DOI:
10.1039/C5SC01225B
(Edge Article)
Chem. Sci., 2015,
6, 4698-4703
Cytocompatible in situ cross-linking of degradable LbL films based on thiol–exchange reaction†
Received
6th April 2015
, Accepted 18th May 2015
First published on 18th May 2015
Abstract
Formation of both mechanically durable and programmably degradable layer-by-layer (LbL) films in a biocompatible fashion has potential applications in cell therapy, tissue engineering, and drug-delivery systems, where the films are interfaced with living cells. In this work, we developed a simple but versatile method for generating in situ cross-linked and responsively degradable LbL films, based on the thiol–exchange reaction, under highly cytocompatible conditions (aqueous solution at pH 7.4 and room temperature). The cytocompatibility of the processes was confirmed by coating individual yeast cells with the cross-linked LbL films and breaking the films on demand, while maintaining the cell viability. In addition, the processes were applied to the controlled release of an anticancer drug in the HeLa cells.
Introduction
The recent advancements in the layer-by-layer (LbL) technique have paved the way for applications in the biochemical and biomedical areas particularly dealing with living cells, such as tissue engineering, cell therapy, and drug-delivery systems.1 Natural or synthetic polymers with biocompatibility have been used to incorporate therapeutic proteins in LbL films under physiologically relevant conditions to maintain the structural and functional integrity of the proteins.2 Individual mammalian cells were also coated with an LbL pair of natural fibronectin and gelatin, and three-dimensional tissue models were constructed as a drug-screening platform.3 In cell therapy, the LbL coating of microbial and mammalian cells has been attempted to protect the encased cells from external harmful stressors including enzymatic attack and heat.4 However, most LbL films have been generated primarily based on the electrostatic interactions between oppositely charged polyelectrolytes; hence, their mechanical durability is low, and the structural integrity is perturbed with ease by changes in ionic strength and/or acidity of the aqueous media, limiting the long-term applications of the LbL films in the bio-related areas. The mechanical durability of films was increased by depositing inorganic materials, such as silica and titania, onto the LbL films,5 and very recently copper-free triazole formation has been applied to in situ cross-linking of LbL films to harden the films and achieve pulsatile release of multiple protein drugs.6 However, it is still desirable but challenging to generate durable LbL films, which degrade on demand, in a cytocompatible fashion. For example, the reversible formation of mechanically durable coats on single cells is a prerequisite for the realization of artificial spores7 as well as controlled drug delivery systems. In this paper, we demonstrate a highly cytocompatible method for assembling and disassembling cross-linked LbL films in a programmed manner, based on the in situ thiol–exchange reaction. The disulfide-linked films were formed spontaneously under physiologically mild conditions (in an aqueous solution of pH 7.4 and at room temperature), and they degraded in response to external stimuli, such as glutathione (GSH). The cytocompatible processes enabled reversible surface engineering of individual living cells, in addition to controlled drug release.
Results and discussion
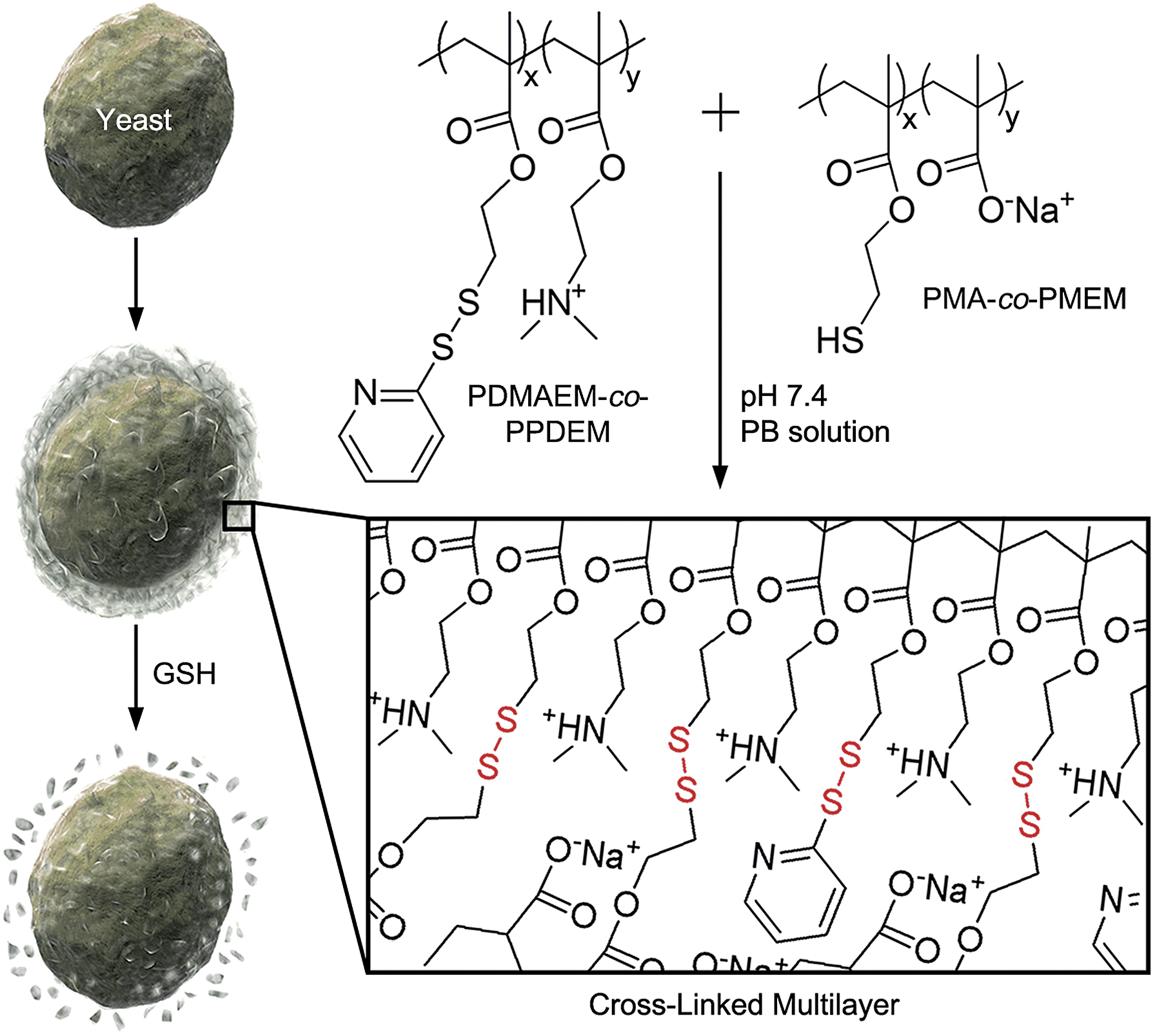
It is reported by one of us that the interconversion reaction between thiol and pyridyl disulfide occurs under mild conditions, and this thiol–exchange reaction has been utilized for the formation of degradable nanogels.8 For the formation of LbL films in this work, we designed two oppositely charged copolymers, positively charged poly(2-(dimethylamino)ethyl methacrylate-co-2-(pyridyl disulfide)ethyl methacrylate) (PDMAEM-co-PPDEM) and negatively charged poly(methacrylate-co-2-mercaptoethyl methacrylate) (PMA-co-PMEM) (Fig. 1). It was envisioned that the cross-linking in the polyelectrolyte films would occur in situ via the thiol–exchange reaction between the pyridyl disulfide in PDMAEM-co-PPDEM and the thiol in PMA-co-PMEM. As a negative control, we also synthesized negatively charged PMA-co-PPDEM that contained no free thiols.
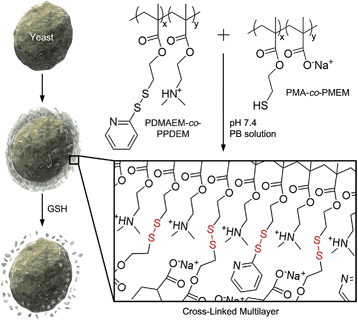 |
| | Fig. 1 Schematic procedure for cell surface engineering with cross-linked LbL multilayers of PDMAEM-co-PPDEM and PMA-co-PMEM (x: 0.33; y: 0.67). | |
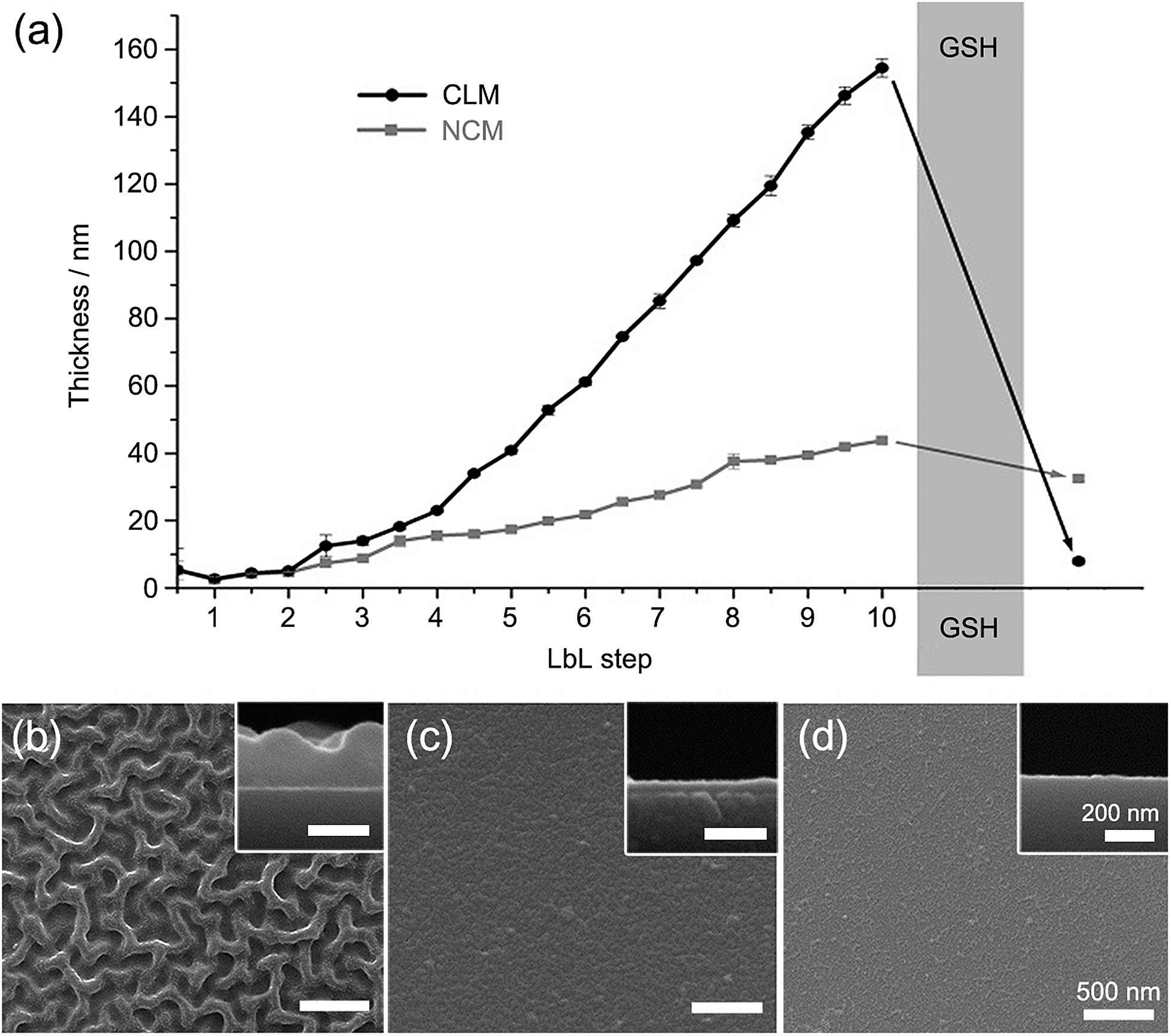
We alternately deposited PDMAEM-co-PPDEM and PMA-co-PMEM onto a silicon wafer under physiologically mild conditions (phosphate-buffered solution, pH 7.4), resulting in the formation of cross-linked multilayers (CLMs). As a comparison, non-cross-linked multilayers (NCMs) were formed with PDMAEM-co-PPDEM and PMA-co-PPDEM under the same conditions. The ellipsometric measurements showed that the film thickness of CLMs increased linearly with the number of depositions from CLM4/4 (Fig. 2a), and the CLM films were much thicker than the NCM films (a multilayer with n layers of positively charged polyelectrolytes and m layers of negatively charged polyelectrolytes is denoted by Mn/m). For example, the thickness of CLM10/10 was ∼160 nm, and that of NCM10/10 was only ∼40 nm. It is noticeable that the efficiency of LbL assembly, in terms of thickness, was enhanced by the cross-linked disulfide bond. The surface of CLM10/10 exhibited woven-fabric structures on the nanometer scale compared with that of NCM10/10 (Fig. 2b and c). The degradability of the CLMs was examined with naturally occurring GSH, which is capable of dissociating disulfide bonds into two thiol groups.8,9 The ellipsometric thickness of CLM10/10 dropped dramatically from ∼160 nm to less than 10 nm after 3 h of exposure to GSH (100 mM), whereas the NCM thickness was not strongly affected (Fig. 2a). The scanning electron microscopy (SEM) analyses also revealed that the CLM thickness decreased, and its surface became smooth because of the degradation (Fig. 2d). The results clearly showed that the multilayered films were formed through in situ cross-linking and could be induced to degrade on demand under biocompatible conditions.
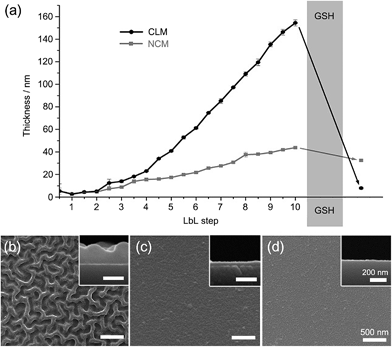 |
| | Fig. 2 (a) Ellipsometric thicknesses of LbL films before and after GSH treatment. GSH was treated to CLM10/10. SEM micrographs of (b) CLM10/10, (c) NCM10/10, and (d) GSH-treated CLM10/10. | |
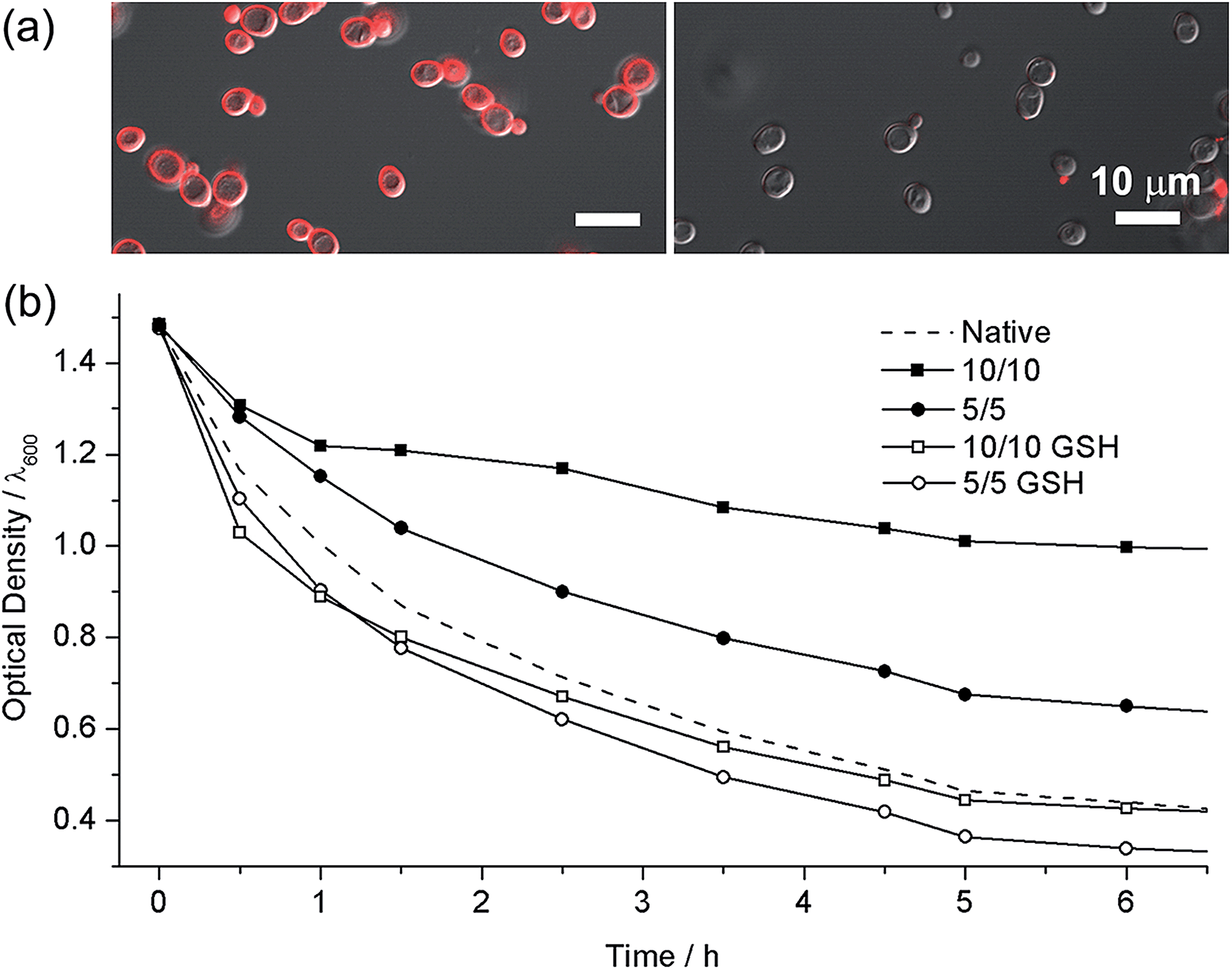
The high biocompatibility of the film-forming and -breaking processes was confirmed with living cells as a model substrate. Individual cells of Saccharomyces cerevisiae (baker's yeast) were coated with CLMs (yeast@CLM). The CLM coating was visualized by functionalizing the LbL multilayer with rhodamine-linked maleimide that reacted with the remaining thiol groups in the CLMs (Fig. 3a). The observation of ring-shaped red fluorescence signals clearly indicated that the cells were uniformly coated with CLMs. The cell-viability results of an assay based on fluorescein diacetate (FDA), which assesses the activity of intracellular esterases and the membrane integrity, were strikingly high compared with the previous reports of single-cell coating:5,10 94% viability for the native yeast, 93% viability for yeast@CLM5/5, and 88% viability for yeast@CLM10/10. Although disulfide linkages have been used for cross-linking the LbL microcapsules, the cross-linking reactions were not cytocompatible because they occurred at low pH (∼pH 4) or in the presence of toxic oxidants for the oxidation of thiol to disulfide.9,11 The high viability of yeast@CLM in our system was caused by the cytocompatible LbL processes including in situ cross-linking at pH 7.4. In addition, the cell viability did not significantly decrease even after shell degradation was induced by 3 h of incubation with GSH, as confirmed by the disappearance of the ring-shaped red fluorescence signals (Fig. 3a): 92% viability for yeast@CLM5/5 and 85% viability for yeast@CLM10/10 after film degradation. The film stability and degradability were also investigated by the thickness-dependent protection of the cells against lyticase, a cell-wall-lysing enzyme complex. Yeast@CLM displayed significant resistance to lysis compared with the native yeast (Fig. 3b). The resistance to lyticase was also controlled by adjusting the CLM thickness; the optical density of yeast@CLM10/10 remained higher than that of yeast@CLM5/5 for 6 h. This result implied that the CLM coating was mechanically tough and capable of protecting living cells by suppressing the penetration of lyticase.10b After the degradation of the protective CLM coating, both yeast@CLM5/5 and yeast@CLM10/10 exhibited decreased resistances, which were similar to that of the native yeast.
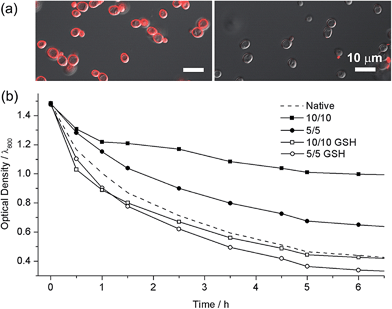 |
| | Fig. 3 (a) Confocal fluorescence micrographs of (left) yeast@CLM5/5 and (right) GSH-treated yeast@CLM5/5. The CLMs were visualized with rhodamine-maleimide. (b) Survival of native yeast, yeast@CLM5/5, GSH-treated yeast@CLM5/5, yeast@CLM10/10, and GSH-treated yeast@CLM10/10, when exposed to lyticase. The optical density was measured at 600 nm. | |
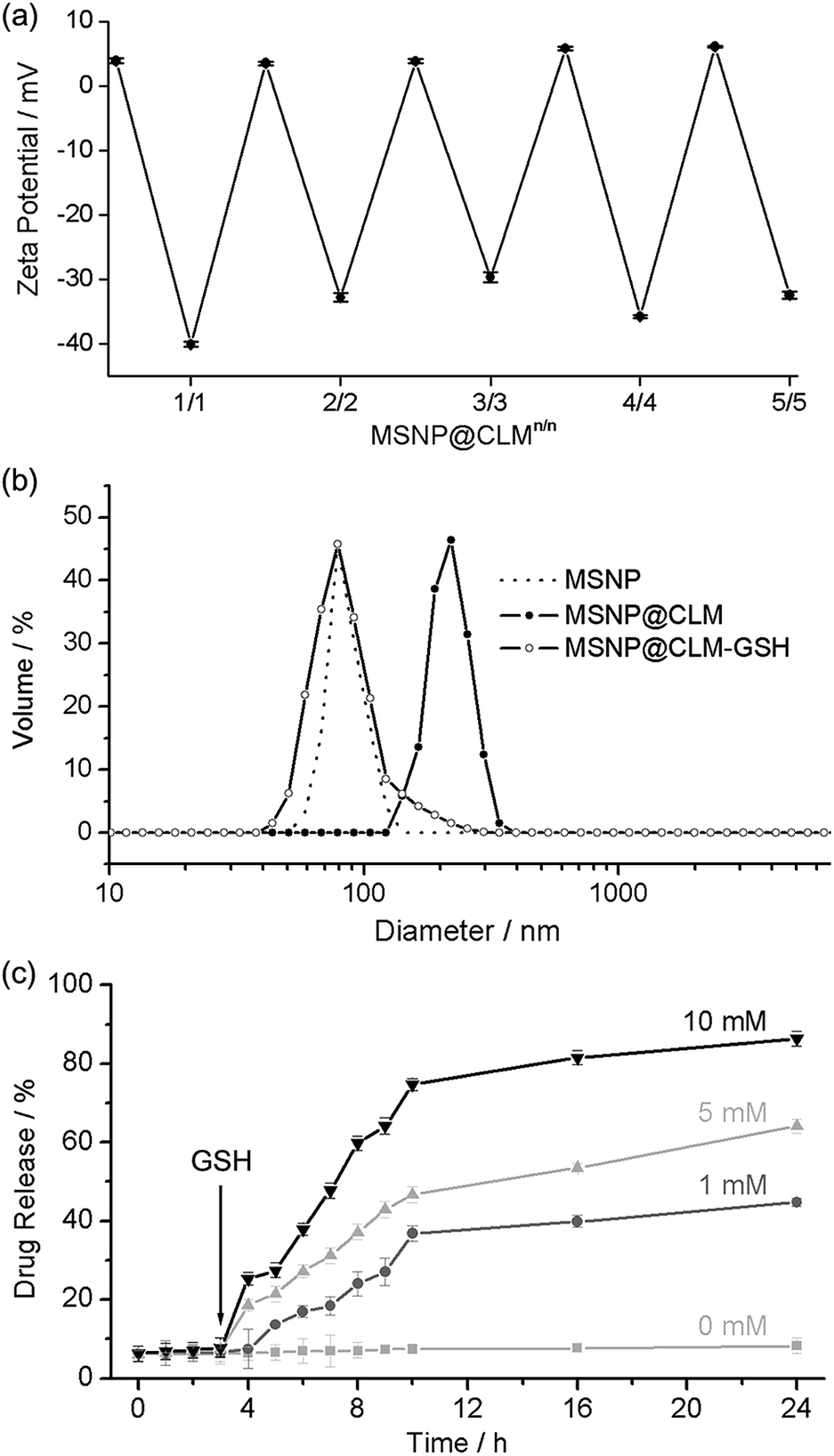
After confirming the high cytocompatibility, the in situ cross-linking and degradation were characterized further by using spherical nanoparticles. Mesoporous silica nanoparticles (MSNPs) of 30 nm in diameter were coated with CLMs (MSNP@CLM). The zeta-potential values of MSNP@CLM (from 1/0 to 5/5) periodically oscillated between positive and negative values, indicating the successful formation of multilayers (Fig. 4a and Table S1†). The in situ cross-linking was confirmed by analyzing the byproduct of the thiol–exchange reaction, pyridinethione, with a UV-vis spectrophotometer (Fig. S1 and Table S2†). Dynamic light scattering (DLS) analysis indicated that the hydrodynamic diameter increased from 90 nm for the original MSNPs to 185 nm for MSNP@CLM5/5 and that GSH-induced degradation restored the original particle size, again confirming the degradation of the CLMs (Fig. 4b and Table S3†). In addition to the characterizations, the degradability of the CLMs was applied to a stimulus-responsive drug-delivery platform. Doxorubicin hydrochloride (Dox), an anticancer drug, was loaded into the MSNPs, and the CLMs were then formed. The loading of Dox to the MSNPs was measured to be up to 6.7% (entrapment efficiency: 7.2%; extinction coefficient: 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 M−1 cm−1), based on UV-vis spectroscopy. The in vitro release profile showed that no release of Dox occurred without the addition of GSH and that the rate of release increased as the concentration of GSH was increased (Fig. 4c). A cell culture assay with HeLa cells also confirmed the GSH-induced degradation of the CLMs and the release of Dox into the cells. Although MSNP@CLM5/5 without Dox loading showed no death of the cancerous cells up to a high concentration of 2 mg mL−1 of MSNP@CLM (Fig. S2†), Dox-containing MSNP@CLM5/5 significantly caused cell death (Fig. S3†). These observations indicated that the CLMs degraded inside the cells in response to a high intracellular GSH concentration in HeLa cells, in addition to the cytocompatibility of CLM films.
500 M−1 cm−1), based on UV-vis spectroscopy. The in vitro release profile showed that no release of Dox occurred without the addition of GSH and that the rate of release increased as the concentration of GSH was increased (Fig. 4c). A cell culture assay with HeLa cells also confirmed the GSH-induced degradation of the CLMs and the release of Dox into the cells. Although MSNP@CLM5/5 without Dox loading showed no death of the cancerous cells up to a high concentration of 2 mg mL−1 of MSNP@CLM (Fig. S2†), Dox-containing MSNP@CLM5/5 significantly caused cell death (Fig. S3†). These observations indicated that the CLMs degraded inside the cells in response to a high intracellular GSH concentration in HeLa cells, in addition to the cytocompatibility of CLM films.
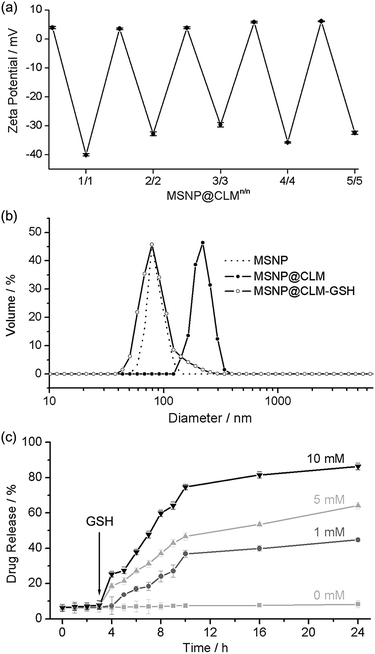 |
| | Fig. 4 (a) Zeta-potential values measured during multilayer preparation. (b) Hydrodynamic diameters of MSNPs, MSNP@CLM5/5, and GSH-treated MSNPt@CLM5/5. (c) Stimulus-triggered drug-release profiles with different GSH concentrations. GSH was added at 3 h. | |
Conclusions
In summary, we developed a highly cytocompatible LbL process for generating the multilayers that were cross-linked in situ and degraded on demand. It has been challenging to achieve both mechanical stability and responsive degradability of LbL films in a cytocompatible fashion, although these features are strongly desired for recent research activities involving the interfacing of living cells with functional materials, in addition to the controlled release of biogenic drugs.1,3,4,7,12 We believe that the cytocompatible formation and degradation of LbL films demonstrated herein will provide a versatile platform for surface engineering of living cells and the control of cellular metabolisms.
Experimental procedures
Materials
2-(Dimethylamino)ethyl methacrylate (DMAEM, Sigma-Aldrich), methacrylic acid (MA, Sigma-Aldrich), sodium chloride (NaCl, ≥99.5%, Jin Chemical Pharmaceutical), sodium phosphate dibasic (≥99%, Sigma-Aldrich), sodium phosphate monobasic (≥99%, Sigma-Aldrich), 2-cyano-2-propyl benzodithioate (Aldrich), azobisisobutyronitrile (AIBN, Aldrich), diethyl ether (Sigma-Aldrich), N,N-dimethylformamide (DMF, Aldrich), dithiothreitol (DTT, Sigma-Aldrich), fluorescein diacetate (FDA, Sigma), acetone (≥99.8%, Merck), L-glutathione reduced (GSH, Sigma-Aldrich), rhodamine red® C2 maleimide (rhodamine-maleimide, Life Technology), doxorubicin hydrochloride (Dox, Ontario Chemical Inc.), cetyltrimethylammonium bromide (CTAB, Aldrich), triethanolamine (Sigma), tetraethyl orthosilicate (TEOS, Aldrich), glycerol (Sigma), Dulbecco's modified Eagle's medium (DMEM, Welgene), penicillin-streptomycin (Welgene), 0.25% trypsin-EDTA (Welgene), fetal bovine serum (FBS, Welgene), yeast extract-peptone-dextrose (YPD, Duchefa-Biochemie), lyticase from Arthrobacter luteus (lyticase, ≥ 2000 units per mg protein, Sigma), and Tris–EDTA (TE) buffer solution (pH 7.5, Intron Biotechnology) were used as received. 2-(Pyridyl disulfide)ethyl methacrylate (PDEM) was synthesized by following a previously reported procedure.13 Deionized water (DI water, 18.3 MΩ cm) from the Human Ultrapure System (Human Corp.) was used.
Polymer synthesis
(a) Poly(2-(dimethylamino)ethyl methacrylate-co-2-(pyridyl disulfide)ethyl methacrylate) (PDMAEM-co-PPDEM).
DMAEM (1.44 g, 9.15 mmol), PDEM (1 g, 3.92 mmol), 2-cyano-2-propyl benzodithioate (15 mg, 0.068 mmol), and AIBN (3.22 mg, 0.020 mmol) were dissolved in DMF (10 mL), and the resulting mixture solution was degassed by performing three freeze–pump–thaw cycles. The reaction vessel was sealed and then placed in a pre-heated oil bath at 95 °C. After 20 h, the resulting mixture was precipitated by adding cold diethyl ether (200 mL). To remove unreacted monomers, the precipitate was further dissolved in DMF (5 mL) and re-precipitated in cold diethyl ether (100 mL) to yield purified PDMAEM-co-PPDEM as a waxy liquid. GPC (PS standard): Mn = 26.8 kDa, PDI = 1.08, 1H NMR (600 MHz, DMSO) δ = 8.46, 7.84–7.75, 7.27, 4.34, 3.01–2.71, 2.04–1.65, 1.24–0.87. The molar ratio between DMAEM and PDEM was determined by integrating the methyl proton in DMAEM and the aromatic proton in the pyridine moiety and was found to be 0.67:0.33 (DMAEM:PDEM).
(b) Poly(methacrylate-co-mercaptoethyl methacrylate) (PMA-co-PMEM).
MA (1.575 g, 18.30 mmol), PDEM (2 g, 7.84 mmol), 2-cyano-2-propyl benzodithioate (29 mg, 0.13 mmol), and AIBN (6.45 mg, 0.040 mmol) were dissolved in DMF (10 mL), and the resulting mixture solution was degassed by performing three freeze–pump–thaw cycles. The reaction vessel was sealed and then placed in a pre-heated oil bath at 95 °C for 24 h. The mixture was precipitated in cold diethyl ether (200 mL). To remove unreacted monomers, the precipitate was further dissolved in DMF (5 mL) and re-precipitated in cold diethyl ether (100 mL) to yield purified PMA-co-PPDEM as a waxy liquid. GPC (PS standard): Mn = 229 kDa, PDI = 3.64, 1H NMR (600 MHz, DMSO) δ = 8.47, 7.82–7.77, 7.25, 4.15, 3.08, 2.04–1.65, 1.24–0.87. The molar ratio between MA and PDEM was determined by integrating the methyl proton in MA and the aromatic proton in the pyridine moiety and was found to be 0.67:0.33 (MA:PDEM). To prepare PMA-co-PMEM, PMA-co-PPDEM was added to a DMF solution of excess DTT (4 equiv. to the pyridyl disulfide group), and after 12 h diethyl ether was added to precipitate PMA-co-PMEM. To prevent the oxidation of thiol to disulfide, freshly prepared polymer solutions were used for LbL.
Preparation of CLM and NCM films on silicon wafers
After cleaning a silicon wafer (1 cm × 0.8 cm) with O2 plasma for 10 min, the wafer was immersed in the phosphate-buffered (PB) solution (pH 7.4) of PDMAEM-co-PPDEM (1 mg mL−1) for 5 min. After rinsing with 0.15 M NaCl aqueous solution for 1 min, the rinsed wafer was immersed in the PB solution (50 mM, pH 7.4) of PMA-co-PMEM (1 mg mL−1) for 5 min, resulting in the formation of CLM1/1. The substrate was rinsed with the PB solution after each LbL step. The LbL processes were repeated to generate CLMn/m (a multilayer with n layers of positively charged PDMAEM-co-PPDEM and m layers of negatively charged PMA-co-PMEM is denoted by Mn/m). On the other hand, the NCMs were formed by alternately immersing a silicon wafer in the solution of PDMAEM-co-PPDEM and in that of PMA-co-PPDEM. The formed CLM and NCM films were characterized with a spectroscopic ellipsometer (Elli-SE, Ellipso Technology) and Sirion FEI XL FEG/SFEG microscope (FEI).
Encapsulation of yeast cells and characterizations
(a) Encapsulation.
A single colony of yeast cells was picked from a YPD broth agar plate, and suspended in the YPD broth and cultured in a shaking incubator at 30 °C for 30 h. After washing with 0.15 M aqueous NaCl solution, the cells were alternately immersed in the PB solution (pH 7.4) of PDMAEM-co-PPDEM (1 mg mL−1) and that of PMA-co-PMEM (1 mg mL−1) for 5 min for each step. The cells were washed with the 0.15 M NaCl solution after each LbL step.
(b) Functionalization.
The CLM layer was functionalized with a rhodamine group by placing yeast@CLM in the rhodamine-maleimide solution, which had been prepared by filtering an aqueous solution of rhodamine-maleimide (1 mg mL−1) and then adding the resulting solution to the PB solution (100 mM, pH 7.4) in a 1:1 (v/v) ratio. Rhodamine-functionalized yeast@CLM was characterized with LSM 700 META microscope (Carl Zeiss).
(c) Viability test.
The viability of yeast cells was measured by examining the activity of intracellular esterase and the membrane integrity with FDA. The FDA stock solution (10 mg mL−1) was first prepared by dissolving FDA in acetone, because FDA was poorly soluble in water. 2 μL of the stock solution was mixed with 1 mL of yeast cell suspension (10 mM PB solution, pH 6.5). The suspension was incubated for 30 min at room temperature while shaking, and then the cells were collected by centrifugation, washed with 0.15 M NaCl aqueous solution, and characterized with a confocal laser-scanning microscope (LSM 700 META, Carl Zeiss).
(d) Lysis test.
Before the lysis test, the optical density of native yeasts, yeast@CLM5/5, or yeast@CLM10/10, was adjusted to ∼1.0 at 600 nm by dilution with 0.85% aqueous NaCl solution. The lyticase stock solution was first prepared by dissolving lyticase (∼3.8 mg) in a mixture of glycerol (500 μL) and TE buffer solution (500 μL). 10 μL of the stock solution was added to the yeast suspension (TE buffer solution), and the suspension was incubated while shaking at 37 °C. A small amount of the mixture was picked at the predetermined time, and the optical density was measured at 600 nm with a UV-visible spectroscope (UV-2550, Shimadzu).
Synthesis and functionalizations of MSNPs
(a) Synthesis.
MSNPs were synthesized using the soft-templating method.14 A mixture of 1.54 g of CTAB, 0.347 g of triethanolamine and 100 mL of DI water was stirred at 80 °C for 1 h, and then 14.58 g of TEOS was quickly added into the surfactant solution. The mixture was stirred at 80 °C with a stirring speed of 1200 rpm for another 2 h. The resulting precipitates were filtered and washed twice with DI water, and then dried at 100 °C overnight. Finally, the dried sample was calcinated at 550 °C for 5 h in air.
(b) Dox loading.
About 5 mg of MSNPs were dispersed in 1 mL of aqueous solution containing 5 mg of Dox, and the mixture was gently stirred for 3 h at room temperature. The samples were centrifuged, and the precipitate was washed by a re-dispersion cycle to remove unloaded Dox and collected for further use. The UV-vis spectrum was used to determine the doxorubicin loading using the following equations.15
| Entrapment efficiency (%) = (mass of drug in MSNPs)/(initial mass of drug) |
| Drug loading (%) = (mass of drug in MSNPs)/(mass of drug loaded MSNPs) |
(c) LbL.
A suspension of the Dox-loaded MSNPs (10 wt%) in the PB solution (50 mM, pH 7.4) was washed with the PB solution via several centrifugation/re-dispersion cycles. The resulting suspension was re-dispersed in 1 mL of the PB solution and combined with 1 mL of PDMAEM-co-PPDEM (2 mg mL−1) in the PB solution (50 mM, pH 7.4), and the adsorption of PDMAEM-co-PPDEM was allowed to proceed for 15 min with constant stirring in a magnetic stirrer. After centrifugation, the particles were washed with the PB solution (two times) and with distilled water, and re-dispersed in 1 mL of the PB solution, to which was added 1 mL of PMA-co-PMEM (2 mg mL−1) in the PB solution. The mixture was allowed to form the cross-linked disulfide bonds between PDMAEM-co-PPDEM and PMA-co-PMEM for 15 min with constant stirring. The same processes were repeated to generate CLMn/m with PDMAEM-co-PPDEM and PMA-co-PMEM. The exchange reaction between thiol and pyridyl disulfide was analyzed with the left-out polymer solution from the supernatant by using UV-vis absorbance.16
(d) Dox release profile.
The in vitro release of Dox from MSNP@CLM5/5 was investigated with different GSH concentrations: 0, 1, 5, and 10 mM. The release of Dox from MSNP@CLM5/5 was measured using fluorescence with excitation at 480 nm and emission at 580 nm.
(e) Cell viability.
The cell viability was tested with MSNP, MSNP@CLM5/5 and Dox-loaded MSNP-@CLM5/5 using the Alamar blue assay. The HeLa cell lines were grown in the DMEM medium containing 10% FBS and 1% penicillin-streptomycin at 37 °C in a 5% CO2 humidified atmosphere. The cells were seeded onto a 96-well plate at a density of 5 × 103 cells per well in 100 μL of DMEM containing 10% FBS, incubated for 24 h (37 °C, 5% CO2), and analyzed after 24 h.
Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (MSIP) (2013R1A1A1008102 and 2012R1A3A2026403) and the Ministry of Education (201135BC00024). We thank S. Shin for assistance with the polymer synthesis and S. H. Joo for kindly providing mesoporous silica nanoparticles.
Notes and references
-
(a)
M. Matsusaki and M. Akashi, in Cell Surface Engineering, ed. R. Fakhrullin, I. S. Choi and Y. Lvov, RSC, Cambridge, 2014, pp. 216–239 Search PubMed;
(b) Y. Yan, M. Björnmalm and F. Caruso, Chem. Mater., 2014, 26, 452–460 CrossRef CAS;
(c) R. F. Fakhrullin, A. I. Zamaleeva, R. T. Minullina, S. A. Konnova and V. N. Paunov, Chem. Soc. Rev., 2012, 41, 4189–4206 RSC;
(d) P. T. Hammond, Mater. Today, 2012, 15, 196–206 CrossRef CAS;
(e) P. T. Hammond, Nanomedicine, 2012, 7, 619–622 CrossRef CAS PubMed;
(f) A. L. Becker, A. P. R. Johnston and F. Caruso, Small, 2010, 6, 1836–1852 CAS.
-
(a) N. J. Shah, M. N. Hyder, J. S. Moskowitz, M. A. Quadir, S. W. Morton, H. J. Seeherman, R. F. Padera, M. Spector and P. T. Hammond, Sci. Transl. Med., 2013, 5, 191ra83 Search PubMed;
(b) Z. Poon, D. Chang, X. Zhao and P. T. Hammond, ACS Nano, 2011, 5, 4284–4292 CrossRef CAS PubMed;
(c) N. J. Shah, M. L. Macdonald, Y. M. Beben, R. Padera, R. E. Samuel and P. T. Hammond, Biomaterials, 2011, 32, 6183–6193 CrossRef CAS PubMed;
(d) A. Sexton, P. G. Whitney, S.-F. Chong, A. N. Zelikin, A. P. R. Johnston, R. De Rose, A. G. Brooks, F. Caruso and S. J. Kent, ACS Nano, 2009, 3, 3391–3400 CrossRef CAS PubMed;
(e) R. De Rose, A. N. Zelikin, A. P. R. Johnston, A. Sexton, S.-F. Chong, C. Cortez, W. Mulholland, F. Caruso and S. J. Kent, Adv. Mater., 2008, 20, 4698–4703 CrossRef CAS PubMed.
-
(a) M. Matsusaki, H. Ajiro, T. Kida, T. Serizawa and M. Akashi, Adv. Mater., 2012, 24, 454–474 CrossRef CAS PubMed;
(b) A. Nishiguchi, H. Yoshida, M. Matsusaki and M. Akashi, Adv. Mater., 2011, 23, 3506–3510 CrossRef CAS PubMed.
-
(a) J. H. Park, S. H. Yang, J. Lee, E. H. Ko, D. Hong and I. S. Choi, Adv. Mater., 2014, 26, 2001–2010 CrossRef CAS PubMed;
(b) M. K. Drachuk, V. Gupta and V. Tsukruk, Adv. Funct. Mater., 2013, 23, 4437–4453 CrossRef PubMed;
(c) R. F. Fakhrullin and Y. M. Lvov, ACS Nano, 2012, 6, 4557–4564 CrossRef CAS PubMed.
-
(a)
J. H. Park, J. Lee, B. J. Kim and S. H. Yang, in Cell Surface Engineering, ed. R. Fakhrullin, I. S. Choi and Y. Lvov, RSC, Cambridge, 2014, pp. 48–79 Search PubMed;
(b) J. Lee, J. Choi, J. H. Park, M.-H. Kim, D. Hong, H. Cho, S. H. Yang and I. S. Choi, Angew. Chem., Int. Ed., 2014, 53, 8056–8059 CrossRef CAS PubMed;
(c) E. H. Ko, Y. Yoon, J. H. Park, S. H. Yang, D. Hong, K.-B. Lee, H. K. Shon, T. G. Lee and I. S. Choi, Angew. Chem., Int. Ed., 2013, 52, 12279–12282 CrossRef CAS PubMed;
(d) S. H. Yang, E. H. Ko and I. S. Choi, Langmuir, 2012, 28, 2151–2155 CrossRef CAS PubMed;
(e) S. H. Yang, E. H. Ko, Y. H. Jung and I. S. Choi, Angew. Chem., Int. Ed., 2011, 50, 6115–6118 CrossRef CAS PubMed;
(f) S. H. Yang, K.-B. Lee, B. Kong, J.-H. Kim, H.-S. Kim and I. S. Choi, Angew. Chem., Int. Ed., 2009, 48, 9160–9163 CrossRef CAS PubMed;
(g) J. H. Park, I. S. Choi and S. H. Yang, Chem. Commun., 2015, 51, 5523–5525 RSC;
(h) D. Hong, H. Lee, E. H. Ko, J. Lee, H. Cho, M. Park, S. H. Yang and I. S. Choi, Chem. Sci., 2015, 6, 203–208 RSC.
- B. B. Hsu, K. S. Jamieson, S. R. Hagerman, E. Holler, J. Y. Ljubimova and P. T. Hammond, Angew. Chem., Int. Ed., 2014, 53, 8093–8098 CrossRef CAS PubMed.
-
(a)
D. Hong, E. H. Ko and I. S. Choi, in Cell Surface Engineering, ed. R. Fakhrullin, I. S. Choi and Y. Lvov, RSC, Cambridge, 2014, pp. 142–161 Search PubMed;
(b) D. Hong, M. Park, S. H. Yang, J. Lee, Y.-G. Kim and I. S. Choi, Trends Biotechnol., 2013, 31, 442–447 CrossRef CAS PubMed;
(c) S. H. Yang, D. Hong, J. Lee, E. H. Ko and I. S. Choi, Small, 2013, 9, 178–186 CrossRef CAS PubMed.
-
(a) J.-H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS PubMed;
(b) J.-H. Ryu, S. Jiwpanich, R. Chacko, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 8246–8247 CrossRef CAS PubMed.
-
(a) Y. Yan, Y. Wang, J. K. Heath, E. C. Nice and F. Caruso, Adv. Mater., 2011, 23, 3916–3921 CrossRef CAS PubMed;
(b) O. Kulygin, A. D. Price, S.-F. Chong, B. Stadler, A. N. Zelikin and F. Caruso, Small, 2010, 6, 1558–1564 CrossRef CAS PubMed;
(c) S. Sivakumar, V. Bansal, C. Cortez, S.-F. Chong, A. N. Zelikin and F. Caruso, Adv. Mater., 2009, 21, 1820–1824 CrossRef CAS PubMed;
(d) A. L. Becker, A. N. Zelikin, A. P. R. Johnston and F. Caruso, Langmuir, 2009, 25, 14079–14085 CrossRef CAS PubMed;
(e) A. N. Zelikin, Q. Li and F. Caruso, Chem. Mater., 2008, 20, 2655–2661 CrossRef CAS;
(f) A. N. Zelikin, A. L. Becker, A. P. R. Johnston, K. L. Wark, F. Turatti and F. Caruso, ACS Nano, 2007, 1, 63–69 CrossRef CAS PubMed.
-
(a) J. Lee, S. H. Yang, S.-P. Hong, D. Hong, H. Lee, H.-Y. Lee, Y.-G. Kim and I. S. Choi, Macromol. Rapid Commun., 2013, 34, 1351–1356 CrossRef CAS PubMed;
(b) S. H. Yang, S. M. Kang, K.-B. Lee, T. D. Chung, H. Lee and I. S. Choi, J. Am. Chem. Soc., 2011, 133, 2795–2797 CrossRef CAS PubMed.
-
(a) O. Shimoni, A. Postma, Y. Yan, A. M. Scott, J. K. Heath, E. C. Nice, A. N. Zelikin and F. Caruso, ACS Nano, 2012, 6, 1463–1472 CrossRef CAS PubMed;
(b) S.-F. Chong, J. H. Lee, A. N. Zelikin and F. Caruso, Langmuir, 2011, 27, 1724–1730 CrossRef CAS PubMed;
(c) Y. Yan, A. P. R. Johnston, S. J. Dodds, M. M. J. Kamphuis, C. Ferguson, R. G. Parton, E. C. Nice, J. K. Heath and F. Caruso, ACS Nano, 2010, 4, 2928–2936 CrossRef CAS PubMed;
(d) A. N. Zelikin, J. F. Quinn and F. Caruso, Biomacromolecules, 2006, 7, 27–30 CrossRef CAS PubMed.
-
(a) S. A. Konnova, A. A. Danilushkina, G. I. Fakhrullina, F. S. Akhatova, A. R. Badrutdinov and R. F. Fakhrullin, RSC Adv., 2015, 5, 13530–13537 RSC;
(b) E. A. Naumenko, M. R. Dzamukova, G. I. Fakhrullina, F. S. Akhatova and R. F. Fakhrullin, Curr. Opin. Pharmacol., 2014, 18, 84–90 CrossRef CAS PubMed;
(c) R. F. Fakhrullin, A. G. Bikmullin and D. K. Nurgaliev, ACS Appl. Mater. Interfaces, 2009, 1, 1847–1851 CrossRef CAS PubMed.
- S. Ghosh, S. Basu and S. Thayumanavan, Macromolecules, 2006, 39, 5595–5597 CrossRef CAS.
- K. Zhang, L. L. Xu, J. G. Jiang, N. Calin, K. F. Lam, S. J. Zhang, H. H. Wu, G. D. Wu, B. Albela, L. Bonneviot and P. Wu, J. Am. Chem. Soc., 2013, 135, 2427–2430 CrossRef CAS PubMed.
- B. Chang, J. Guo, C. Liu, J. Qian and W. Yang, J. Mater. Chem., 2010, 20, 9941–9947 RSC.
- D. C. González-Toro, J. H. Ryu, R. T. Chacko, J. Zhuang and S. Thayumanavan, J. Am. Chem. Soc., 2012, 134, 6964–6967 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc01225b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence