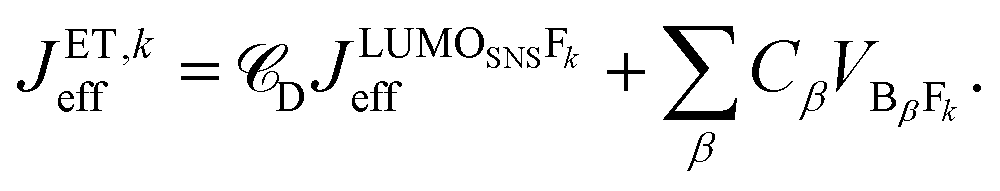DOI:
10.1039/C5SC01104C
(Edge Article)
Chem. Sci., 2015,
6, 4196-4206
Charge transfer versus molecular conductance: molecular orbital symmetry turns quantum interference rules upside down†
Received
27th March 2015
, Accepted 8th May 2015
First published on 11th May 2015
Abstract
Destructive quantum interference has been shown to strongly reduce charge tunneling rates across molecular bridges. The current consensus is that destructive quantum interference occurs in cross-conjugated molecules, while linearly conjugated molecules exhibit constructive interference. Our experimental results on photoinduced charge transfer in donor-bridge-acceptor systems, however, show that hole transfer is ten times faster through a cross-conjugated biphenyl bridge than through a linearly conjugated biphenyl bridge. Electronic structure calculations reveal that the surprisingly low hole transfer rate across the linearly conjugated biphenyl bridge is caused by the presence of destructive instead of constructive interference. We find that the specific molecular orbital symmetry of the involved donor and acceptor states leads to interference conditions that are different from those valid in single molecule conduction experiments. Furthermore, the results indicate that by utilizing molecular orbital symmetry in a smart way new opportunities of engineering charge transfer emerge.
1 Introduction
Charge transfer in organic molecules is a process that is of fundamental and practical importance in several areas of science and technology. Important technological innovations, such as organic solar cells,1,2 water splitting devices,3 or single molecule electronics,4–9 heavily rely on the control of intramolecular charge transfer reactions. A detailed understanding of these reactions along complex molecules is therefore of prime importance.
The experimental methods to probe the transfer of charge along molecular systems can be roughly divided into two types: charge transfer in donor-bridge-acceptor (DBA) molecules and single molecule conduction experiments in metal-bridge-metal (MBM) junctions.10 In DBA systems, the charge transfer process is directly followed in time, typically on a picosecond time scale, using for example femtosecond pump-probe spectroscopy.11 These experiments have been widely used to study the different parameters governing charge transfer in DBA systems.12–14 In a single molecule conduction experiment, the donor and the acceptor moieties of a DBA molecule are replaced by anchoring groups that are used to connect the molecular bridge to metallic electrodes. A steady-state current flowing through the molecular bridge is then measured upon application of a bias voltage between the two electrodes. Such an experimental set-up is particularly interesting for the design of molecular electronic devices but also to probe charge transport at the single molecule level.15
Although the two techniques are different in nature, similar behaviors are often observed. Both the charge transfer rate constant and the electronic conductance of short molecular bridges show a pronounced exponential distance dependence16,17 and a cross-over to a nearly distance-independent regime for longer bridges.18–22 This cross-over has been explained in both cases as a change from a tunneling mechanism to a thermally activated hopping mechanism.23–26 Recently, the possibility to modulate the conductance or the charge transfer rate by controlling the conjugation of the bridge has received a lot of attention. It has been theoretically predicted that linearly and cross-conjugated bridges placed in a MBM junction should present marked differences in their charge transport properties.27,28 These theoretical predictions have shown that the appearance of destructive quantum interference significantly reduces the electronic conduction of cross-conjugated bridges. On the contrary, constructive quantum interference occurs along linearly conjugated bridges, leading to relatively good charge transfer properties.29,30 These interference effects arise from the possibility for the tunneling charges to take multiple pathways via the different molecular orbitals of the bridge to cross the junction. These pathways can cancel each other in certain cases, thus leading to destructive quantum interference. Depending on which pathways cancel each other, different types of destructive interference can be observed. Over the last few years the majority of theoretical studies have described the here called HOMO–LUMO quantum interference in conjugated molecules.31–35 There, the destructive interference comes from pairwise cancelling contributions from occupied and unoccupied orbitals of the bridge. The experimental observation of this type of destructive quantum interference in MBM junctions has recently been reported by different groups.36–39 These reports have shown that the conductance of cross-conjugated bridges is a few orders of magnitude lower than the conductance of linearly conjugated bridges. Another type of destructive interference in MBM junctions has been shown both in theory and experiment to originate from cancelling contributions of degenerate orbitals.40,41 On the other hand, only a few articles have reported signatures of quantum interference in DBA systems.42–44
Because of the common phenomena, the two techniques are often treated in an analogous way and a clear theoretical connection has been demonstrated.28,45–48 Nitzan has theoretically shown that the rate constant of charge transfer in a DBA molecule is directly related to the conductance of that DBA molecule in the tunneling regime through the effective electronic coupling through the bridge.49 Although this relation has been explicitly derived for the case that the entire DBA molecule is connected to electrodes, it is often assumed that both quantities can be attributed to the properties of the molecular bridge alone. As a consequence, if destructive quantum interference occurs along a given bridge in a MBM junction, an exceptionally low charge transfer rate is generally expected for a DBA molecule containing the same bridge. Such simultaneous occurrence of quantum interference in MBM junctions and DBA molecules for specific bridges has been experimentally reported.42,43 However, as we demonstrate in this article, connecting a molecular bridge directly to electrodes instead of connecting it to donor and acceptor moieties may lead to considerably different trends in the conductance of MBM junctions compared to the charge transfer rate of DBA systems. This is because the symmetry of electronic states on the donor and acceptor is generally very different from the states to which the same molecular bridge couples in the case of metal electrodes. Here, we present a combined experimental and theoretical study of photoinduced charge transfer through a series of DBA molecules with the linearly and cross-conjugated bridges shown in Fig. 1. Contrary to expectations, we observe faster hole transfer for cross than for linear conjugation. A comparison with molecular conductance calculations of the same molecular bridges in MBM junctions confirms, however, the few orders of magnitude lower conductance for the cross-conjugated bridges than for the linearly conjugated bridge. Based on extensive electronic structure calculations, the differences between DBA and MBM systems are traced back to the particular symmetry of the donor and acceptor states that selects certain pathways of the molecular bridge for the charge transfer reaction in the DBA system. These selected pathways can give rise to quantum interference. In case of hole transfer along the linearly conjugated DBA molecule, this leads to destructive quantum interference – instead of constructive interference if no pathway selection takes place as in the MBM junction. Our results thus demonstrate that a given molecular bridge can behave differently when connected to electrodes or to donor and acceptor moieties as a consequence of pathway selection. Yet, this pathway selection offers new possibilities for tuning charge transfer in DBA systems based on the symmetry of molecular orbitals.
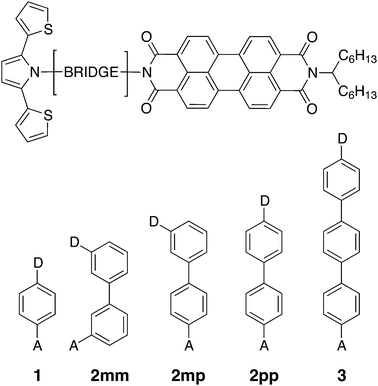 |
| | Fig. 1 Chemical structures of the linearly conjugated DBA systems 1, 2pp, and 3, the doubly cross-conjugated 2mm, and the singly cross-conjugated 2mp biphenyl bridge. | |
2 Results and discussion
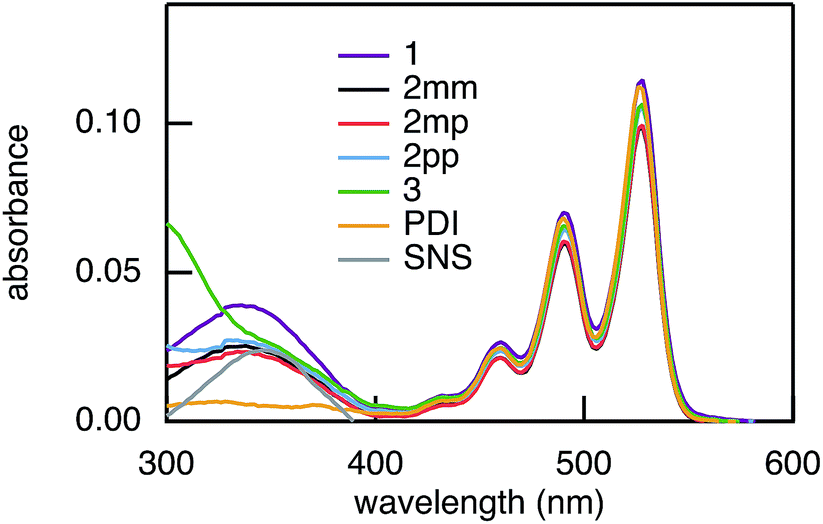
Photoinduced electron and hole transfer were studied through the DBA molecules shown in Fig. 1. These molecules are composed of a pyrrole derivative (SNS) as electron donor, a perylenediimide (PDI) electron acceptor and different n-phenylene bridges. We consider here three linearly conjugated bridges: 1, 2pp and 3 and two cross conjugated bridges: the doubly cross-conjugated 2mm and the singly cross-conjugated 2mp. To rule out a possible effect of the bridge energetics on the charge transfer rates, cross-conjugation is introduced using a chemically equivalent biphenyl bridge and substituting the donor and the acceptor to the bridge at different positions. In 2mm the donor and the acceptor are both connected to the biphenyl bridge in meta position. In 2mp, the donor is in meta position while the acceptor is in para position. Finally, both the donor and the acceptor are para substituted in 2pp. Because of their equal energetics, we particularly focus on the three biphenyl bridges 2mm, 2mp and 2pp. The study of 1 and 3 allows to put our results for the biphenyl bridges in perspective.
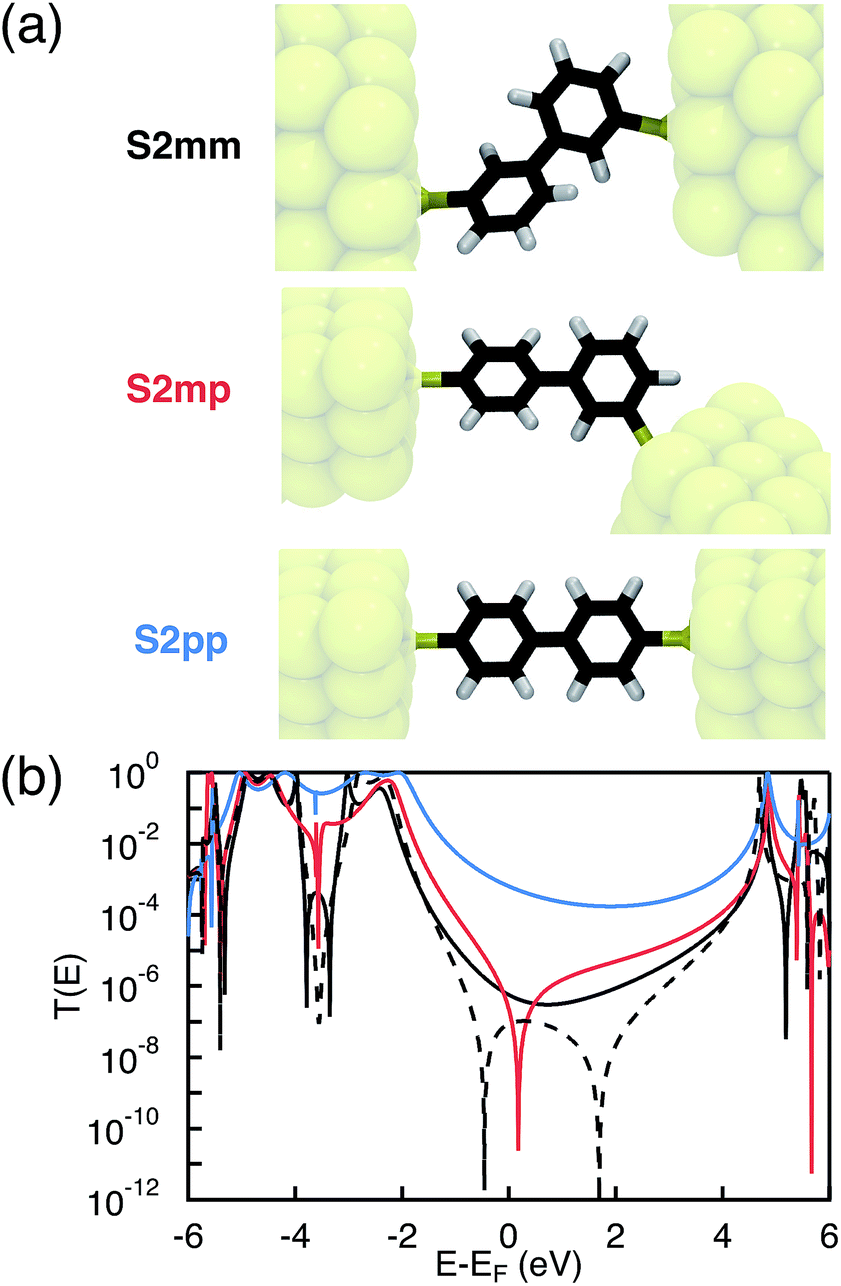
As mentioned in the introduction, we particularly focus on the comparison of photoinduced charge transfer across the three biphenyl bridges of the DBA systems 2mm, 2mp, and 2pp with conductance properties of the same bridges embedded in a MBM junction. Therefore, before presenting the results on photoinduced charge transfer, we will briefly discuss the trend in molecular conductance of these bridges in a MBM junction. To this end, the donor and acceptor moieties were replaced by thiol groups to anchor the molecular bridges to gold electrodes, resulting in the three MBM junctions S2mm, S2mp, and S2pp as shown in Fig. 2(a).
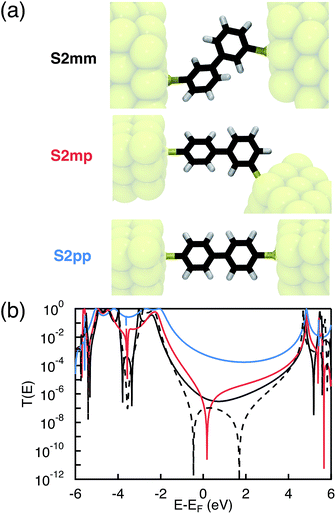 |
| | Fig. 2 Geometries (a) and transmission coefficients (b) of the biphenyl MBM junctions S2mm, S2mp, and S2pp. The cross-conjugated S2mp and S2mm present a much lower transmission than the linearly conjugated S2pp due to quantum interference effects. The black dashed line shows the transmission coefficient of S2mm when limiting the interactions between the two phenyls to through-bond couplings between the connecting carbon atoms. In this case, sharp interference features are present. These clear features however disappear when through-space interactions between the two rings are also considered (plain line). | |
The conductance of a molecular bridge in a MBM junction is (in the low bias limit) proportional to the square of the electronic transmission coefficient T(E) of the junction at the Fermi energy EF of the electrode: 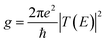 . This transmission coefficient represents the probability that an electron with an energy E will cross the junction. Using the non-equilibrium Green function approach, T(E) can be written as:
. This transmission coefficient represents the probability that an electron with an energy E will cross the junction. Using the non-equilibrium Green function approach, T(E) can be written as:
| | 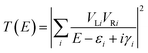 | (1) |
where
VLi (
VRi) is the coupling between the left (right) electrode and the
i-th transmission channel of the bridge, which roughly corresponds to the molecular orbital of the bridge. Its energy and lifetime are given by
εi and
γi respectively. A value of
Γ = 1 eV was used in the calculations. The summation over all molecular orbitals of the bridge explicitly accounts for all possible pathways that an electron can take to cross the junction. All these quantities were calculated at the density functional theory (DFT) level of theory using the Amsterdam Density Functional software
50 with the DZP basis set and the M06-2X
51 functional. The calculations were performed within the wide band limit approximation;
i.e. by coupling the
Pz orbital of each sulfur atom to a fictitious electrode. As seen in
Fig. 2(b),
T(
E), and hence the conductance, of
S2mm and
S2mp near the Fermi energy of the electrode is few orders of magnitude lower than
T(
E) of
S2pp. A clear signature of destructive quantum interference,
i.e. a zero of the transmission function, appears in the conductance of
S2mp. This sharp interference dip is due to the HOMO–LUMO interference,
i.e. an exact pairwise cancellation of the sum in
eqn (1), where the contribution of the LUMO exactly cancels the one of the HOMO
etc. at the energy where the dip occurs. In
S2mm, no such HOMO–LUMO interference occurs as explained in more detail in S.5. of ESI.
† However, when limiting the interactions between the two phenyls to interactions between the connecting carbon atoms another type of interference is present. All contributions cancel each other globally leading to the sharp interference pattern observed for the dashed line of
Fig. 2(b). Taking into account all non-nearest neighbor interactions washes away this perfect cancellation to some extent. Consequently, the calculated
T(
E) of
S2mm has a similar magnitude as
S2mp, except for the dip near the Fermi energy. Compared to the two cross-conjugated MBM junctions, the transmission of
S2pp is relatively large near the Fermi energy due to constructive contributions from HOMO and LUMO. As demonstrated in ESI,
† the trends in
T(
E) are preserved when including an atomistic description of small gold clusters in the calculations. The calculated transmission coefficients of the biphenyl MBM junctions confirm the generally accepted rule
29,34 that constructive quantum interference in the linearly conjugated
S2pp should lead to a much higher conductance with respect to the cross-conjugated
S2mm and
S2mp. Therefore, we initially also expected to obtain much lower charge transfer rates for the equivalent DBA systems
2mm and
2mp than for
2pp.
2.1 Experimental electron and hole transfer rates
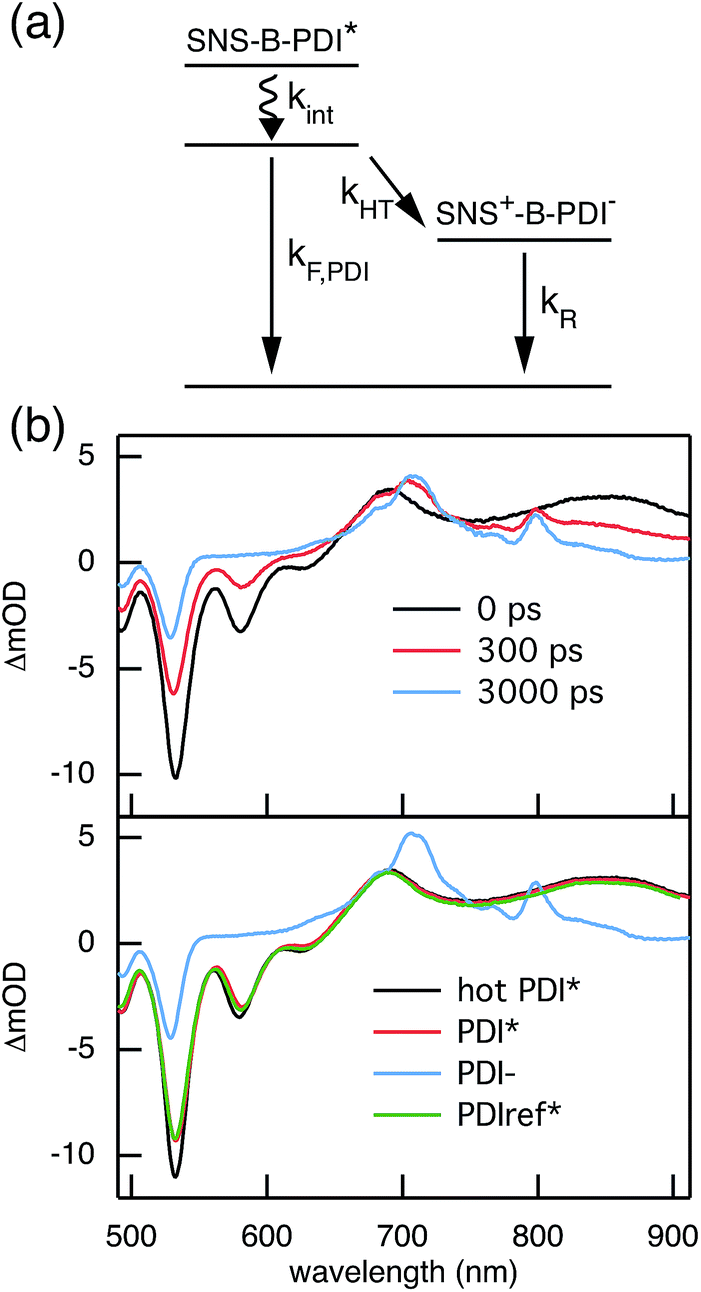
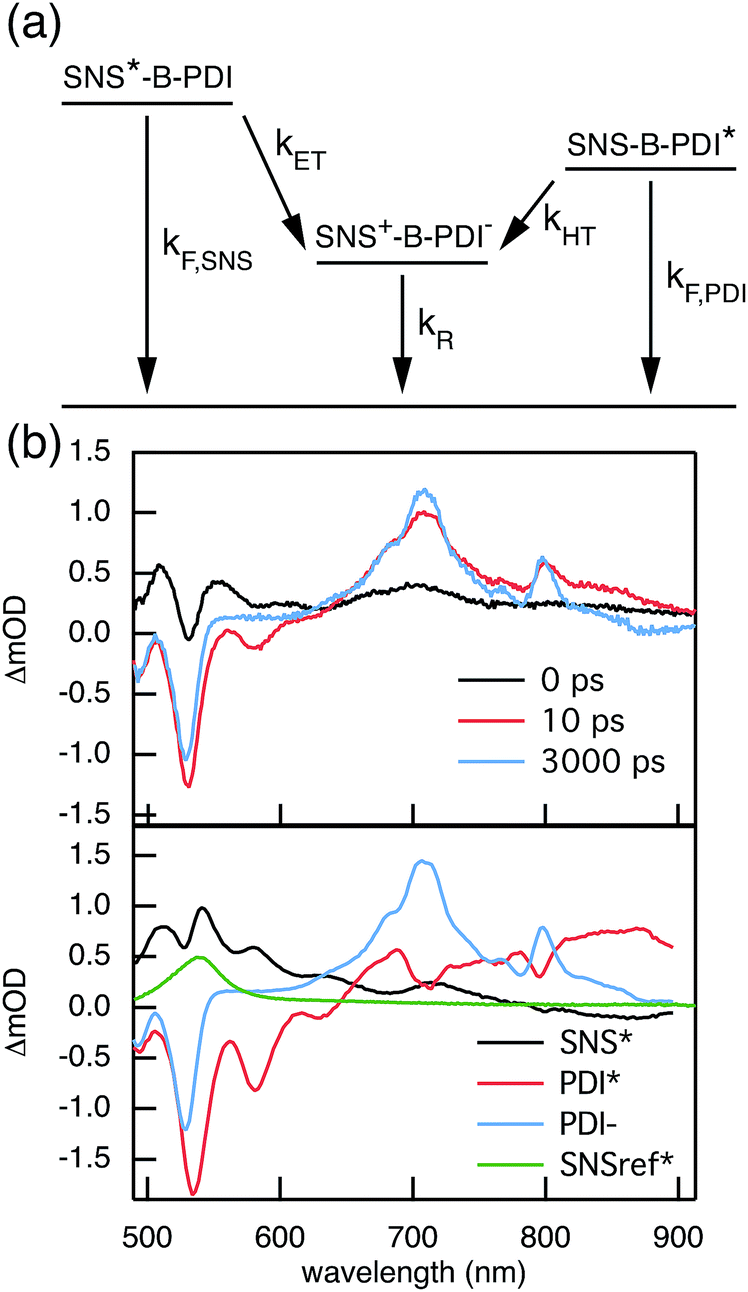
The rates of electron and hole transfer along the DBA molecules shown in Fig. 1 were experimentally determined using femtosecond transient absorption spectroscopy using a tunable Yb:KGW laser system with a time resolution of ca. 200 fs in a spectral window of 490–910 nm (details in ESI†). We call electron transfer the process where the electron donor (SNS) is excited and the excited electron transfers to the electron acceptor (PDI). Hole transfer is the process where the electron acceptor is excited and the generated hole transfers to the electron donor. In Fig. 3 the ground-state absorbance spectra of the studied molecules and the separate PDI and SNS fragments are shown. The maximum absorbance of PDI located at 527 nm shows no overlap with the absorbance of SNS. This allows selective excitation of the electron acceptor and thus the exclusive study of hole transfer. Upon excitation, the excited PDI can subsequently decay via hole transfer forming PDI−˙. This can be seen in Fig. 4(b) where the temporal evolution of the difference absorbance (ΔOD) spectra of 2pp is shown. Immediately upon excitation, the ΔOD spectrum matches the one of PDI*, which is obtained from an independent measurement of neat PDI (PDIref*). Within a few nanoseconds, this spectrum transforms into a neat spectrum of PDI−˙ (blue spectrum in Fig. 4(b)). Because of the overlap of the two spectra, the rate constant of the formation of PDI−˙ could not be deduced from kinetic traces at a single wavelength. Therefore, global and target analysis was performed using the open source software Glotaran52 (details in ESI†). The two-dimensional datasets obtained upon excitation at 527 nm were modelled with the sequential kinetic scheme describing the underlying photophysical processes depicted in Fig. 4(a). The initially excited PDI (hot PDI*) undergoes internal relaxation within the first picoseconds53 with rate constant kint. Subsequently, hole transfer occurs. For all samples, the rate constant of hole transfer kHT is at least one order of magnitude higher than the competing radiative decay of PDI*. The rate constant of the radiative decay kF,PDI was fixed to 2.5 × 10−4 ps−1 in the fits for all samples. This value was obtained from an independent measurement of neat PDI and is in agreement with previous observations.53 Although the photoinduced spectrum of SNS+˙ is outside the spectral range of our experimental setup and could thus not be observed, the rate constant of PDI−˙ formation could be set equal to kHT because previous work by Weiss et al.54 has shown that no hole transfer occurs to the phenylene bridge. All rate constants obtained from target analysis are listed in Table 1. The ΔOD spectra of hot PDI*, PDI*, and PDI−˙, which are also obtained from target analysis, are shown in the bottom graph of Fig. 4(b).
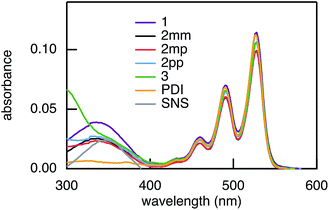 |
| | Fig. 3 Ground-state absorbance spectra of 1, 2mm, 2mp, 2pp, and 3, and of the donor and acceptor reference compounds SNS and PDI. | |
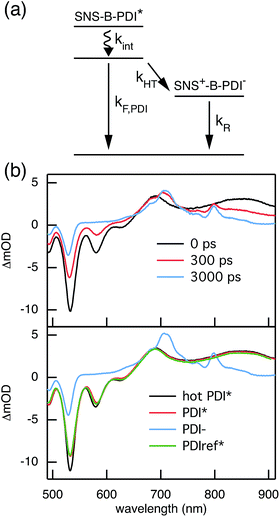 |
| | Fig. 4 (a) Photophysical processes occurring upon excitation at 527 nm. (b) The top graph shows ΔOD spectra at several points in time after exciting 2pp at 527 nm. The bottom graph presents the ΔOD spectra of hot PDI*, PDI*, PDI−˙, and PDIref* that are obtained from the target analysis of transient absorption following excitation at 527 nm using the kinetic scheme depicted in (a). | |
Table 1 Rate constants (ps−1) of all photophysical processes determined from target analysis of transient absorption following excitation at 350 nm and 527 nm respectively. kF,PDI = 2.5 × 10−4 and kF,SNS = 2.7 × 10−3, determined from independent measurements of neat PDI and SNS, were fixed in all fits
|
|
Ex. at 527 nm |
Ex. at 350 nm |
|
k
int
|
k
HT
|
k
R
|
k
HT
|
k
ET
|
k
R
|
|
1
|
0.37 |
0.043 |
1.1 × 10−3 |
0.028 |
0.35 |
8.6 × 10−4 |
|
2mm
|
0.18 |
0.014 |
6.4 × 10−4 |
0.013 |
0.13 |
6.2 × 10−4 |
|
2mp
|
0.31 |
0.0040 |
1.0 × 10−4 |
0.0038 |
0.18 |
5.4 × 10−5 |
|
2pp
|
0.25 |
0.0025 |
7.1 × 10−5 |
0.0024 |
0.25 |
4.3 × 10−5 |
|
3
|
0.23 |
0.0012 |
4.6 × 10−5 |
0.0010 |
0.17 |
1.9 × 10−5 |
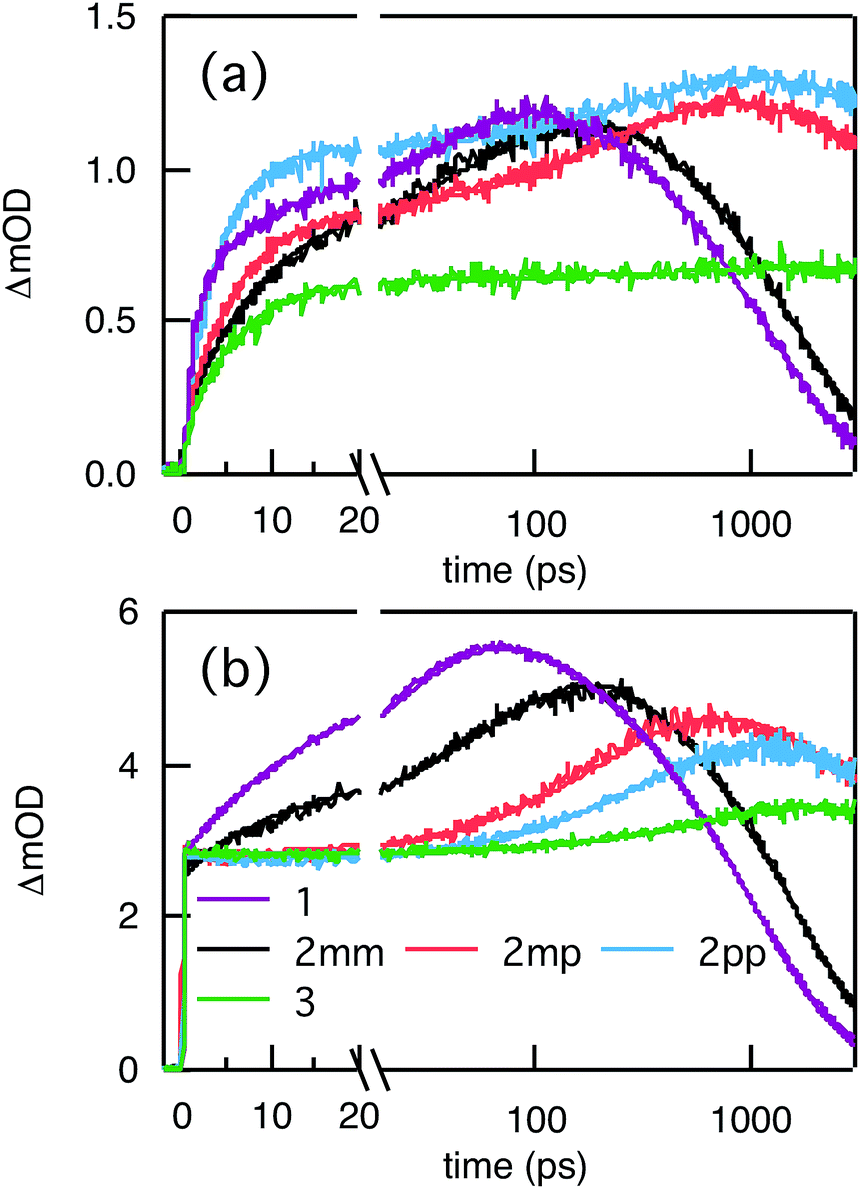
Compared to kHT, determining the rate constant of electron transfer was less straight forward as the maximum absorbance of SNS around 350 nm overlaps with a relatively small absorbance by PDI. Therefore, excitation at 350 nm not only triggers electron transfer from the excited SNS but also hole transfer from the PDI excited state. Modeling the datasets for all samples upon excitation at 350 nm involved therefore a kinetic scheme, in which electron and hole transfer occur parallel to each other, as depicted in Fig. 5(a). The internal relaxation of PDI* is omitted in the fitting procedure for simplicity. The simultaneous excitation at 350 nm can be seen in the ΔOD spectrum at 0 ps in Fig. 5(b), which shows features of the PDI* spectrum on top of a positive band between 500 and 600 nm. The latter band is ascribed to SNS*. After a few nanoseconds a neat PDI−˙ spectrum is observed. Applying target analysis with the kinetic scheme of Fig. 5(a) with fixed rate constants of the radiative decays of PDI* (2.5 × 10−4 ps−1) and SNS* (2.7 × 10−3 ps−1) yields all remaining rate constants (Table 1) as well as the ΔOD spectra of SNS*, PDI*, and PDI−˙ (bottom graph of Fig. 5(b)). While the spectrum of PDI−˙ is identical to the one obtained at 527 nm excitation, spectra of PDI* and SNS* deviate from the excited state spectra of neat PDI and SNS. This is due to the difficulty of disentangeling the two simultaneously excited species. Nevertheless, the coincident values of kHT obtained from target analysis of data from 527 nm and 350 nm-excitation demonstrate the validity of the applied analysis. The quality of the fits is shown in Fig. 6 where the experimental and fitted kinetic traces at 710 nm for both excitations are displayed for all samples. As seen in this figure, excellent fits were obtained for each DBA molecule. These kinetic traces qualitatively reflect the rates of electron and hole transfer in the studied DBA systems. The corresponding rate constants of electron transfer kET and hole transfer kHT are listed in Table 1 and plotted against the donor–acceptor distance in Fig. 7.
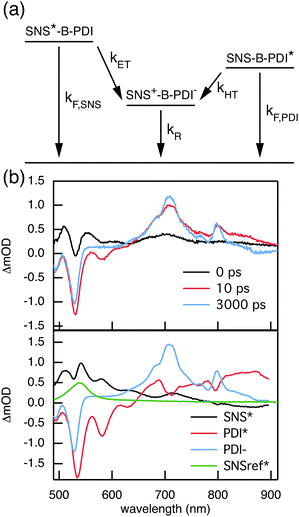 |
| | Fig. 5 (a) Kinetic scheme of the parallel electron and hole transfer processes occurring after excitation at 350 nm. (b) ΔOD spectra at several points in time after exciting 2pp at 350 nm are shown in the top graph. Target analysis of the transient absorption data using the kinetic scheme from (a) yields the ΔOD spectra of SNS*, PDI*, PDI−˙, and SNSref* that are presented in the bottom graph. | |
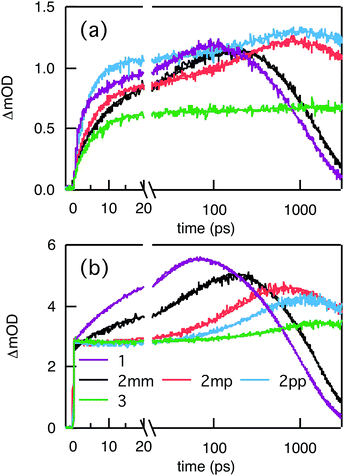 |
| | Fig. 6 Kinetic traces of ΔOD at 710 nm (main absorption band of PDI−˙) upon pulsed laser excitation at 350 nm (a) and 527 nm (b). The thick solid lines are the fits to the data obtained from global and target analysis. | |
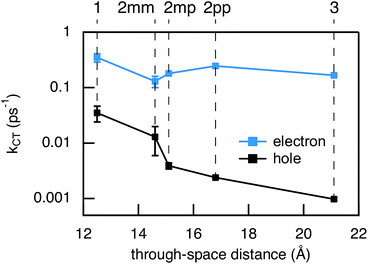 |
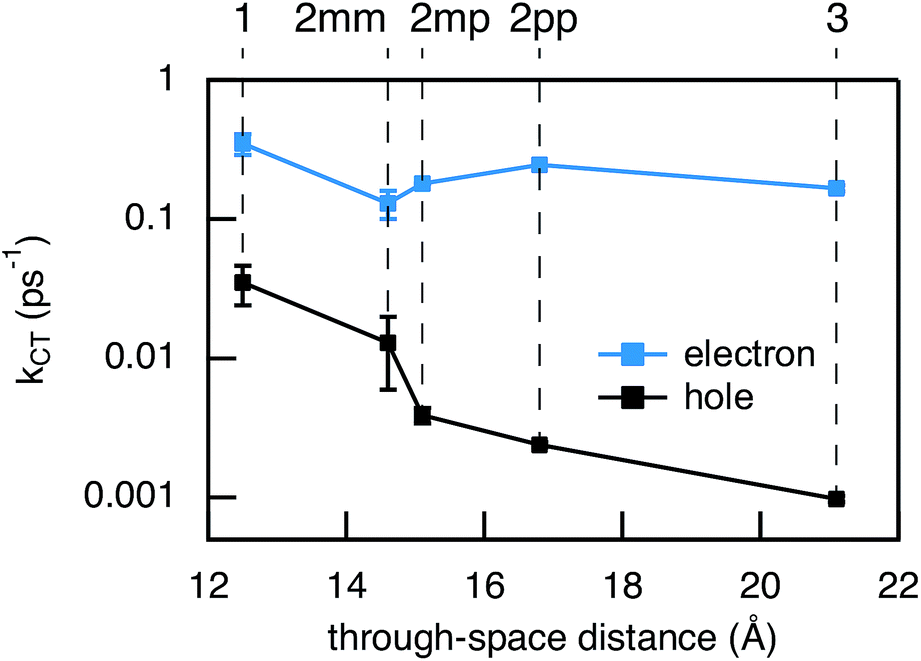
| | Fig. 7 Experimental charge transfer rate constants for electron (blue) and hole (black) transfer vs. the donor–acceptor distance. The donor–acceptor distance is only used for the purpose of visualization. The error bars are determined from multiple measurements. | |
As seen in Fig. 7, the electron transfer rate constants are 1–2 orders of magnitude larger than the hole transfer rates. Additionally, the distance dependence for electron transfer is much weaker than for hole transfer. For electron transfer, the effect of conjugation on the transfer rate seems to comply with the prediction based on T(E) calculations for the equivalent MBM junctions: i.e. the electron transfer rate of 2pp is larger than of 2mm and 2mp. One could conclude that destructive quantum interference hampers the charge transfer along 2mm and 2mp. However, the differences in kET along these three bridges are much smaller than expected in the case of quantum interference effects.
Remarkably, the relation between conjugation and rate constant is reversed when considering hole transfer: kHT of 2mm is larger than of 2mp and 2pp. This trend is exactly opposite to the trend observed for the electronic conduction of these bridges as represented in Fig. 2(b). This unexpected result clearly demonstrates that a given molecular bridge can behave very differently in a MBM junction and in a DBA system.
2.2 Electron and hole transfer parameters
To understand why electron and hole transfer along the different molecules studied here are so different from each other and from the molecular conductance, we compare the experimental values of the rate constants to charge transfer parameters obtained from DFT calculations using the Amsterdam Density Functional software.50 The alkyl chains attached to the PDI unit were replaced by hydrogen atoms in all calculations. The ground state geometries of the different DBA molecules were hence optimized using a DZP basis set and the M06-2X functional.51 To do so, the ground state geometries of the isolated donor-bridge, bridge-acceptor, and the isolated bridge fragments were first fully relaxed. These fragments were then assembled to form the DBA molecules whose geometry were optimized with respect of the rotation angles between the different fragments while keeping the rest of the structure fixed. The resulting structures were used to evaluate the charge transfer rates. In the context of non-adiabatic charge transfer theory, the rate constant kCT for the transition between one initial and one final state is proportional to the square of the effective electronic coupling Jeff:| | 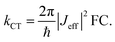 | (2) |
The Franck–Condon factor FC is, in semi-classical Marcus theory,55 expressed as
| | 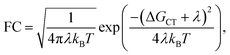 | (3) |
where
λ is the reorganization energy, and Δ
GCT the Gibbs free energy. Although
λ, and Δ
GCT can be evaluated using DFT, we focus on the calculation of the effective coupling
Jeff. This is because a satisfying agreement between theoretical and experimental values of
kCT requires calculation of
λ and Δ
GCT with an accuracy of a few tens of meV, which is difficult to achieve at the DFT level of theory. Instead of a direct comparison between experimental and calculated values for
kCT, we compare the experimental values of
kCT to computed
J2eff that should reflect the same trends. In particular, this should hold for
2mm,
2mp, and
2pp on the grounds of approximately equal energetics. In its most general form, the effective coupling between a localized initial state on the charge donor (I) and a localized final state (F) on the charge acceptor reads
56| | 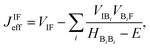 | (4) |
where the Hamiltonian submatrix
HBB describing the bridge is diagonalized.
VIF represents the direct coupling between the initial and final state, while the second term of the equation accounts for the indirect coupling between these two states
via the molecular orbitals of the bridge. In this second term
VIBi (
VBiF) is the direct coupling between the initial (final) state and the
i-th orbital of the bridge;
HBiBi is its energy.
E is the energy of the DBA system when charge transfer occurs. The summation over all bridge orbitals explicitly accounts for the multiple pathways mediating charge transfer similarly to
eqn (1) and therefore inherently accounts for the possible occurrence of interference effects. Strictly speaking,
JIFeff must be evaluated at the transition point where the initial and final states are at resonance. In our calculations, however, we evaluate
JIFeff of the Hamiltonian at ground state geometry and approximate
E to the energy of the initial state. The various direct couplings
VXY and the energies of the
i-th bridge orbitals
HBiBi in
eqn (4) were extracted from the Fock matrix of the DBA molecules obtained with DFT (M06-2X) using the molecular orbitals of the donor, bridge, and acceptor radical fragment molecules as basis set.
57 The coupling between these fragment orbitals and their energy is then given by the off-diagonal and the diagonal matrix elements of the Fock matrix:
HXY = 〈
X∣
H∣
Y〉 and
HBiBi = 〈
Bi|
H|
Bi〉. Because the fragment orbitals are in general not orthogonal, the finale value for the electronic coupling between the fragment orbitals
X and
Y was determined as
VXY =
HXY − 0.5
SXY (
HXX +
HYY), where
S is the overlap matrix. The fragment orbitals were obtained from electronic structure calculations of the isolated radical fragment molecules at the DFT level of theory with the DZP basis set and M06-2X functional.
2.2.1 Nature of the initial state.
The determination of JIFeff requires the identification of the fragment orbitals that form the initial and final states of electron and hole transfer.58–60 Therefore, excitation spectra of the DBA molecules studied here were calculated at the TD-DFT level of theory (DZP/M06-2X) using the same fragment orbital approach as described above. The calculated spectrum obtained for molecule 2pp is shown in Fig. 8. The spectra of all other compounds were similar, as were the experimental absorption spectra shown in Fig. 3. The most prominent features of these spectrum are located at 455 nm and 300 nm, corresponding to excitation of PDI and SNS respectively. The deviation from the experimental values (527 nm and 350 nm) to shorter wavelengths is likely due to stabilization by the solvent that is not taken into account in the calculations. As already discussed in our previous article,60 the excitation of SNS and PDI are very different in nature. The excitation at 455 nm mainly consists of a HOMOPDI–LUMOPDI transition, where the subscript denotes to which fragment the fragment orbitals belong. This means that the excitation is entirely localized on the PDI fragment (weight of >0.95). The initial state for hole transfer can thus be described with reasonable accuracy by the HOMOPDI. On the contrary, the initial excitation at 350 nm exhibits a significant charge transfer character. It is characterized by a one-electron HOMOSNS–LUMOSNS transition and a one-electron transition from the HOMOSNS to the LUMOPhn. Similar results were previously obtained for the linearly conjugated molecules 1, 2pp, and 3.60 Moreover, in the case of 2mm and 2mp, the LUMOSNS exhibits substantial orbital mixing with the LUMO + 1Phn; in case of 3 with the LUMO + 5Phn. The initial state for electron transfer was therefore described by a superposition of LUMOSNS and the relevant orbitals of phenylene: 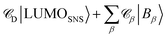 . The weights
. The weights ![[capital script C]](https://www.rsc.org/images/entities/char_e522.gif) x of the fragment orbitals X in this linear combination were directly obtained from the TD-DFT calculations with the fragment orbitals used as basis set and are listed in Table 2.
x of the fragment orbitals X in this linear combination were directly obtained from the TD-DFT calculations with the fragment orbitals used as basis set and are listed in Table 2.
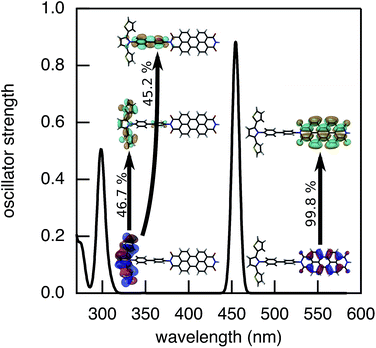 |
| | Fig. 8 Optical excitation of 2pp calculated using TD-DFT with DZP/M06-2X. | |
Table 2 Weights X of the fragment orbitals X in the initial state description for electron transfer
|
|
LUMOSNS |
LUMOPhn |
LUMO + 1Phn |
LUMO + 5Phn |
|
1
|
0.57 |
0.43 |
|
|
|
2mm
|
0.70 |
0.18 |
0.12 |
|
|
2mp
|
0.66 |
0.15 |
0.19 |
|
|
2pp
|
0.52 |
0.48 |
|
|
|
3
|
0.57 |
0.33 |
|
0.10 |
2.2.3 Comparing rate constants and electronic coupling.
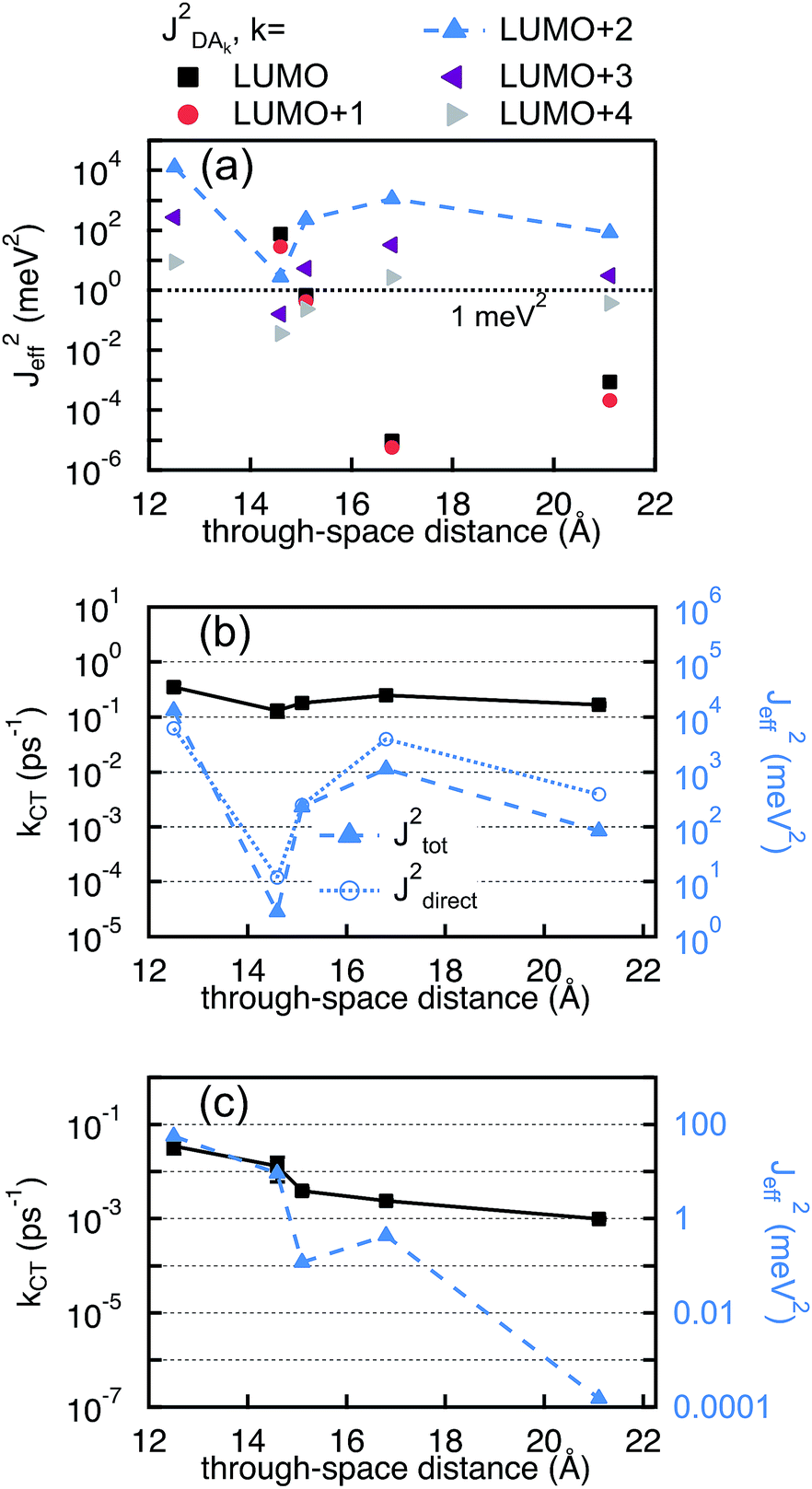
In Fig. 9(b), the computed (JETeff)2 for electron transfer from the initially delocalized state to the LUMO + 2PDI is compared to the experimentally determined kET. Although (JETeff)2 reflects the same trend as the experimental kET, it overestimates the difference in kET within 2mm, 2mp, and 2pp. While (JETeff)2 is more than two orders of magnitude smaller in the doubly cross-conjugated 2mm as compared to the linearly conjugated 2pp, the experimental kET differs only by a factor of two. This discrepancy is most likely due to the additional electron transfer path in 2mm and 2mp to the LUMOPDI as final state that increases kET. Additionally, the occurrence of decoherence, for instance by solvent fluctuations, could be responsible for the comparably smaller difference in kET than in (JETeff)2. We therefore assume that we can reliably interpret our experimental results solely based on the values of the electronic coupling. As explained above, the total effective electronic coupling for electron transfer has two contributions: one bridge-mediated contribution from the SNS to the PDI and one direct contribution between the bridge and the PDI. As can be seen in Fig. 9(b), this direct coupling dominates the total effective coupling explaining the relatively high values obtained for the electron transfer rates and their weak distance dependence. Furthermore, any impact of destructive interference on the electron transfer rate that is described by the bridge-mediated contribution is screened by the direct contribution. Moreover, because of the strong direct contribution, the electron transfer process in our DBA systems cannot be compared to the charge tunneling process in the equivalent MBM junctions where only bridge-mediated contributions play a role.
In contrast to electron transfer, the effective electronic coupling for hole transfer is solely composed of the bridge-mediated contribution because of the localized initial state. For such a tunneling process, comparable trends for charge transfer in DBA and MBM systems are commonly expected.43 In the present study, these expectations imply lower hole transfer rates for 2mm and 2mp as compared to 2pp. However, the comparison between the experimental kHT and (JHTeff)2 in Fig. 9(c) confirms the unexpected reduction in kHT when increasing conjugation from the doubly cross-conjugated 2mm to the linearly conjugated 2pp. This figure shows that only 1 and 2mm exhibit considerable couplings above 1 meV2. (JHTeff)2 for 2mp and 2pp is about two orders of magnitude smaller than for 2mm and (JHTeff)2 drops another four orders of magnitude for 3. It should be noted, that these values are too small to be accurately determined by DFT so that differences between 2mp, 2pp, and 3 should not be considered. The clearly different trend in the hole transfer rates in the DBA systems 2mm, 2mp, and 2pp and the conductance of the equivalent MBM junctions S2mm, S2mp, and S2pp discloses a fundamental differences between the properties of a given bridge embedded in a MBM junction and in a DBA molecule.
2.2.4 Orbital symmetry and pathway selection.
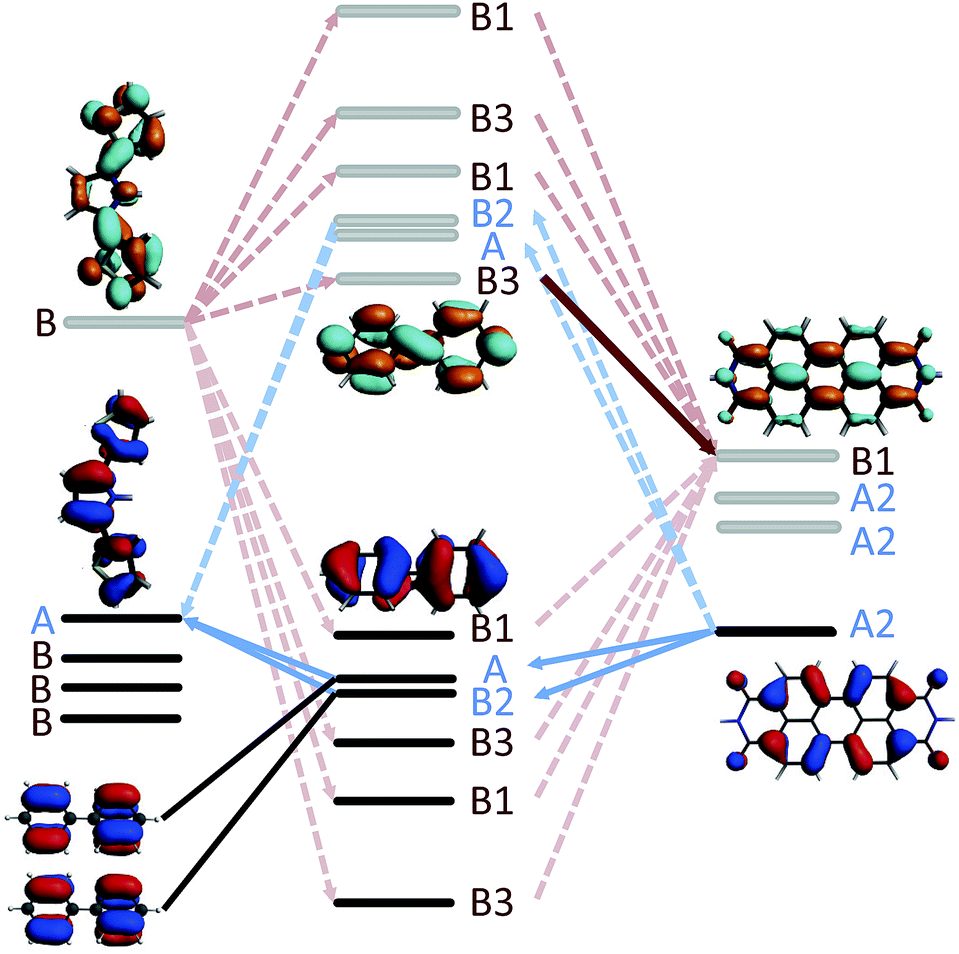
A simple explanation for the unexpectedly low hole transfer rate in the linearly conjugated 2pp can be obtained by examining the symmetries of the relevant fragment orbitals of SNS, the phenylene bridge, and PDI. The DBA molecule 2pp has a C2 point group symmetry with a rotational axis aligned with the biphenyl bridge. As depicted in Fig. 10, the fragment orbitals are therefore symmetric (blue label) or antisymmetric (red label) with respect to rotation around this C2 axis. The initial state for hole transfer, i.e. the HOMOPDI, is symmetric with respect of such rotation. It can therefore only couple only to the HOMOSNSvia the symmetric orbitals of biphenyl. Within the π-network, these are the degenerate HOMO − 1 and HOMO − 2, and the degenerate LUMO + 1 and LUMO + 2 of the bridge. Consequently, as illustrated in Fig. 10, the hole only has four distinct pathways to reach the hole acceptor as most of the fragment orbitals of the bridge are forbidden by symmetry. However, a closer examination of the degenerate HOMO − 1 and HOMO − 2 of the bridge, reveals that these two fragment orbitals have opposite symmetry with respect of a rotation along an axis perpendicular to the C2 axis. Consequently, their contributions to JHTeff exactly cancel each other. Since the same reasoning holds for the degenerate LUMO + 1 and LUMO + 2 of the bridge, no pathways are available for the hole to cross the bridge and to reach the hole acceptor. Hence, JHTeff should be exactly zero because of destructive interference between the degenerate orbitals. The nonzero value of JHTeff reported in Fig. 9 for 2pp are due to the σ-network and deviation of the molecular structure from the perfectly symmetric geometry. The symmetry considerations are valid irrespective of the dihedral angle between the bridge and the donor/acceptor as demonstrated in S.6. of ESI.† A similar analysis holds for 3, explaining the very low value of JHTeff obtained for this compound.
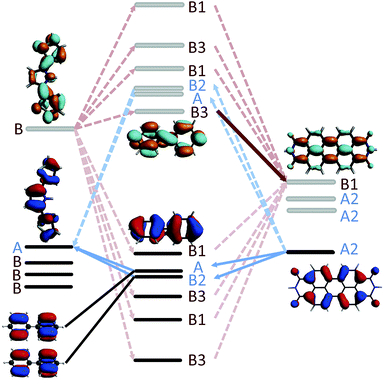 |
| | Fig. 10 Illustration of the available pathways for electron (red) and hole (blue) transfer along 2pp. The occupied orbitals are represented in black, the unoccupied orbitals in gray. As explained in the text the available pathways are entirely dictated by the symmetries of the fragment orbitals involved in the charge transfer. Note, that the fragment orbitals of donor, bridge, and acceptor are shown in one plane for illustrative purposes. The dihedral angle between the donor and bridge is around 60°, between acceptor and bridge around 65°. | |
In the case of 2mp, the PDI and the phenylene bridge still share the same common C2 axis (illustrated and discussed in S.7. of ESI†). As a consequence, the fragment orbitals HOMO − 1, HOMO − 2, LUMO + 1 and LUMO + 2 of the bridge remain the only mediating pathways for hole transfer to the HOMOSNS. However, the meta-substitution of the SNS fragment lifts the degeneracy of HOMO − 1 and HOMO − 2, and LUMO + 1 and LUMO + 2 to some extend, so that the contributions of each pair cancel each other only partially. This partial cancellation results in a nonzero theoretical (JHTeff)2 and might explain the slightly larger rate constant kHT observed in 2mp than in 2pp. Note, that this difference in the computed (JHTeff)2 for 2mp and 2pp can not be seen in the results obtained with DFT (see Fig. 9(c)) because of the already mentioned inaccuracy of DFT calculations and the deviation from the prefect symmetry. Finally, in 2mm, the three fragments do not share a common axis (illustrated and discussed in S.7. of ESI†) and therefore the symmetry restrictions do not apply. This explains the relatively large effective coupling for hole transfer obtained for 2mm in comparison to 2mp and 2pp.
In the case of electron transfer, the symmetry considerations discussed above clarify why the lowest final state (LUMOPDI) is not coupled to the initial state for 1, 2pp, and 3. Both LUMOs of the SNS and of the bridge, i.e. the fragment orbitals that form the initial state of electron transfer, are antisymmetric around the C2 axis of the DBA systems. Since the LUMO of the PDI is symmetric, it can not couple to the initial state. It is hence necessary to account for higher fragment orbitals to obtain a nonzero effective coupling. As discussed above, the LUMO + 2 of PDI is the lowest antisymmetric unoccupied orbital of the electron acceptor that is coupled to the initial state for all studied DBA molecules. It therefore constitutes the primary acceptor state for electron transfer.
3 Conclusions
We have studied the effect of quantum interference on photoinduced electron and hole transfer in donor-bridge-acceptor systems containing chemically equivalent biphenyl bridges in comparison to conductance properties of the same bridges in metal-bridge-metal junctions. The computed transmission coefficients of the selected molecular bridges show pronounced quantum interference effects when embedded in a MBM junction. In particular, the transmission of the cross-conjugated bridges is significantly lower than of the linearly conjugated bridge. When connecting these bridges to donor and acceptor molecules, the experimentally determined hole transfer rate constants exhibit a reverse trend. We were able to reproduce this trends by calculations of the effective electronic coupling using density functional theory. These calculations reveal the importance of molecular orbital symmetries when studying photoinduced charge transfer in DBA systems. In the case of the linearly conjugated bridge, the majority of the pathways for hole transfer are symmetry-forbidden. Moreover, the contributions to the effective coupling of the remaining active pathways cancel each other. Consequently, the surprisingly low charge transfer rate obtained for the linearly conjugated bridge is due to destructive quantum interference. This destructive quantum interference however differs inherently from the HOMO–LUMO interference that is commonly observed in cross-conjugated MBM junctions. The destructive quantum interference obtained in the linearly conjugated DBA system originates from canceling degenerate pathways. Note that this type of destructive quantum interference is also present in the equivalent linearly conjugated MBM junction. However, other pathways provided by the bridge (e.g. HOMO and LUMO) are not switched off by symmetry and surpass this destructive interference.
The comparison of charge transfer through molecular bridges in DBA systems and charge transport in MBM junctions demonstrates that, although both mechanisms can be accurately described by a coherent tunneling mechanism, the symmetry of the molecular orbitals of donor and acceptor leads to a pathway selection of certain bridge orbitals that mediate charge transfer. In DBA systems, quantum interference is thus not determined directly by the conjugation of the bridge alone, but by specific symmetry relations of the involved donor, bridge, and acceptor states. This result demonstrates that one has to be careful when explaining photoinduced charge transfer phenomena using transmission coefficients of MBM junctions in which the specific donor and acceptor moieties are neglected. Moreover, the results presented in this article point to new design principles for DBA molecules based on the symmetry of their molecular orbitals and not only on the energetic characteristics.
Acknowledgements
This work is supported by the Netherlands Organization for Scientific Research (NWO) through a VIDI grant. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement no. 240299.
References
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- C. Bauer, J. Teuscher, J. C. Brauer, A. Punzi, A. Marchioro, E. Ghadiri, J. D. Jonghe, M. Wielopolski, N. Banerji and J. E. Moser, Chimia, 2011, 65, 704–709 CrossRef CAS.
- J. D. Megiatto Jr, D. D. Méndez-Hernández, M. E. Tejeda-Ferrari, A.-L. Teillout, M. J. Llansola-Portolés, G. Kodis, O. G. Poluektov, T. Rajh, V. Mujica, T. L. Groy, D. Gust, T. A. Moore and A. L. Moore, Nat. Chem., 2014, 6, 423–428 CrossRef PubMed.
- A. Aviram and M. A. Ratner, Chem. Phys. Lett., 1974, 29, 277–283 CrossRef CAS.
- M. Bockrath, D. H. Cobden, P. L. McEuen, N. G. Chopra, A. Zettl, A. Thess and R. E. Smalley, Science, 1997, 275, 1922–1925 CrossRef CAS.
- S. J. Tans, A. R. M. Verschueren and C. Dekker, Nature, 1998, 393, 49–52 CrossRef CAS.
- C. Joachim, J. K. Gimzewski and A. Aviram, Nature, 2000, 408, 541–548 CrossRef CAS PubMed.
- M. Ratner, Nat. Nanotechnol., 2013, 8, 378–381 CrossRef CAS PubMed.
- H. Song, Y. Kim, Y. H. Jang, H. Jeong, M. A. Reed and T. Lee, Nature, 2009, 462, 1039–1043 CrossRef CAS PubMed.
- N. J. Tao, Nat. Nanotechnol., 2006, 1, 173–181 CrossRef CAS PubMed.
- B. Albinsson and J. Martensson, J. Photochem. Photobiol., C, 2008, 9, 138–155 CrossRef CAS PubMed.
- G. L. Closs and J. R. Miller, Science, 1988, 240, 440–447 CAS.
- O. S. Wenger, Acc. Chem. Res., 2011, 44, 25–35 CrossRef CAS PubMed.
- J. Sukegawa, C. Schubert, X. Zhu, H. Tsuji, D. M. Guldi and E. Nakamura, Nat. Chem., 2014, 6, 899–905 CrossRef CAS PubMed.
- S. V. Aradhya and L. Venkataraman, Nat. Nanotechnol., 2013, 8, 399–410 CrossRef CAS PubMed.
- M. N. Paddon-Row, Acc. Chem. Res., 1994, 27, 18–25 CrossRef CAS.
- L. Lafferentz, F. Ample, H. Yu, S. Hecht, C. Joachim and L. Grill, Science, 2009, 323, 1193–1197 CrossRef CAS PubMed.
- W. B. Davis, W. A. Svec, M. A. Ratner and M. R. Wasielewski, Nature, 1998, 396, 60–63 CrossRef CAS.
- R. A. Malak, Z. N. Gao, J. F. Wishart and S. S. Isied, J. Am. Chem. Soc., 2004, 126, 13888–13889 CrossRef CAS PubMed.
- S. H. Choi, B. Kim and C. D. Frisbie, Science, 2008, 320, 1482–1486 CrossRef CAS PubMed.
- T. Hines, I. Diez-Perez, J. Hihath, H. Liu, Z.-S. Wang, J. Zhao, G. Zhou, K. Mllen and N. Tao, J. Am. Chem. Soc., 2010, 132, 11658–11664 CrossRef CAS PubMed.
- X. Zhao, C. Huang, M. Gulcur, A. S. Batsanov, M. Baghernejad, W. Hong, M. R. Bryce and T. Wandlowski, Chem. Mater., 2013, 25, 4340–4347 CrossRef CAS.
- M. Bixon and J. Jortner, J. Am. Chem. Soc., 2001, 123, 12556–12567 CrossRef CAS PubMed.
- Y. A. Berlin, A. L. Burin and M. A. Ratner, Chem. Phys., 2002, 275, 61–74 CrossRef CAS.
- F. D. Lewis, H. Zhu, P. Daublain, T. Fiebig, M. Raytchev, Q. Wang and V. Shafirovich, J. Am. Chem. Soc., 2006, 128, 791–800 CrossRef CAS PubMed.
- F. C. Grozema, Y. A. Berlin, L. D. Siebbeles and M. A. Ratner, J. Phys. Chem. B, 2010, 114, 14564–14571 CrossRef CAS PubMed.
- P. Sautet and C. Joachim, Chem. Phys. Lett., 1988, 153, 511–516 CrossRef CAS.
- G. C. Solomon, D. Q. Andrews, T. Hansen, R. H. Goldsmith, M. R. Wasielewski, R. P. V. Duyne and M. A. Ratner, J. Chem. Phys., 2008, 129, 054701 CrossRef PubMed.
- G. C. Solomon, D. Q. Andrews, R. H. Goldsmith, T. Hansen, M. R. Wasielewski, R. P. V. Duyne and M. A. Ratner, J. Am. Chem. Soc., 2008, 130, 17301–17308 CrossRef CAS PubMed.
- D. Q. Andrews, G. C. Solomon, R. P. V. Duyne and M. A. Ratner, J. Am. Chem. Soc., 2008, 130, 17309–17319 CrossRef CAS PubMed.
- T. Hansen, G. C. Solomon, D. Q. Andrews and M. A. Ratner, J. Chem. Phys., 2009, 131, 194704 CrossRef PubMed.
- S. Chen, Y. Zhang, S. Koo, H. Tian, C. Yam, G. Chen and M. A. Ratner, J. Phys. Chem. Lett., 2014, 5, 2748–2752 CrossRef CAS.
- N. Renaud, M. A. Ratner and C. Joachim, J. Phys. Chem. B, 2011, 115, 5582–5592 CrossRef CAS PubMed.
- T. Markussen, R. Stadler and K. S. Thygesen, Nano Lett., 2010, 10, 4260–4265 CrossRef CAS PubMed.
- A. A. Kocherzhenko, F. C. Grozema and L. D. A. Siebbeles, J. Phys. Chem. C, 2010, 114, 7973–7979 CAS.
- M. Mayor, H. B. Weber, J. Reichert, M. Elbing, C. V. Hanisch, D. Beckmann and M. Fischer, Angew. Chem., Int. Ed. Engl., 2003, 42, 5834–5838 CrossRef CAS PubMed.
- C. R. Arroyo, S. Tarkuc, R. Frisenda, J. S. Seldenthuis, C. H. Woerde, R. Eelkema, F. C. Grozema and H. S. Van der Zant, Angew. Chem., Int. Ed. Engl., 2013, 52, 3152–3155 CrossRef CAS PubMed.
- C. M. Guedon, H. Valkenier, T. Markussen, K. S. Thygesen, J. C. Hummelen and S. J. Van der Molen, Nat. Nanotechnol., 2012, 7, 304–308 CrossRef PubMed.
- S. V. Aradhya, J. S. Meisner, M. Krikorian, S. Ahn, R. Parameswaran, M. L. Steigerwald, C. Nuckolls and L. Venkataraman, Nano Lett., 2012, 12, 1643–1647 CrossRef CAS PubMed.
- R. Härtle, M. Butzin, O. Rubio-Pons and M. Thoss, Phys. Rev. Lett., 2011, 107, 046802 CrossRef.
- S. Ballmann, R. Hartle, P. B. Coto, M. Elbing, M. Mayor, M. R. Bryce, M. Thoss and H. B. Weber, Phys. Rev. Lett., 2012, 109, 056801 CrossRef.
- C. Patoux, C. Coudret, J. P. Launay, C. Joachim and A. Gourdon, Inorg. Chem., 1997, 36, 5037–5049 CrossRef CAS.
- A. B. Ricks, G. C. Solomon, M. T. Colvin, A. M. Scott, K. Chen, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2010, 132, 15427–15434 CrossRef CAS PubMed.
- M. L. Kirk, D. A. Shultz, D. E. Stasiw, D. Habel-Rodriguez, B. Stein and P. D. Boyle, J. Am. Chem. Soc., 2013, 135, 14713–14725 CrossRef CAS PubMed.
- D. M. Adams, L. Brus, C. E. D. Chidsey, S. Creager, C. Creutz, C. R. Kagan, P. V. Kamat, M. Lieberman, S. Lindsay, R. A. Marcus, R. M. Metzger, M. E. Michel-Beyerle, J. R. Miller, M. D. Newton, D. R. Rolison, O. Sankey, K. S. Schanze, J. Yardley and X. Y. Zhu, J. Phys. Chem. B, 2003, 107, 6668–6697 CrossRef CAS.
- D. Segal, A. Nitzan, W. B. Davis, M. R. Wasielewski and M. A. Ratner, J. Phys. Chem. B, 2000, 104, 3817–3829 CrossRef CAS.
- A. Nitzan and M. A. Ratner, Science, 2003, 300, 1384–1389 CrossRef CAS PubMed.
- Y. A. Berlin and M. A. Ratner, Radiat. Phys. Chem., 2005, 74, 124–131 CrossRef CAS PubMed.
- A. Nitzan, J. Phys. Chem. A, 2001, 105, 2677–2679 CrossRef CAS.
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. V. Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. Van Stokkum, J. Stat. Software, 2012, 49, 1–22 Search PubMed.
- C. Hippius, I. H. M. Van Stokkum, E. Zangrando, R. M. Williams and F. Wurthner, J. Phys. Chem. C, 2007, 111, 13988–13996 CAS.
- E. A. Weiss, M. J. Ahrens, L. E. Sinks, A. V. Gusev, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 5577–5584 CrossRef CAS PubMed.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CrossRef CAS.
- J. W. Evenson and M. Karplus, J. Chem. Phys., 1992, 96, 5272–5278 CrossRef CAS PubMed.
- K. Senthilkumar, F. C. Grozema, F. M. Bickelhaupt and L. D. A. Siebbeles, J. Chem. Phys., 2003, 119, 9809–9817 CrossRef CAS PubMed.
- S. Skourtis and A. Nitzan, J. Chem. Phys., 2003, 119, 6271–6276 CrossRef CAS PubMed.
- T. R. Prytkova, D. N. Beratan and S. S. Skourtis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 802–807 CrossRef CAS PubMed.
- N. Gorczak, S. Tarkuc, N. Renaud, A. J. Houtepen, R. Eelkema, L. D. Siebbeles and F. C. Grozema, J. Phys. Chem. A, 2014, 118, 3891–3898 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on the synthesis and characterization of 1, 2mm, 2mp, 2pp, and 3, details on the experimental methodology, xyz-coordinates for all structures used in the calculations, details on the presence of destructive interference in S2mm, details on the orbital symmetry considerations. See DOI: 10.1039/c5sc01104c |
| ‡ Present address: Department of Biomedical Engineering, Near East University, TRNC Mersin 10, Turkey. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence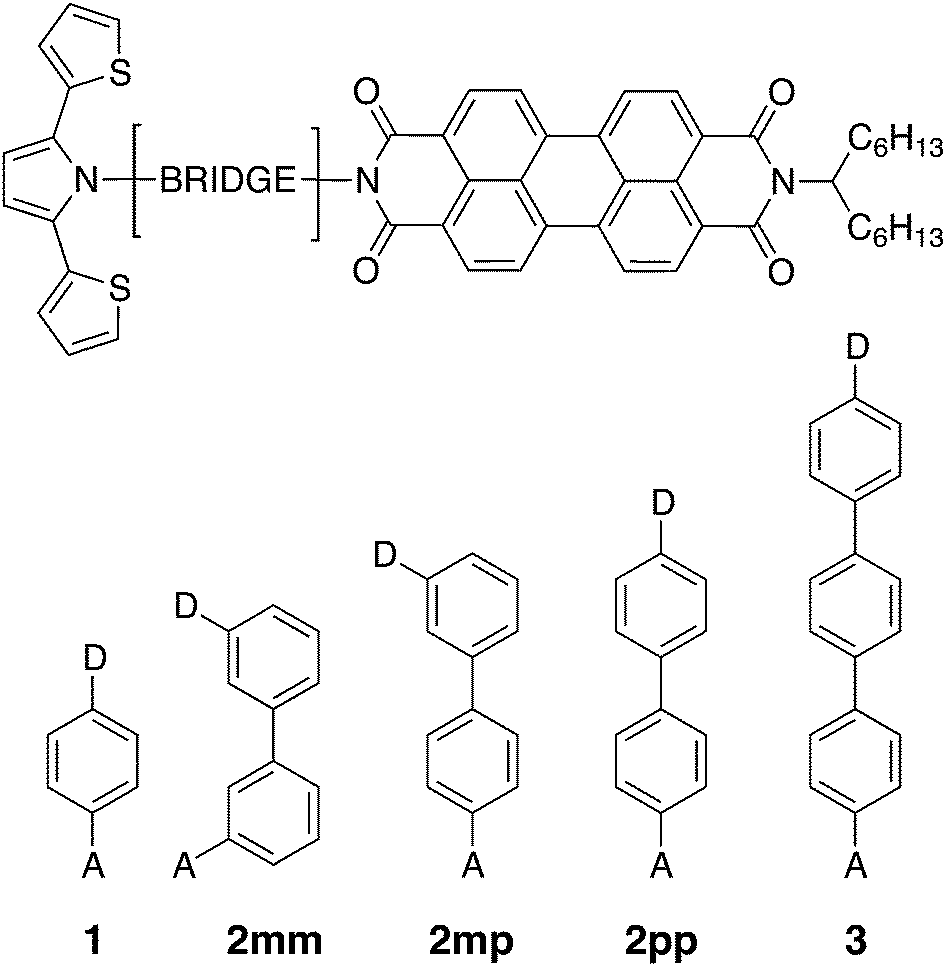
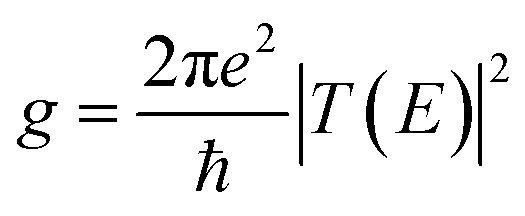 . This transmission coefficient represents the probability that an electron with an energy E will cross the junction. Using the non-equilibrium Green function approach, T(E) can be written as:
. This transmission coefficient represents the probability that an electron with an energy E will cross the junction. Using the non-equilibrium Green function approach, T(E) can be written as: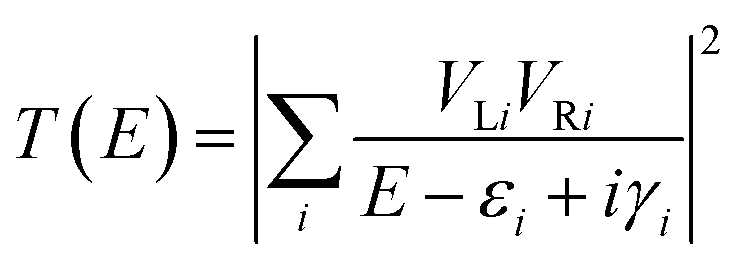
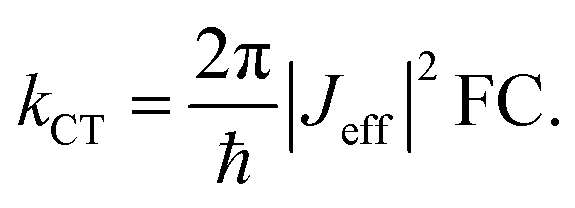
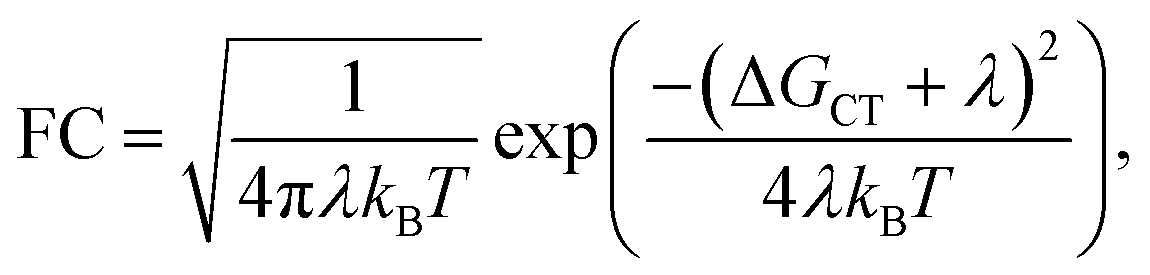
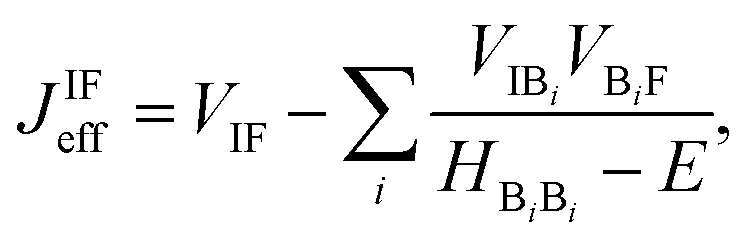
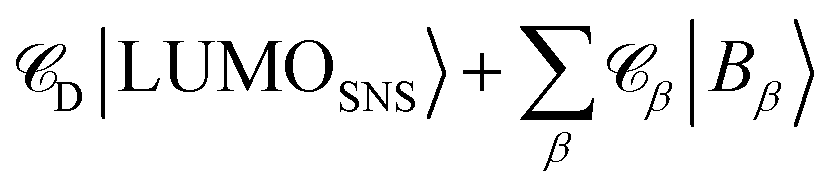 . The weights
. The weights 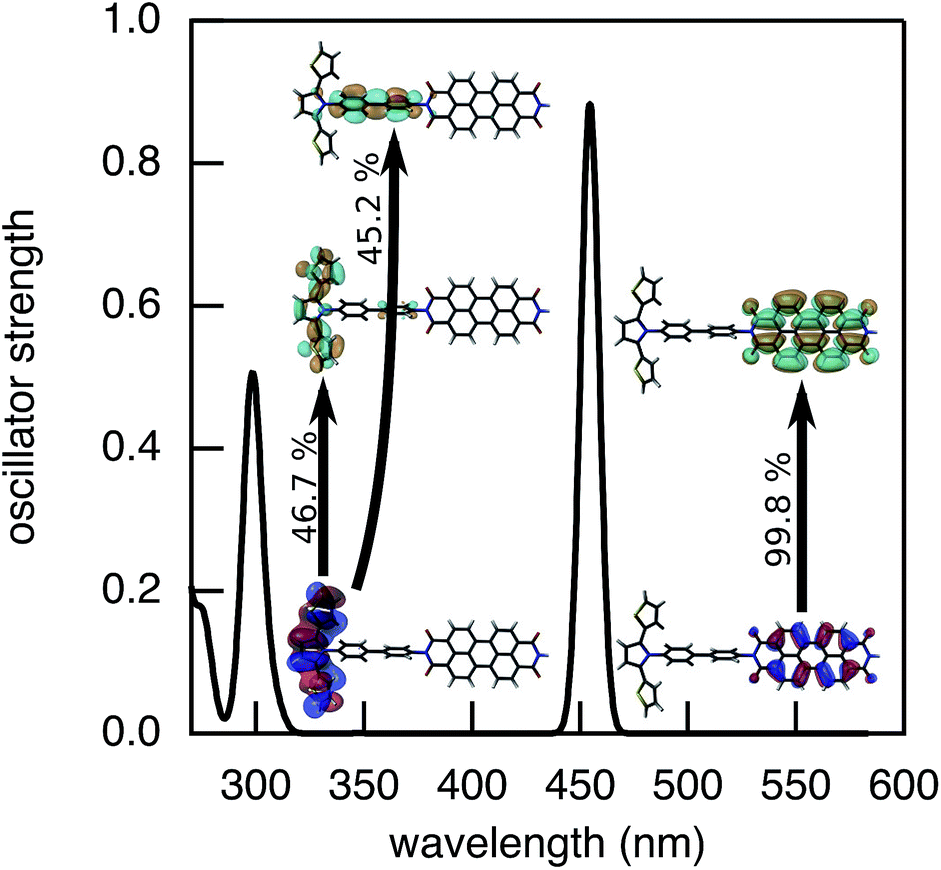
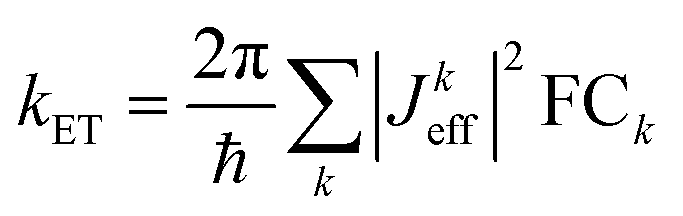 . It depends therefore on the magnitude of the respective Jkeff (and the respective unknown FC factor) which final state will dominate. Since the initial state is a linear combination of SNS and bridge states, its coupling to an acceptor state is composed of two terms: (i) the bridge-mediated coupling
. It depends therefore on the magnitude of the respective Jkeff (and the respective unknown FC factor) which final state will dominate. Since the initial state is a linear combination of SNS and bridge states, its coupling to an acceptor state is composed of two terms: (i) the bridge-mediated coupling 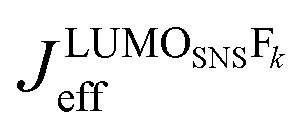 between the donor and acceptor and (ii) the direct coupling VBβFk between the bridge and the acceptor states:
between the donor and acceptor and (ii) the direct coupling VBβFk between the bridge and the acceptor states: