
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePost-synthetic halide conversion and selective halogen capture in hybrid perovskites†
D.
Solis-Ibarra‡
,
I. C.
Smith
and
H. I.
Karunadasa
*
Department of Chemistry, Stanford University, Stanford, CA 94305, USA. E-mail: hemamala@stanford.edu
First published on 5th May 2015
Abstract
Reaction with halogen vapor allows us to post-synthetically exchange halides in both three- (3D) and two-dimensional (2D) organic–inorganic metal-halide perovskites. Films of 3D Pb–I perovskites cleanly convert to films of Pb–Br or Pb–Cl perovskites upon exposure to Br2 or Cl2 gas, respectively. This gas–solid reaction provides a simple method to produce the high-quality Pb–Br or Pb–Cl perovskite films required for optoelectronic applications. Reactivity with halogens can be extended to the organic layers in 2D metal-halide perovskites. Here, terminal alkene groups placed between the inorganic layers can capture Br2 gas through chemisorption to form dibromoalkanes. This reaction's selectivity for Br2 over I2 allows us to scrub Br2 to obtain high-purity I2 gas streams. We also observe unusual halogen transfer between the inorganic and organic layers within a single perovskite structure. Remarkably, the perovskite's crystallinity is retained during these massive structural rearrangements.
Introduction
We have investigated the post-synthetic reactivity of both organic and inorganic components in hybrid metal-halide perovskites. These hybrids combine the properties of molecules and extended solids, allowing for different reactivity to be realized in the same material. We have found that halogen gas can be used to cleanly exchange the halides in the inorganic framework, while preserving the perovskite's structure and crystallinity (Fig. 1). We used this reactivity to post-synthetically exchange the halides of the three-dimensional (3D) perovskites: (MA)[PbX3] (MA = CH3NH3+, X = Br, and I). The Pb–I perovskite has recently emerged as a promising absorber for high-efficiency and low-cost solar cells.1,2 With a bandgap of 1.6 eV,3 these absorbers have so far yielded solar cells with voltages up to 1.15 V.4 Halide substitution in this structure affords bandgaps of 3.11 and 2.35 eV for X = Cl5 and Br,5 respectively, while mixed-halide perovskites show systematic shifts in bandgap with halide ratio.6 This electronic flexibility has enabled further applications in wavelength-tunable light-emitting diodes7,8 and lasers9,10 using Pb–Br and Pb–Cl perovskites. In order to access higher voltages for solar cells, and for the construction of the higher bandgap absorber in a tandem solar cell, there is considerable interest in (MA)[PbBr3] absorbers.11 These optoelectronic devices require uniform films with good substrate coverage. Such high-quality films of (MA)[PbI3] can be formed by many methods, including depositing (MA)I with either PbI2 or PbCl2 precursors or by converting PbI2 films to the perovskite through exposure to (MA)I solution or vapor.2,12 However, similar processing does not yield continuous films of (MA)[PbBr3] and (MA)[PbCl3]. We find that by using Pb–I films as templates, conversion reactions with Br2 and Cl2 gas afford uniform and high-coverage films of Pb–Br and Pb–Cl perovskites, respectively, while generating only volatile by-products. Because different routes to forming high-coverage Pb–I films on various substrates have already been developed, converting these films to (MA)[PbX3] (X = Br and Cl) using halogen vapor provides a very general method for obtaining the high-quality Pb–Br and Pb–Cl perovskite films required for optoelectronic devices.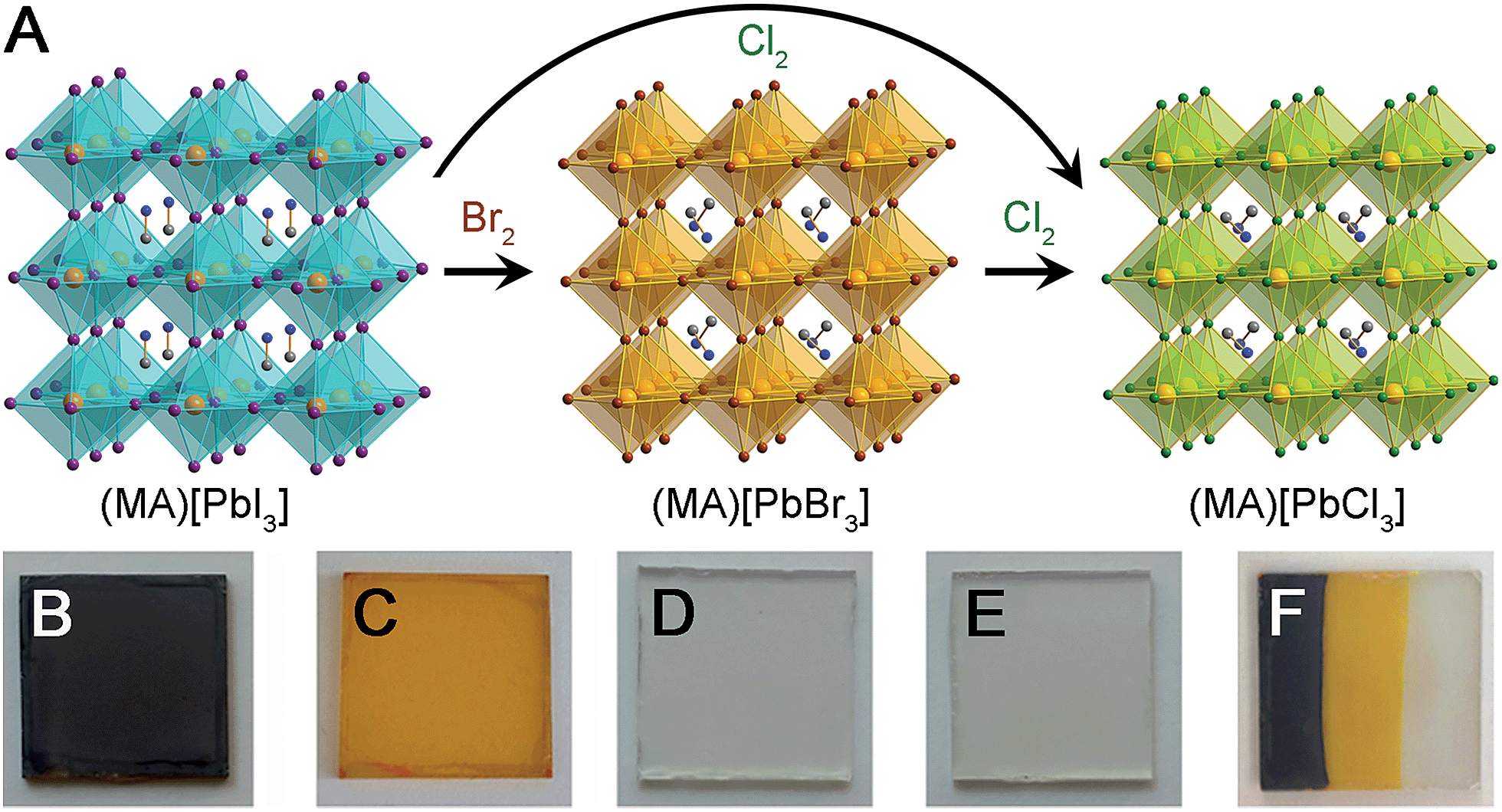 | ||
| Fig. 1 (A) Reaction of 3D perovskites (MA)[PbX3] (MA = CH3NH3+, X = I and Br) with Br2 and Cl2 gas. Pb: orange, I: purple, Br: brown, Cl: green, N: blue, C: gray. Photographs of (B) a (MA)[PbI3] film, (C) a (MA)[PbBr3] film prepared by exposing a (MA)[PbI3] film to Br2 gas, (D) a (MA)[PbCl3] film prepared by sequentially exposing a (MA)[PbI3] film to Br2 and Cl2 gas, and (E) a (MA)[PbCl3] film prepared by exposing a (MA)[PbI3] film to Cl2 gas. (F) A film of all three perovskites formed by exposing parts of a (MA)[PbI3] film to Br2 and then to Cl2 using a mask. | ||
This reactivity with halogens can be extended to the organic groups in 2D hybrid perovskites. We recently described reversible and irreversible I2 capture in the organic layers of 2D perovskites.13 These nonporous materials topotactically expand, by up to 36% in volume, to capture iodine gas by forming covalent C–I bonds in the organic layers. Most porous sorbents capture gases through physisorption, where selectivity is achieved through pore size or by weak electrostatic interactions.14,15 A less explored alternative is chemisorption in nonporous solids, where chemical reactivity dictates substrate selectivity. Here, we report on selective chemisorption of Br2 and IBr over I2 in 2D perovskites. This selectivity can be used to scrub Br2 and IBr to obtain high-purity I2 gas streams—a separation that is difficult to achieve in the gas phase. Furthermore, perovskites that react with halogens in both the inorganic and organic layers show unusual halogen transfer between the inorganic and organic layers of the same 2D perovskite. Notably, the materials remain crystalline despite these very large atomic rearrangements.
Experimental section
Aqueous solutions were prepared using deionized water. Organic solvents were of reagent grade or higher purity and were not dried prior to use except for N,N-dimethylformamide (DMF), which was dried and degassed using a JC Meyer solvent purification system. The 2D perovskites (BEA)2[PbBr4] and (PEA)2[CuCl4] (BEA = but-3-en-1-ammonium and PEA = prop-2-en-1-ammonium) were synthesized according to our previous report.13 All other reagents were purchased from commercial vendors and used as received. Prior to the halogenation reactions, the 2D perovskites were ball-milled for 3 h at 500 rpm in 12 mL zirconia vials equipped with 5 mm agate balls using a Fritsch Pulverisette 7 planetary mill.Preparation of (MA)[PbI3] films
Solid PbI2 (230 mg, 0.500 mmol) was dissolved in 1 mL of dry DMF. After filtering through paper, the solution (60 μL) was deposited on a clean fluorine-doped tin oxide (FTO) coated glass substrate and spun at 2000 rpm for 30 s. Immediately after spinning began, a light flow of nitrogen was directed at the substrate to aid the formation of a flat, uniform film. A dry isopropanol solution of (CH3NH3)I ((MA)I, 63.0 mM) was added dropwise to the PbI2 film spun at 3000 rpm (approx. 200 μL, 8 drops). The sample was allowed to dry between each drop to avoid material loss from the spinning substrate. The films were then annealed at 100 °C for 15 minutes. The phase purity of the (MA)[PbI3] films was confirmed by powder X-ray diffraction (PXRD) (Fig. S1†) and the film coverage was determined through scanning electron microscopy (Fig. 2A). | ||
| Fig. 2 SEM images of 3D perovskite films. Films prepared from PbX2 and (MA)X solutions (X = I, Br, or Cl): (A) (MA)[PbI3], (B) (MA)[PbBr3], and (C) (MA)[PbCl3]. Films formed by reaction of (MA)[PbI3] films with X2 gas (X = Br or Cl): (D) (MA)[PbBr3] and (E) (MA)[PbCl3]. The yellow scale bar shows 1 μm. | ||
Conversion of (MA)[PbI3] to (MA)[PbBr3] using Br2 gas
Liquid bromine (ca. 3 mL) was added to a 50 mL round-bottom flask. A gas-flow meter was used to allow 20 μL min−1 of dry N2 carrier gas to flow over the Br2 vapor. This gas stream was combined with a dry N2 stream (flow rate of 8 mL min−1), creating a dilution of ca. 400 times that of saturated Br2 vapor. This diluted Br2 vapor was then passed over a (MA)[PbI3] film for 8 minutes, converting the film into an orange/yellow (MA)[PbBr3] film. The phase purity of the (MA)[PbBr3] film was confirmed by PXRD (Fig. S2 and S3†) and the film coverage was determined through scanning electron microscopy (Fig. 2D).Conversion of (MA)[PbX3] (X = I or Br) to (MA)[PbCl3] using Cl2 gas
Pure Cl2 gas (flow rate of 20 μL min−1) was combined with dry N2 (flow rate of 10 mL min−1) to dilute the Cl2 by a factor of 500. This diluted gas stream was then passed over a (MA)[PbX3] (X = I or Br) film for 5 minutes, converting the film to a colorless (MA)[PbCl3] film. The phase purity of the (MA)[PbCl3] film was confirmed by PXRD (Fig. S3 and S4†) and the film coverage was determined through scanning electron microscopy (Fig. 2E).Bromination of 2D perovskites
Perovskite powder samples (2–100 mg) were placed in a darkened glass jar containing approximately 0.5 g of liquid Br2, and the jar was closed with a PTFE-lined cap. The samples were placed in glass vials to avoid contact between the solids and liquid Br2. After reaction with Br2 (over 12–48 h), the solids were held at reduced pressure for 0.5 h to remove surface-adsorbed bromine and analyzed by PXRD (Fig. S5 and S6†) and solid-state IR spectroscopy (Fig. S7 and S8†).Chlorination of 2D perovskites
Perovskite powder samples (2–100 mg) were placed in a gas-tight glass chamber and evacuated for approximately 10 minutes. The chamber was then filled with Cl2 gas at 1 atm, sealed, and placed in the dark for 12–48 h. After reaction with Cl2, the solids were held at reduced pressure for 0.5 h and analyzed by PXRD (Fig. S9–S11†) and solid-state IR spectroscopy (Fig. S12†).Results and discussion
Post-synthetic halide conversion in 3D perovskites
Thin-film solar cells require absorbers of uniform thickness and good substrate coverage in order to maximize light absorption and avoid electrical shunts. Owing to its higher bandgap, compared to that of the Pb–I perovskite, the Pb–Br perovskite has been targeted as an absorber for higher voltage solar cells and as the higher-bandgap absorber in a tandem device. However, the typical deposition methods used for Pb–I perovskites2,12 do not afford high-quality Pb–Br perovskite films. For example, codeposition of PbBr2 and (MA)Br from N,N-dimethylformamide leads to discontinuous films of (MA)[PbBr3] (Fig. 2B). Indeed, the low short-circuit current attained from devices containing these absorbers has been attributed to poor perovskite film uniformity.11 The film quality and device performance have been improved by incorporating chloride16 or HBr17 to the precursor solutions and solar cells with (MA)[PbBr3] absorbers have shown high open-circuit voltages (VOC) of up to 1.51 V.17 We investigated if exposing high-coverage Pb–I perovskite films to halogen vapor could provide a general method for easily obtaining the high-quality (MA)[PbX3] (X = Br and Cl) films that are required for photovoltaic and other optoelectronic devices such as light-emitting diodes7,8 and lasers.9,10Dark red/brown films of (MA)[PbI3] (Fig. 1B) turn orange when exposed to Br2 vapor (Fig. 1C) and the powder X-ray diffraction (PXRD) pattern of the product confirms complete conversion to (MA)[PbBr3] (Fig. S2 and S3†). Similarly, (MA)[PbI3] films turn colourless when exposed to Cl2 gas (Fig. 1E) and PXRD data confirm quantitative formation of (MA)[PbCl3] (Fig. S3 and S4†). Notably, films of (MA)[PbX3] (X = Br or Cl) formed through this conversion process (and without subsequent annealing) are consistently more crystalline than the parent films of annealed (MA)[PbI3] (Fig. S1†). Films of (MA)[PbBr3] can also be cleanly converted to (MA)[PbCl3] films through reaction with Cl2 gas (Fig. 1D and S4†). These transformations occur in a few seconds with pure halogen vapor (see Video in the ESI†). Dilute halogen gas streams produce higher-quality films of the product (Fig. 2D and E). Importantly, the films of (MA)[PbCl3] and (MA)[PbBr3] that are formed through reaction with dilute halogen vapor are consistently more uniform and have higher substrate coverage than films produced through sequential deposition or codeposition of PbX2 and (MA)X (X = Cl or Br) (Fig. 2B and C). By using a simple mask, we can also convert parts of a (MA)[PbI3] film to (MA)[PbBr3] and then to (MA)[PbCl3] by sequentially exposing unmasked areas to Br2 and Cl2 vapor (Fig. 1F).
These conversion reactions could be understood in terms of halogen reduction potentials. Standard reduction potentials for I2, Br2, and Cl2 are 0.54, 1.07, and 1.36 V vs. SHE, respectively.18 Therefore, Br2 gas can oxidize the I− ions in the inorganic sheets to I2, while generating Br− ions that coordinate to the Pb2+ centers, thereby transforming (MA)[PbI3] to (MA)[PbBr3]. Similarly, Cl2 gas can oxidize either I− or Br− to form the halogen, and cleanly convert (MA)[PbI3] or (MA)[PbBr3] to (MA)[PbCl3] with only gas-phase by-products.
Bromine capture in 2D perovskites
Similar to the reactivity we observed with I2 gas,13 exposing the 2D alkene perovskite (BEA)2[PbBr4] (BEA = but-3-en-1-ammonium, Fig. 3B and 4) to Br2 gas results in a new crystalline phase with an extended crystallographic c axis. Unlike I2 absorption, however, Br2 absorption is irreversible owing to the greater stability of the dibromo-alkane compared to the diiodo-alkane. We confirmed the formation of BEA–Br2 within the hybrid perovskite first by solid-state vibrational spectroscopy and then by solution-state NMR spectroscopy and mass spectrometry of the digested material (Fig. S7 and S13†). We also obtained single crystals of the product (BEA–Br2)2[PbBr4] (Fig. 3A) by allowing a concentrated solution of (BEA–Br2)2[PbBr4] in aqueous HBr to evaporate over 5 days. The crystal structure19 confirms that the transformation occurs as a topotactic expansion of the inorganic layers as Br2 adds across the terminal alkenes. Here, the c axis expands by 3.2 Å (23.9%) and the unit-cell volume increases by 204 Å3 (22.5%) (Table 1). The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths (1.306(1) and 1.303(1) Å) in (BEA)2[PbBr4] elongate to form the single C–C bonds in (BEA–Br2)2[PbBr4] (1.456(1) and 1.462(1) Å). The organo-bromine atoms in (BEA–Br2)2[PbBr4] form one-dimensional chains that traverse the material with Br⋯Br contacts of 3.645(1) Å. We were previously unable to obtain the crystal structure of (BEA–I2)2[PbBr4] due to its instability to I2 release.13 The close match between its PXRD pattern and the PXRD pattern predicted from the crystal structure of (BEA–Br2)2[PbBr4] confirms that very similar structures are formed in both cases (Fig. S14†). The reactivity of (BEA)2[PbBr4] with Br2 can be extended to hybrid perovskites containing different metals, halides, and alkenes. For example, (PEA)2[CuCl4] (PEA = prop-2-en-1-ammonium) reacts with Br2 gas to form (PEA-Br2)2[CuCl4], as corroborated through single-crystal and PXRD data (Fig. S6 and S15†).
C bond lengths (1.306(1) and 1.303(1) Å) in (BEA)2[PbBr4] elongate to form the single C–C bonds in (BEA–Br2)2[PbBr4] (1.456(1) and 1.462(1) Å). The organo-bromine atoms in (BEA–Br2)2[PbBr4] form one-dimensional chains that traverse the material with Br⋯Br contacts of 3.645(1) Å. We were previously unable to obtain the crystal structure of (BEA–I2)2[PbBr4] due to its instability to I2 release.13 The close match between its PXRD pattern and the PXRD pattern predicted from the crystal structure of (BEA–Br2)2[PbBr4] confirms that very similar structures are formed in both cases (Fig. S14†). The reactivity of (BEA)2[PbBr4] with Br2 can be extended to hybrid perovskites containing different metals, halides, and alkenes. For example, (PEA)2[CuCl4] (PEA = prop-2-en-1-ammonium) reacts with Br2 gas to form (PEA-Br2)2[CuCl4], as corroborated through single-crystal and PXRD data (Fig. S6 and S15†).
 | ||
| Fig. 3 Reaction of (BEA)2[PbBr4] with Br2 and Cl2 gas. Crystal structures of (A) (BEA–Br2)2[PbBr4], (B) (BEA)2[PbBr4], and (C) (BEA–Br2)2[PbCl4]. Pb–Br octahedra: orange, Pb–Cl octahedra: green, Pb: orange, Br: brown, Cl: green, N: blue, C: gray. Hydrogen and disordered atoms are omitted for clarity.19 | ||
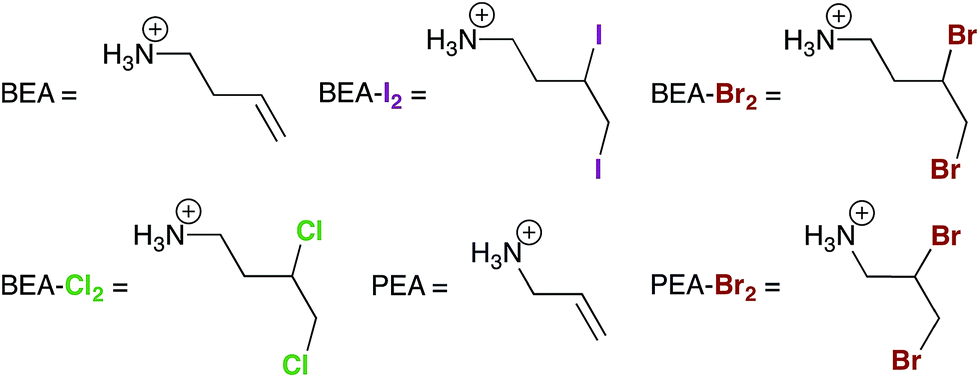 | ||
| Fig. 4 Abbreviations used for the organic cations in the 2D perovskites. | ||
Br2 and IBr scrubbing
Reversible I2 absorption and irreversible Br2 absorption by (BEA)2[PbBr4] prompted us to study the perovskite's utility for scrubbing Br2 from I2 gas streams. This separation is traditionally performed by reducing the halogens and selectively precipitating their halide salts.3 However, this process is labor and energy intensive, especially if the halogen and not the halide is the desired product. This gas-phase separation is further complicated because I2 and Br2 coexist in equilibrium with their reaction product IBr.20 We reasoned that irreversible Br2 addition to (BEA)2[PbBr4] should shift the gas-phase equilibrium towards I2, thereby removing Br2 and IBr from the gas-phase mixture (Fig. 5). | ||
| Fig. 5 Schematic representation for Br2 and IBr scrubbing from I2 gas streams using (BEA)2[PbBr4]. | ||
To test its capacity for I2–Br2 separation, we exposed solid (BEA)2[PbBr4] to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 mole ratios of I2–Br2 gas. Analysis of the resulting products by mass spectrometry and NMR showed the presence of (BEA–Br2)2[PbBr4] and trace amounts of (BEA–IBr)2[PbBr4] (BEA-IBr = 3-bromo-4-iodobutan-1-ammonium and 4-bromo-3-iodobutan-1-ammonium). We obtained average values of 87(2)% and 86(4)% for the total Br2 recovered as (BEA–Br2)2[PbBr4] for the 2:1 and 1:1 mixtures, respectively (Table S1†). This results in a final Br2 concentration of less than 20 ppm in the gas phase. We note that Br2 adsorption to the container walls and small losses during transfer results in an underestimation of the total captured Br2. This separation cannot easily be performed using the alkene molecules alone. The alkyl amines are liquids and the chloride salts of the alkene-ammonium molecules deliquesce under the same conditions used for I2–Br2 separation with the perovskites. These results illustrate that chemisorption in nonporous materials can be used to separate molecules of similar size.
:1 and 2:1 mole ratios of I2–Br2 gas. Analysis of the resulting products by mass spectrometry and NMR showed the presence of (BEA–Br2)2[PbBr4] and trace amounts of (BEA–IBr)2[PbBr4] (BEA-IBr = 3-bromo-4-iodobutan-1-ammonium and 4-bromo-3-iodobutan-1-ammonium). We obtained average values of 87(2)% and 86(4)% for the total Br2 recovered as (BEA–Br2)2[PbBr4] for the 2:1 and 1:1 mixtures, respectively (Table S1†). This results in a final Br2 concentration of less than 20 ppm in the gas phase. We note that Br2 adsorption to the container walls and small losses during transfer results in an underestimation of the total captured Br2. This separation cannot easily be performed using the alkene molecules alone. The alkyl amines are liquids and the chloride salts of the alkene-ammonium molecules deliquesce under the same conditions used for I2–Br2 separation with the perovskites. These results illustrate that chemisorption in nonporous materials can be used to separate molecules of similar size.
Chlorine capture in 2D perovskites
Exposure of (BEA)2[PbBr4] to Cl2 gas also afforded a new crystalline phase. However, analysis of the digested product by mass spectrometry revealed that the reaction product contained BEA–Br2 and not BEA–Cl2. The PXRD pattern of the reaction product with Cl2 matches that of the Pb–Cl perovskite (BEA–Br2)2[PbCl4] (Fig. 3C and S9†), indicating that chloride was incorporated into the inorganic layers. This transformation could occur as a multi-step process. The Br− ions in the inorganic layers could first be oxidized by Cl2 to form Br2 and Cl− ions. The Cl− ions could then be incorporated into the inorganic sheets while Br2 molecules add across the double bonds of the alkenes, thereby facilitating halogen transfer between the inorganic and organic layers. Trapping of the Br2 molecules, generated during halogen conversion in the inorganic sheets, by the terminal alkenes supports our proposed mechanism for halide conversion in 3D perovskites. Interestingly, this transformation also occurs with retention of crystallinity (Fig. S10†). We obtained the crystal structure of (BEA–Cl2)2[PbBr4] (formed through reaction of (BEA–Cl2)Br and PbBr2, Fig. S16†) to confirm that it is a stable, isolable material, though we do not see it in the reaction product of (BEA)2[PbBr4] and Cl2 gas. We also determined that unsaturated organic groups were not required for this halide conversion. For example, exposure of (BEA–Br2)2[PbBr4] to Cl2 gas resulted in its conversion to (BEA–Br2)2[PbCl4] (Fig. 3 and S11†).Terminal alkenes can react with both Cl2 and Br2 to form dihalo-alkanes. The absence of BEA–Cl2 in the alkene perovskite after exposure to Cl2 can be understood in terms of reaction kinetics, and is consistent with experimental rates of alkene chlorination and bromination in solution.21–23 Computational studies on the relative stabilities of the halonium ions24–26 have attributed the faster rates of alkene bromination to the greater stability of the bromonium intermediate compared to the chloronium intermediate. Therefore, although C–Cl bond enthalpies (ca. 85 kcal mol−1) are greater than C–Br bond enthalpies (ca. 72 kcal mol−1),27 the kinetics of alkene halogenation and the redox chemistry that occurs in the perovskite allows us to exclusively isolate BEA–Br2 between the inorganic sheets. Therefore, (BEA)2[PbBr4] can be used to capture both Br2 and Cl2 gas. Here, Cl2 is trapped in the inorganic sheets, while Br2 is trapped in the organic layers.
Conclusions
We show that halogen gas can be used to exchange the halides in the inorganic components of hybrid perovskites while generating only gaseous by-products. For this conversion to occur, the halogen gas must have a more positive reduction potential than the halide in the perovskite. This reaction provides a convenient method of converting 3D Pb–I perovskite films to Pb–Br or Pb–Cl perovskite films of high quality, without further annealing or purification steps. The ability to post-synthetically exchange halides in hybrid perovskites using redox chemistry, while preserving the crystalline structure, is a testament to their remarkable self-annealing properties. Recently, halides in hybrid perovskites have been exchanged by immersing a perovskite film in a solution containing an excess of a halide salt,28 although this method does not appear to preserve film morphology. We further show that the organic functionalities in 2D perovskites can be designed for selective chemisorption to enable separation of gases of similar size. In further studies, we will attempt to tune these sorbents for timed-release of the substrate to inexpensively regenerate the capture material. In future work we will also investigate partially converting the halides in 3D perovskite films as a function of film depth. Because the Pb–Br and Pb–Cl perovskites are more moisture resistant than the Pb–I perovskite, a thin but continuous top layer of the Pb–Br/Cl perovskite may protect the underlying Pb–I absorber from moisture. The long-term stability, under device operating conditions, of perovskite films containing these stratified halide compositions requires further study. We recently described instabilities in the homogenous mixed-halide perovskites (MA)[Pb(BrxI1−x)3] that led to the reversible formation of trap states under continuous white-light illumination.29 The ability to partially convert only the top of a Pb–I perovskite film to a Pb–Br perovskite may also aid carrier extraction in perovskite solar cells. Because the halide composition predominantly affects the top of the valence band, a stable halide gradient at the Pb–I/Pb–Br interface could help shuttle holes to the cathode (Fig. S17†).Acknowledgements
This research was funded by the National Science Foundation CAREER award (DMR-1351538) and the Global Climate and Energy Project (GCEP). Powder and single-crystal XRD studies were performed at the Stanford Nanocharacterization Laboratory (SNL) and at beamlines 11.3.1 and 12.2.2 at the Advanced Light Source (ALS). The ALS is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. We thank Adam Jaffe, Simon Teat, and Kevin Gagnon for assistance with crystallography.Notes and references
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS PubMed.
- P. Gao, M. Gratzel and M. K. Nazeeruddin, Energy Environ. Sci., 2014, 7, 2448–2463 CAS.
- Y. Yasuhiro, N. Toru, E. Masaru, W. Atsushi and K. Yoshihiko, Appl. Phys. Express, 2014, 7, 032302 CrossRef.
- A. Ishii, A. K. Jena and T. Miyasaka, APL Mater., 2014, 2, 091102 CrossRef PubMed.
- N. Kitazawa, Y. Watanabe and Y. Nakamura, J. Mater. Sci., 2002, 37, 3585–3587 CrossRef CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CAS.
- Z.-K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS PubMed.
- Y.-H. Kim, H. Cho, J. H. Heo, T.-S. Kim, N. Myoung, C.-L. Lee, S. H. Im and T.-W. Lee, Adv. Mater., 2015, 27, 1248–1254 CrossRef CAS PubMed.
- F. Deschler, M. Price, S. Pathak, L. E. Klintberg, D.-D. Jarausch, R. Higler, S. Hüttner, T. Leijtens, S. D. Stranks, H. J. Snaith, M. Atatüre, R. T. Phillips and R. H. Friend, J. Phys. Chem. Lett., 2014, 5, 1421–1426 CrossRef CAS.
- G. Xing, N. Mathews, S. S. Lim, N. Yantara, X. Liu, D. Sabba, M. Grätzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480 CrossRef CAS PubMed.
- E. Edri, S. Kirmayer, D. Cahen and G. Hodes, J. Phys. Chem. Lett., 2013, 4, 897–902 CrossRef CAS.
- T. C. Sum and N. Mathews, Energy Environ. Sci., 2014, 7, 2518–2534 CAS.
- D. Solis-Ibarra and H. I. Karunadasa, Angew. Chem., Int. Ed., 2014, 53, 1039–1042 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- E. Edri, S. Kirmayer, M. Kulbak, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2014, 5, 429–433 CrossRef CAS.
- J. H. Heo, D. H. Song and S. H. Im, Adv. Mater., 2014, 26, 8179–8183 CrossRef CAS PubMed.
- P. Vanýsek, CRC Handbook of Chemistry and Physics, Taylor and Francis, LLC, 95th edn, 2014 Search PubMed.
- ESI†.
- M. Schmeisser, in Handbook of Preparative Inorganic Chemistry, Elsevier, 1963, pp. 272–333, DOI:10.1016/B978-0-12-395590-6.50013-2.
- M. L. Poutsma, J. Am. Chem. Soc., 1965, 87, 4285–4293 CrossRef CAS.
- M. F. Ruasse and E. Lefebvre, J. Org. Chem., 1984, 49, 3210–3212 CrossRef CAS.
- M. F. Ruasse and B. L. Zhang, J. Org. Chem., 1984, 49, 3207–3210 CrossRef CAS.
- V. I. Teberekidis and M. P. Sigalas, Tetrahedron, 2002, 58, 6171–6178 CrossRef CAS.
- V. I. Teberekidis and M. P. Sigalas, Tetrahedron, 2003, 59, 4749–4756 CrossRef CAS.
- V. I. Teberekidis and M. P. Sigalas, Tetrahedron, 2005, 61, 3967–3976 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- N. Pellet, J. Teuscher, J. Maier and M. Grätzel, Chem. Mater., 2015, 27, 2181–2188 CrossRef CAS.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1048945–1048947. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01135c |
| ‡ Current address: Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México. |
| This journal is © The Royal Society of Chemistry 2015 |