-free conditions, highly oxidation-sensitive substrates (Table 2, entries 7, 13 and Table 3, entries 3–6, 8, 15) which are problematic in oxidative dehydrogenations, seldom interfered with the catalytic dehydrogenation reaction and also provided excellent yields. Usually these readily oxidizable ketones are problematic in oxidative dehydrogenations, for example, we attempted to prepare the phenols derived from the substrates (Table 2, 1g and Table 3, 1'q, 1's, 1't) using Stahl's aerobic catalytic system.6a,8 The results showed these ketones provided little or no desired products, instead a large amount of miscellaneous oxidation products were detected. When the substrate containing a tetrahydrofuran ring was examined, we were pleased to find that not only the cyclohexanone moiety but also the tetrahydrofuran ring was dehydrogenated (Table 2, entry 10). This observation combined with our previous study, palladium(0) catalyzed acceptorless dehydrogenation,11 suggests the present catalytic dehydrogenation system may be applied to the dehydrogenation of substituted tetrahydrofurans to synthesize furans.
|
|
|||||
|---|---|---|---|---|---|
| Entry substrate | Phenol (time/yield) | Entry substrate | Phenol (time/yield) | Entry substrate | Phenol (time/yield) |
| a R1, R2, R3, R4, R5: Ar, Me, H. The reaction conditions are as follows: cyclic ketone (1.0 mmol), Pd/C (5 mol% Pd), K2CO3 (20 mol%), DMA (N,N-dimethylacetamide, 2 mL), 150 °C, 1 atm of gas mixture atmosphere (30 vol% H2 and 70 vol% N2). Isolated product yields are reported. | |||||
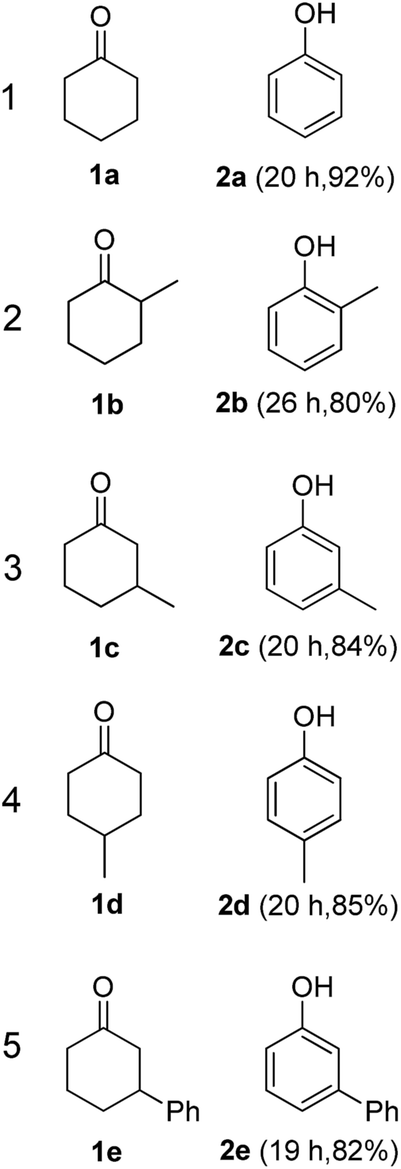
|
|
|
|||
|
|
|||||
|---|---|---|---|---|---|
| Entry substrate | Phenol (time/yield) | Entry substrate | Phenol (time/yield) | Entry substrate | Phenol (time/yield) |
| a R1, R2, R3, R4, R5: Ar, Me, H. The reaction conditions are as follows: cyclic ketone (1.0 mmol), Pd/C (5 mol% Pd), K2CO3 (20 mol%), DMA (N,N-dimethylacetamide, 2 mL), 150 °C, 1 atm of gas mixture atmosphere (20 vol% H2 and 80 vol% N2). Isolated product yields are reported. | |||||
|
|
|
|||
Elucidation of the detailed mechanistic pathway will undoubtedly shed light on the pathway and allow further optimization of this dehydrogenation system. In the present case, a detailed mechanism for the overall catalytic process cannot yet be deduced, but an outline of a potential mechanism is provided in Scheme 2. The reaction started with a keto–enol tautomerism equilibrium under the alkaline conditions. The metal Pd(0) reacted with hydrogen gas to generate the HPd(II)H species. The HPdH species may undergo dehydrogenative palladation of the β-position of cyclohexanone (either the keto or enol tautomer) to generate the intermediate (A). The palladation with Pd(0) may be difficult compared with use of the HPd(II)H species. This may be why H2 can activate the palladium(0) catalyst. The intermediate (A) was subsequently converted to a key intermediate (B), meanwhile H2 was released due to the directing effect of the electron-donating property of hydroxyl groups. The dissociation of the intermediate (B) led to Pd(0) and the intermediate (C). Sequentially, the second dehydrogenation of the 2-cyclohexenone (C), a process similar to the first dehydrogenation, resulted in the generation of compound (D), which tautomerized into the target product. Gas chromatography (GC) was used to detect the reaction gas atmosphere. As shown in Fig. 2, we clearly detected the generation of H2 during the reaction. This demonstrated the hydrogen that departed from the substrate formed hydrogen gas.
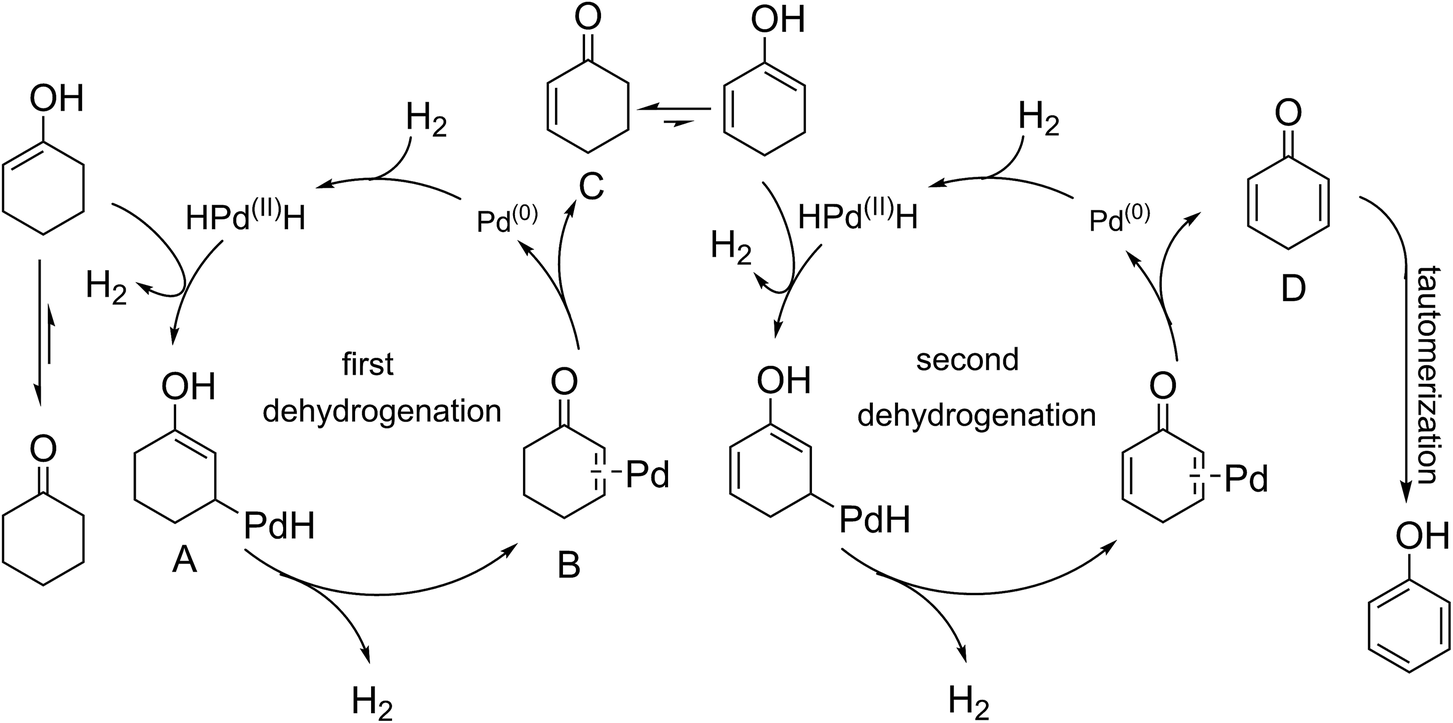 | ||
| Scheme 2 Proposed mechanistic pathway of palladium-catalyzed dehydrogenation of cyclohexanone to phenol and H2. | ||
Conclusions
Dehydrogenation, excluding deprotonation, is an oxidation process. Therefore, to obtain efficient dehydrogenation processes, a lot of attention was naturally turned to developing effective catalysis with oxidants while largely ignoring the advantages inherent in catalysis with a reducing agent (H2). Our findings unlock opportunities for markedly different synthetic strategies. These reactions achieve high isolated yields and they are intrinsically devoid of the complications caused by over-oxidation in oxidative dehydrogenation, while the catalyst tolerates useful substrates with diverse functional groups, including aromatic which are susceptible to oxidation and heteroatom substituents with strong coordinating ability. This dehydrogenation does not need oxidants and hydrogen acceptors, and manifests a high atom economy. Moreover, the only by-product (H2) of these reactions is a clean energy carrier. Palladium on active carbon is a commonly used recycled industrial catalyst. Thus this Pd/C, in combination with H2, catalytic dehydrogenation process displays considerable promise for its practical application in an industrial setting.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant 21072055) and the Ministry of Science and Technology of China (grant 2008AA06Z306).Notes and references
- (a) J. H. P. Tyman, Synthetic and Natural Phenols, Elsevier, New York, 1996 Search PubMed; (b) Z. Rappoport, The Chemistry of Phenols, Wiley-VCH, New York, 2003 CrossRef PubMed.
- (a) R. E. Maleczka Jr, F. Shi, D. Holmes and M. R. Smith, J. Am. Chem. Soc., 2003, 125, 7792 CrossRef PubMed; (b) Z. Guo and A. G. Schultz, Org. Lett., 2001, 3, 1177 CrossRef CAS PubMed; (c) D. H. Lee, K. H. Kwon and C. S. Yi, J. Am. Chem. Soc., 2012, 134, 7325 CrossRef CAS PubMed; (d) R. B. Bedford, S. J. Coles, M. B. Hursthouse and M. E. Limmert, Angew. Chem., Int. Ed., 2003, 42, 112 CrossRef CAS PubMed; (e) T. Truong and O. Daugulis, Chem. Sci., 2013, 4, 531 RSC; (f) C. L. Ciana, R. J. Phipps, J. R. Brandt, F. M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458 CrossRef CAS PubMed; (g) K. Fujimoto, Y. Tokuda, H. Maekawa, Y. Matsubara, T. Mizuno and I. Nishiguchi, Tetrahedron, 1996, 52, 3889 CrossRef CAS.
- (a) C. A. Fyfe, The Chemistry of the Hydroxyl Group, Wiley-Interscience, New York, 1971, pp. 83–127 Search PubMed; (b) C. Hoarau and T. R. R. Pettus, Synlett, 2003, 127 CAS; (c) P. Hanson, J. R. Jones, A. B. Taylor, P. H. Walton and A. W. Timms, J. Chem. Soc., Perkin Trans. 2, 2002, 1135 RSC; (d) T. George, R. Mabon, G. Sweeny, J. B. Sweeney and A. J. Tavassoli, J. Chem. Soc., Perkin Trans. 1, 2000, 2529 RSC.
- A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553 CrossRef CAS.
- L. Ackermann, A. R. Kapdi, S. Fenner, C. Kornhaaß and C. Schulzke, Chem.–Eur. J., 2011, 17, 2965 CrossRef CAS PubMed.
- (a) Y. Izawa, D. Pun and S. S. Stahl, Science, 2011, 333, 209 CrossRef CAS PubMed; (b) Y. Izawa, C. Zheng and S. S. Stahl, Angew. Chem., Int. Ed., 2013, 52, 3672 CrossRef CAS PubMed; (c) T. Moriuchi, K. Kikushima, T. Kajikawa and T. Hirao, Tetrahedron Lett., 2009, 50, 7385 CrossRef CAS PubMed; (d) C. S. Yi and D. W. Lee, Organometallics, 2009, 28, 947 CrossRef CAS PubMed; (e) J. Muzart, Eur. J. Org. Chem., 2010, 2010, 3779 CrossRef PubMed.
- Encyclopedia of Reagents for Organic Synthesis, ed. D. Crich, Wiley, New York, 2010 Search PubMed.
- (a) D. Pun, T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8213 CrossRef CAS PubMed; (b) T. Diao, D. Pun and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8205 CrossRef CAS PubMed.
- C. A. Rice-Evans, N. J. Miller and G. Paganga, Trends Plant Sci., 1997, 2, 152 CrossRef.
- (a) R. Kawahara, K. I. Fujita and R. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 3643 CrossRef CAS PubMed; (b) W. Fang, Q. Zhang, J. Chen, W. Deng and Y. Wang, Chem. Commun., 2010, 46, 1547 RSC; (c) S. Musa, I. Shaposhnikov, S. Cohen and D. Gelman, Angew. Chem., Int. Ed., 2011, 123, 3595 CrossRef PubMed; (d) C. Gunanathan, L. J. Shimon and D. Milstein, J. Am. Chem. Soc., 2009, 131, 3146 CrossRef CAS PubMed; (e) J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840 CrossRef CAS PubMed; (f) M. Nielsen, H. Junge, A. Kammer and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 5711 CrossRef CAS PubMed; (g) B. Gnanaprakasam, J. Zhang and D. Milstein, Angew. Chem., Int. Ed., 2010, 49, 1468 CrossRef CAS PubMed; (h) B. Gnanaprakasam, E. Balaraman, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 12240 CrossRef CAS PubMed; (i) B. Gnanaprakasam and D. Milstein, J. Am. Chem. Soc., 2011, 133, 1682 CrossRef CAS PubMed; (j) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790 CrossRef CAS PubMed; (k) H. Zeng and Z. Guan, J. Am. Chem. Soc., 2011, 133, 1159 CrossRef CAS PubMed; (l) J. R. Khusnutdinova, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2014, 136, 2998 CrossRef CAS PubMed; (m) M. Montag, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2012, 134, 10325 CrossRef CAS PubMed.
- X. Jin, Y. Liu, Q. Lu, D. Yang, J. Sun, S. Qin, J. Zhang, J. Shen, C. Chu and R. Liu, Org. Biomol. Chem., 2013, 11, 3776 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and 1H and 13C NMR of new materials. See DOI: 10.1039/c5sc01044f |
| This journal is © The Royal Society of Chemistry 2015 |