
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThorium–ligand multiple bonds via reductive deprotection of a trityl group†
Danil E.
Smiles
a,
Guang
Wu
a,
Nikolas
Kaltsoyannis
*b and
Trevor W.
Hayton
*a
aDepartment of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA 93106, USA. E-mail: hayton@chem.ucsb.edu
bChristopher Ingold Laboratories, Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: n.kaltsoyannis@ucl.ac.uk
First published on 30th April 2015
Abstract
Reaction of [Th(I)(NR2)3] (R = SiMe3) (2) with KECPh3 (E = O, S) affords the thorium chalcogenates, [Th(ECPh3)(NR2)3] (3, E = O; 4, E = S), in moderate yields. Reductive deprotection of the trityl group from 3 and 4 by reaction with KC8, in the presence of 18-crown-6, affords the thorium oxo complex, [K(18-crown-6)][Th(O)(NR2)3] (6), and the thorium sulphide complex, [K(18-crown-6)][Th(S)(NR2)3] (7), respectively. The natural bond orbital and quantum theory of atoms-in-molecules approaches are employed to explore the metal–ligand bonding in 6 and 7 and their uranium analogues, and in particular the relative roles of the actinide 5f and 6d orbitals.
Introduction
The study of actinide–ligand multiple bonds has intensified in recent years due to the need to understand the extent of both f-orbital participation and covalency in actinide–ligand bonding.1–9 In this regard, the past ten years have seen considerable progress in the synthesis of oxo,10–13 imido,14–22 carbene,23–29 and nitrido complexes of uranium.30–35 More recently, several terminal phosphinidene36,37 and chalcogenido (S, Se, Te) complexes of uranium have also been isolated,38–42 demonstrating that this chemistry can be extended to the heavier main group elements.Despite these advancements, multiply-bonded complexes of the other actinides remain rare.43 Only one thorium terminal oxo complex is known, namely, [η5-1,2,4-tBu3C5H2]2Th(O)(dmap) (dmap = 4-dimethylaminopyridine), which was recently reported by Zi and co-workers.44 In addition, a handful of terminal imido complexes have been isolated,45 including [Cp*2Th(NAr)(THF)] (Ar = 2,6-dimethylphenyl), which was reported by Eisen and co-workers in 1996.46 A few thorium carbene complexes are also known, but in each example the carbene ligand is incorporated into a chelating ligand, which kinetically stabilizes the Th![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond.47–49 Also of note, terminal thorium sulphides have been invoked as reaction intermediates,44 but have not been isolated. This paucity of examples can be rationalized by the higher energy of the thorium 5f orbitals, relative to uranium, which likely weakens metal–ligand π-bonding.50 However, this hypothesis requires further verification, highlighting the need for new complexes that feature thorium–ligand multiple bonds.
C bond.47–49 Also of note, terminal thorium sulphides have been invoked as reaction intermediates,44 but have not been isolated. This paucity of examples can be rationalized by the higher energy of the thorium 5f orbitals, relative to uranium, which likely weakens metal–ligand π-bonding.50 However, this hypothesis requires further verification, highlighting the need for new complexes that feature thorium–ligand multiple bonds.
Recently, we reported that selective removal of the trityl protecting group from the U(IV) alkoxide, [U(OCPh3)(NR2)3] (R = SiMe3), allowed for the isolation of the oxo complex, [K(18-crown-6)][U(O)(NR2)3].41 Significantly, the uranium centre does not undergo a net oxidation state change during the transformation. Inspired by this result, we endeavoured to synthesize the analogous thorium oxo complex, and its sulphido congener, using this deprotection protocol. Thorium was chosen for this study, in part, to address the scarcity of multiply-bonded complexes of the other actinides, but also because Th4+ is effectively redox inactive, which makes the traditional synthetic routes to multiple bonds (such as oxidative atom transfer) more challenging. Herein, we describe the synthesis of a thorium sulphide and a thorium oxo, along with an analysis of their electronic structures by density functional theory.
Results and discussion
Reaction of ThCl4(DME)2 with 3 equiv. of NaNR2 (R = SiMe3) in THF affords colourless crystals of [Th(Cl)(NR2)3] (1) in 56% yield, upon crystallization from Et2O/hexanes. This material was previously prepared by Bradley51 and Andersen;52 however, it was never structurally characterized. Crystals of complex 1 suitable for X-ray crystallographic analysis were grown from a concentrated diethyl ether (Et2O) solution stored at −25 °C for 24 h. Determination of the solid-state structure revealed the anticipated pseudotetrahedral geometry about the thorium centre (see ESI† for full details). In addition, this material has a melting point of 208–210 °C, nearly identical to that reported by Andersen and co-workers.52 Interestingly, crystallization of the reaction mixture from THF/pentane resulted in isolation of the “ate” complex, [Na(THF)4.5][Th(Cl)2(NR2)3], as determined by X-ray crystallography (see the ESI†). However, this material can readily be converted into 1 upon extraction into, and recrystallization from, Et2O.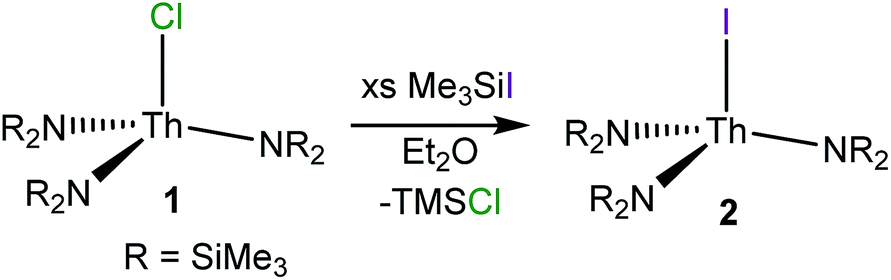 | (1) |
Subsequent reaction of complex 1 with 12 equiv. of Me3SiI, in diethyl ether, affords [Th(I)(NR2)3] (2) as a white powder in 95% yield (eqn (1)). A similar procedure was recently used to prepare the related cerium iodide complex, [Ce(I)(NR2)3].53 Crystals of 2 suitable for X-ray diffraction analysis were grown from a concentrated diethyl ether solution stored at −25 °C for 24 h. Complex 2 crystallizes in the hexagonal setting of the rhombohedral space group R3c, and its solid state molecular structure is shown in Fig. S19.† Complex 2 is isostructural with its chloride analogue 1. Its Th–N distance (2.299(4) Å) is identical to that of 1, while the Th–I bond (3.052(1) Å) is longer than the Th–Cl bond of 1 (2.647(1) Å), consistent with the larger single bond covalent radius of I− (1.33 Å) vs. Cl− (0.99 Å).54 The 1H and 13C NMR spectra of 2 each exhibit a single resonance, at 0.45 ppm and 5.13 pm, respectively, assignable to the methyl groups of the silylamide ligands (Fig. S5 and S6†).
We previously reported the synthesis of a U(IV) alkoxide complex, [U(OCPh3)(NR2)3], via reaction of KOCPh3 and [U(I)(NR2)3],41 and with 2 in hand, we endeavoured to synthesise the analogous thorium alkoxide. Thus, addition of 1 equiv. of KOCPh3 to a cold (−25 °C) suspension of 2 in toluene affords a colourless solution, concomitant with the deposition of fine white powder. A colourless oil is obtained upon work-up, and storage of this oil at −25 °C for 24 h affords [Th(OCPh3)(NR2)3] (3) as a colourless crystalline solid in 33% yield (eqn (2)). Similarly, reaction of complex 2 with 1 equiv. of KSCPh3, in toluene, results in the formation of [Th(SCPh3)(NR2)3] (4) in 57% yield, after crystallization from hexanes (eqn (2)).
 | (2) |
We were unable to obtain X-ray quality crystals of 3; however, complex 4 was amenable to an X-ray diffraction analysis. This material crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , and features a pseudotetrahedral geometry about the thorium centre (Fig. 1). The Th–S bond length in 4 (2.704(1) Å) is similar to those of other thorium thiolate complexes (ca. 2.74).55,56 In addition, the Th–S–C angle (136.72(1)°) is rather small, suggesting that there is minimal 3p π-donation from S to Th. Other thorium thiolates also feature similarly acute Th–S–C angles.55,56
, and features a pseudotetrahedral geometry about the thorium centre (Fig. 1). The Th–S bond length in 4 (2.704(1) Å) is similar to those of other thorium thiolate complexes (ca. 2.74).55,56 In addition, the Th–S–C angle (136.72(1)°) is rather small, suggesting that there is minimal 3p π-donation from S to Th. Other thorium thiolates also feature similarly acute Th–S–C angles.55,56
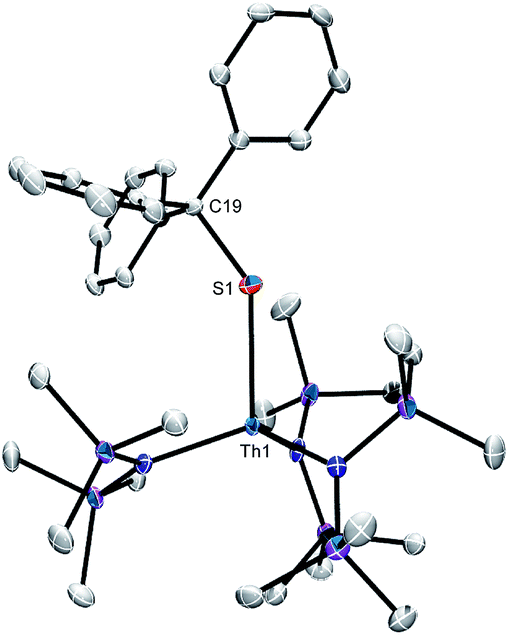 | ||
| Fig. 1 Solid state molecular structure of 4 with 50% probability ellipsoids. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (deg.): Th1–S1 = 2.704(1), S1–C19 = 1.866(4), av. Th–N = 2.31, av. N–Th–N = 112.2, Th1–S1–C19 = 136.72(1). | ||
The 1H NMR spectrum of 3 exhibits a singlet at 0.39 ppm, in benzene-d6, assignable to the methyl groups of the silylamide ligands. In addition, it features resonances at 7.09, 7.18, and 7.39 ppm, in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:6 ratio, respectively, corresponding to the p-, m-, and o-aryl protons of the trityl-alkoxide ligand (Fig. S7†), consistent with the proposed formulation. Not surprisingly, the 1H NMR spectrum of 4, in benzene-d6, is almost identical to that of 3, and also features resonances assignable to three silylamide ligands and one trityl moiety (Fig. S9†).
:6:6 ratio, respectively, corresponding to the p-, m-, and o-aryl protons of the trityl-alkoxide ligand (Fig. S7†), consistent with the proposed formulation. Not surprisingly, the 1H NMR spectrum of 4, in benzene-d6, is almost identical to that of 3, and also features resonances assignable to three silylamide ligands and one trityl moiety (Fig. S9†).
Interestingly, the 1H NMR spectrum of the trityl-alkoxide reaction mixture exhibits resonances due to a second, minor Th-containing product. This was subsequently identified to be the bis(alkoxide) complex, [Th(OCPh3)2(NR2)2] (5), which is likely formed by reaction of 3 with another equivalent of KOCPh3. The 1H NMR spectrum of 5 features a sharp singlet at 0.26 ppm, in benzene-d6, which is assignable to the methyl groups of the silylamide ligands (Fig. S11†). This resonance is slightly upfield from that observed for complex 3, which allows 5 to be distinguished from that complex. Complex 5 was also characterized by X-ray crystallography (see ESI†). Interestingly, there is no evidence for the formation of the analogous uranium complex in the reaction of KOCPh3 with [U(I)(NR2)3],41 consistent with the reduced ionicity of the U–N bond vs. the Th–N bond (see also below), which increases the barrier for ligand scrambling in uranium. Complex 1 can also be used as a precursor to 3, but in this case even greater amounts of complex 5 are formed during the reaction.
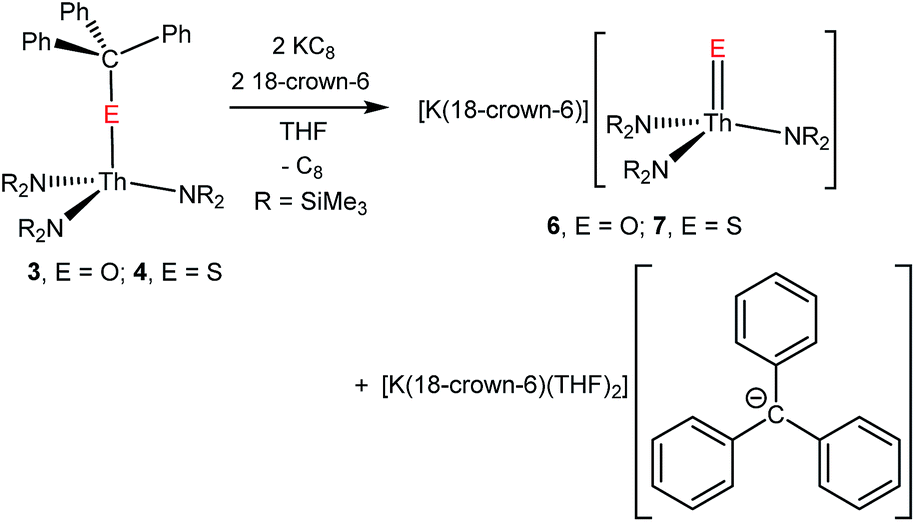
Prompted by our aforementioned success at selectively cleaving the C–O bond in [U(OCPh3)(NR2)3] to afford a uranium oxo complex,41 we explored the reductive cleavage of the C–E (E = O, S) bonds in complexes 3 and 4. Gratifyingly, reduction of 3 with 2 equiv. of KC8, in the presence of 18-crown-6, in THF, results in formation of a vibrant red solution, indicative of the presence of [CPh3]−.41,57 Extraction of the reaction mixture into diethyl ether, followed by filtration, permits removal of the [K(18-crown-6)(THF)2][CPh3] by-product, which is insoluble in this solvent. Work-up of the filtrate affords the thorium oxo complex, [K(18-crown-6)][Th(O)(NR2)3], (6) as colourless blocks in 23% yield (eqn (3)). Similarly, reaction of 4 with 2 equiv. of KC8, in the presence of 18-crown-6, in THF, results in the formation of the thorium sulphide, [K(18-crown-6)][Th(S)(NR2)3] (7), which can be isolated as colourless needles in 62% yield after a similar work-up (eqn (3)). The 1H NMR spectra of 6 and 7, in benzene-d6, both feature two sharp resonances (6:0.64 and 3.09 ppm; 7:0.74 and 3.17 ppm) in a 54:24 ratio, assignable to the methyl groups of the silylamide ligands and the methylene groups of the 18-crown-6 moiety, respectively (Fig. S13 and S15†), consistent with their proposed formulations. Unfortunately, the ThE vibrational modes in 6 and 7 could not be definitively identified by either IR or Raman spectroscopies.
 | (3) |
Complex 6 crystallizes in the orthorhombic spacegroup Pbca, as a diethyl ether solvate, 6·0.5Et2O, while complex 7 crystallizes in the triclinic spacegroup P, with two independent molecules in the asymmetric unit. Their solid state molecular structures are shown in Fig. 2, and selected bond lengths and angles can be found in Table 1. Both complexes feature pseudotetrahedral geometries about their metal centres, along with dative interactions between the chalcogenido ligands and the K+ ion of the [K(18-crown-6)] moiety. The Th–O bond length (1.983(7) Å) in 6 is slightly longer than the Th–O distance in the other structurally characterized thorium oxo (Th–O = 1.929(4) Å),44 but is significantly shorter than a typical Th–O single bond (ca. 2.20 Å),58–63 suggestive of multiple bond character within the Th–O interaction. Interestingly, the Th–O distance in 6 is 0.09 Å longer than the analogous distance in [K(18-crown-6)][U(O)(NR2)3] (1.890(5) Å),41 a difference that is greater than the difference in the 4+ ionic radii of these two metals (0.05 Å).64 The Th–S bond lengths in 7 (2.519(1) and 2.513(1) Å) are significantly shorter than a typical Th–S single bond (ca. 2.74 Å),44,55,56,65 and are again suggestive of multiple bond character within the Th–S interaction. In addition, the Th–S distances in 7 are 0.07 Å longer than the analogous distances in [K(18-crown-6)][U(S)(NR2)3] (2.4463(6) and 2.4513(6) Å),41 which is in-line with the anticipated difference based on ionic radii considerations alone.64
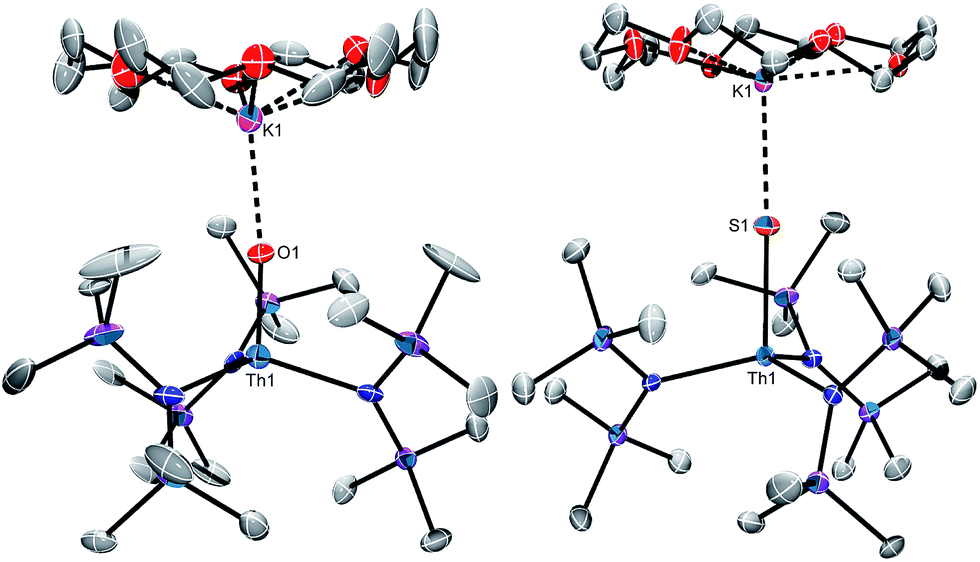 | ||
| Fig. 2 Solid state molecular structures of 6·0.5Et2O (left) and 7 (right) with 50% probability ellipsoids. One molecule of 7, a diethyl ether solvate, and hydrogen atoms have been omitted for clarity. | ||
| M = Th | M = U | |||
|---|---|---|---|---|
| E = O (6) | E = S (7)b | E = O (6-U)a | E = S (7-U)a,b | |
| a Taken from ref. 41. b Two molecules in the asymmetric unit. | ||||
| M–E | 1.983(7), 1.980, (1.980) | 2.513(1), (2.530), 2.519(1), 2.546 | 1.890(5), 1.921, (1.922) | 2.4463(6), (2.467), 2.4513(6), 2.428 |
| E–K | 2.645(7), 2.606, (2.573) | 3.039(2), (2.987), 3.122(2), 3.083 | 2.640(5), 2.641, (2.601) | 3.0684(8), (3.005), 3.1551(8), 3.105 |
| M–N (av.) | 2.42, 2.429, (2.412) | 2.36, 2.371, (2.359) | 2.36, 2.367, (2.349) | 2.30, 2.299, (2.286) |
| M–E–K | 167.5(4), 179.9, (179.9) | 177.95(6), (179.6), 149.20(6), 150.4 | 170.0(3), 176.2, (175.1) | 178.16(3), (179.8), 148.98(3), 150.4 |
| N–M–N (av.) | 115.6 | 116.6 | 117.0 | 116.8 |
In order to gain further insight into the electronic structure and bonding of 6 and 7, as well as the uranium analogues 6-U and 7-U, we turned to quantum chemistry in the form of density functional theory (DFT). We began by optimising the geometries of the four target molecules using the PBE functional; selected bond lengths and angles are given in Table 1. For complexes 6 and 6-U the agreement between experiment and theory is very good, with differences in bond length of no more than 0.04 Å. DFT predicts both molecules to be almost linear along the M–O–K vector, 179.9° and 176.2° for 6 and 6-U respectively, in reasonable agreement with the experimental angles of 167.5(4)° and 170.0(3)°, respectively. In contrast, 7 and 7-U have two molecules in the asymmetric unit, with very different M–S–K angles. The PBE optimised structures agree very well with the experimental data for the molecules with the smaller M–S–K angles; the deviation from the experimental angles is only ca. 1.5°. In addition, a constrained geometry optimisation of the Th–S–K angle in 7, from the optimised angle of 150.4°, yields converged geometries up to Th–S–K = 170.4°, at which point the molecule is only 2.6 kJ mol−1 less stable than in the fully optimised structure. Given this shallow bending potential, we wondered if the differences between the two molecules in the asymmetric units of 7 and 7-U might arise from dispersion forces, and hence re-optimised all four targets with these included via the Grimme D3 corrections. The data for these structures are collected in Table 1 and show that, with the exception of a slight shortening of the O–K distance, there is almost no difference between the PBE and PBE + D3 structures for 6 and 6-U. By contrast, the inclusion of dispersion corrections significantly modifies the geometries of 7 and 7-U, most notably the M–S–K angle, which increases by ca. 30° to linear in both cases, and the E–K distances which, in agreement with experiment, shorten by almost 0.1 Å between the bent and linear structures. For the latter, calculation predicts the M–E bond length reduction on going from Th to U to be ca. 0.06 Å in both the oxo and sulphido cases, essentially the same as the difference in ionic radius between Th4+ and U4+, hence underestimating by ca. 0.03 Å the experimentally determined M–O bond length reduction on going from 6 to 6-U.
We have analysed the electronic structures of all four targets using the Natural Bond Orbital (NBO) and Quantum Theory of Atoms-in-Molecules (QTAIM) approaches and, in order to allow for better comparison, decided to focus on the linear forms of 7 and 7-U, i.e. the electronic structures have been analysed at the PBE + D3 geometries for all four molecules. Complexes 6 and 7 are, of course, closed shell species and hence there is no net spin density for these systems; for 6-U and 7-U, however, NBO finds net spin densities of 2.092 and 2.085 respectively, as expected for U(IV). In all four cases, NBO finds the M–E interaction to be a triple bond; the σ + 2π Th–O natural localised molecular orbitals (NLMOs) in 6 are shown in Fig. 3, and the compositions of the π NLMOs are collected in Table 2 for all four targets. In all cases the orbitals are largely chalcogen-based, a little more so for thorium than uranium. There is clearly more metal involvement in these orbitals in the sulphur systems than the oxygen, and while this is predominantly d-based for thorium there is an almost equal contribution of d and f in 6-U and 7-U.
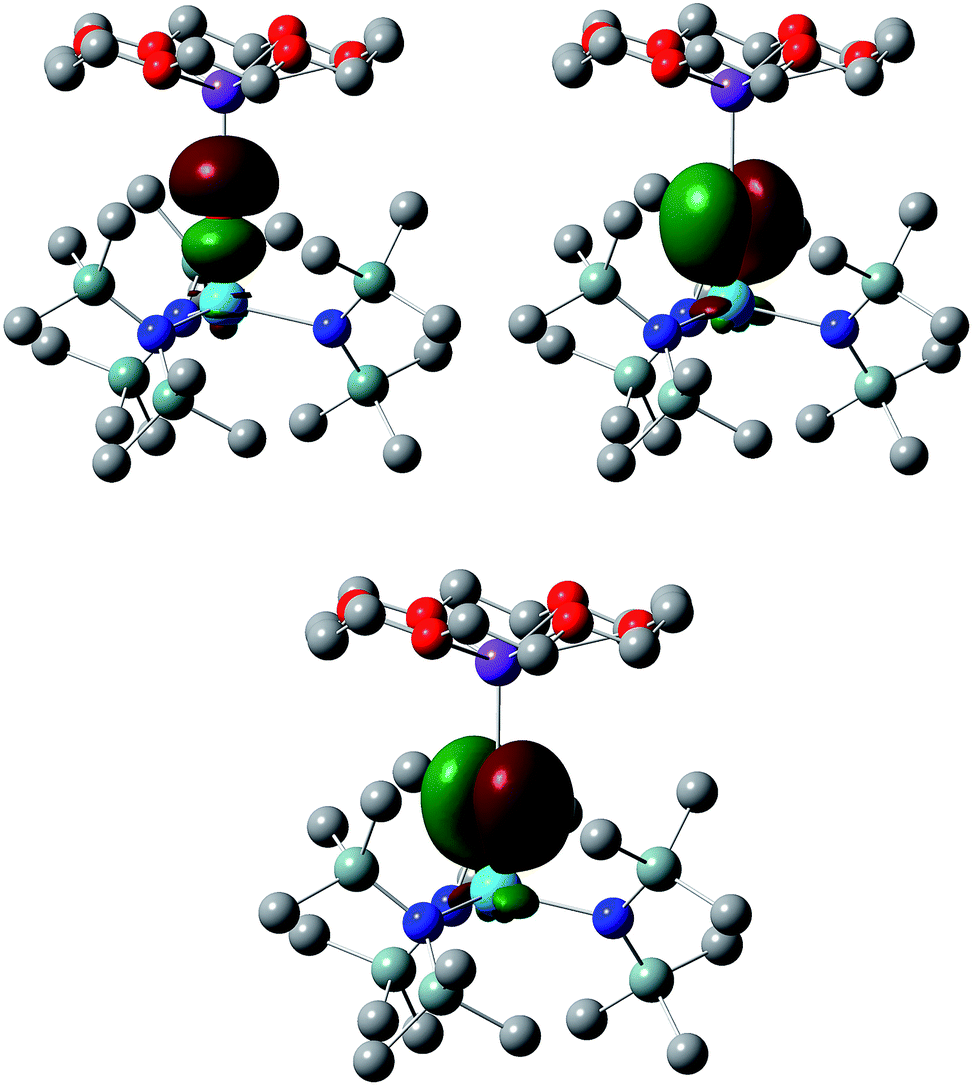 | ||
| Fig. 3 σ and π Th–O NLMOs in 6. Hydrogen atoms omitted for clarity. Isosurface = 0.04. | ||
| E | M | |
|---|---|---|
| 6 | 86.86 (99.97 p) | 11.75 (65.36 d, 34.48 f) |
| 7 | 81.69 (99.94 p) | 16.67 (61.31 d, 38.41 f) |
| 6-U | 83.72 (99.96 p) | 15.18 (48.17 d, 51.73 f) |
| 7-U | 79.44 (99.96 p) | 18.89 (51.10 d, 48.78 f) |
NBO finds the M–N interactions to have double bond character. Three dimensional representations of one set of Th–N NLMOs in 6 are shown in Fig. 4, and the averaged compositions of the σ and π character orbitals are collected in Table 3 for all four targets. As with the M–E bonding, these NLMOs are all strongly polarized toward the nitrogen. There is slightly more uranium contribution than thorium in analogous NLMOs. For the σ orbitals, the metal contributions are significantly more d-based than f (more so for thorium than uranium), while for the π component there is much more even metal d/f content, with a little more f than d for the uranium NLMOs and vice versa for thorium.
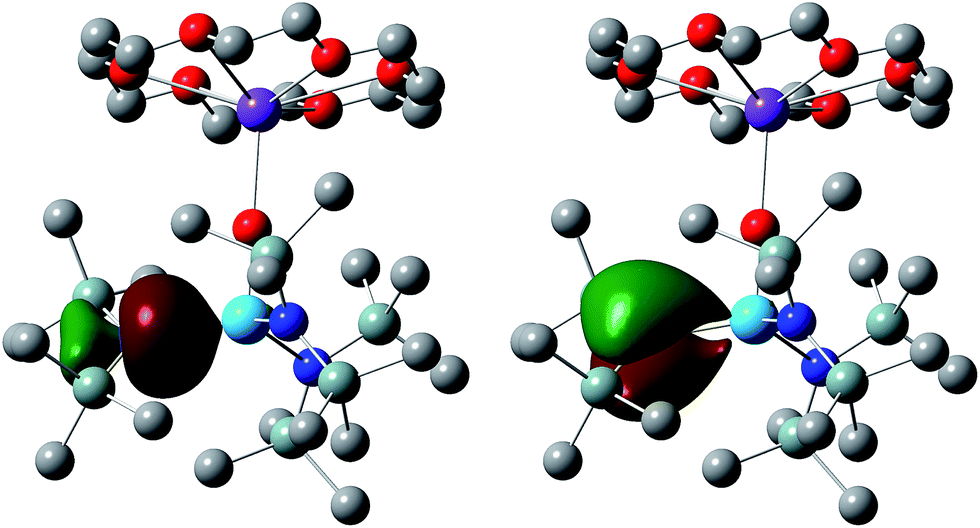 | ||
| Fig. 4 One set of σ and π Th–N NLMOs in 6. Hydrogen atoms omitted for clarity. Isosurface = 0.04. | ||
| N | M | ||
|---|---|---|---|
| 6 | σ | 89.52 (34.93 s, 65.07 p) | 6.47 (3.67 s, 3.42 p, 77.66 d, 15.25 f) |
| π | 86.22 (99.98 p) | 5.94 (54.52 d, 45.15 f) | |
| 7 | σ | 88.50 (39.52 s, 60.49 p) | 7.89 (5.86 s, 2.32 p, 74.52 d, 17.30 f) |
| π | 85.70 (99.98 p) | 7.37 (55.36 d, 44.47 f) | |
| 6-U | σ | 87.51 (33.25 s, 66.74 p) | 8.49 (3.02 s, 2.29 p, 64.65 d, 30.05 f) |
| π | 85.37 (99.96 p) | 7.04 (48.66 d, 51.06 f) | |
| 7-U | σ | 87.79 (36.69 s, 63.30 p) | 10.67 (11.38 s, 1.34 p, 60.06 d, 27.22 f) |
| π | 83.87 (99.99 p) | 9.49 (43.73 d, 56.12 f) |
The deviations of the actinide natural atomic orbital populations (Natural Population Analysis (NPA)) from their formal values are given in Table 4. Typically, deviations from formal populations are taken as a measure of covalency, and such an approach is valid for the early actinides. Table 4 shows that the 7s and 7p orbitals are little involved in bonding. The 6d orbitals have larger deviations from the formal population than the 5f; these are very similar for the two sulphur compounds (1.49 and 1.50), and reduced for the two oxygen compounds, with slightly more 6d in the uranium system than the thorium (1.17 vs. 1.12). A similar situation is found for the 5f populations; the deviations of the sulphur compounds are very similar for thorium and uranium and larger than for the oxygen compounds, for which the uranium 5f population is a little larger than the thorium 5f. In summary, and in agreement with the analysis of the NLMO compositions, these data suggest greater covalency in the sulphur than the oxygen compounds, greater 6d covalency than 5f and, for the latter orbitals, slightly larger covalency in uranium than thorium.
| 5f | 6d | 7 s | 7p | |
|---|---|---|---|---|
| 6 | 0.73 | 1.12 | 0.08 | 0.04 |
| 7 | 0.98 | 1.49 | 0.10 | 0.02 |
| 6-U | 0.85 | 1.17 | 0.08 | 0.03 |
| 7-U | 0.97 | 1.50 | 0.17 | 0.02 |
Table 5 presents the calculated atomic partial charges, using the QTAIM and NPA approaches. While the absolute values differ between methods, the trends are the same and suggest strongly polar M–E and M–N bonding. Taking the difference in charge between the metal and the surrounding atoms as a measure of ionicity, the data indicate that the bonding in the thorium compounds is more ionic than the uranium, and that bonding in the oxygen systems is more ionic than the sulphur, in agreement with the compositions of the NLMOs, which are more thorium localized than uranium, and more oxygen localized than sulphur.
| QTAIM | NPA | QTAIM | NPA | |||
|---|---|---|---|---|---|---|
| 6 | q(M) | 2.451 | 1.847 | 7 | 2.342 | 1.260 |
| q(E) | −1.221 | −1.159 | −1.126 | −0.793 | ||
| q(K) | 0.896 | 0.879 | 0.876 | 0.874 | ||
| q(N) av | −2.175 | −1.722 | −2.152 | −1.693 | ||
| q(M)–q(E) | 3.672 | 3.006 | 3.468 | 2.053 | ||
| q(M)–q(N) | 4.626 | 3.569 | 4.494 | 2.953 | ||
| 6-U | q(M) | 2.242 | 1.819 | 7-U | 2.094 | 1.289 |
| q(E) | −1.112 | −0.991 | −1.04 | −0.720 | ||
| q(K) | 0.901 | 0.874 | 0.877 | 0.874 | ||
| q(N) av | −2.141 | −1.684 | −2.101 | −1.626 | ||
| q(M)–q(E) | 3.354 | 2.810 | 3.134 | 2.009 | ||
| q(M)–q(N) | 4.383 | 3.503 | 4.195 | 2.915 |
We have pioneered the use of the QTAIM in the study of actinide covalency2,4 and bond strength,66,67 and Table 6 collects selected bond critical point (BCP) electron (ρ) and energy (H) densities and ellipticities (ε), and delocalisation indices (δ(A, B) – QTAIM measures of bond order). The ellipticity data reinforce the NBO results, indicating cylindrical (or, for 6-U, near cylindrical) triple-bond symmetry for the M–O interactions, and significantly non-cylindrical double-bond symmetry for M–N.68 The M–O BCP electron densities for 6 and 6-U are very large for actinide bonds, bordering the 0.2 au covalency threshold, and the M–N BCP ρ data are typical.67,69 For both M–O and M–N, the BCP data are larger in an absolute sense in 6-Uvs.6. This is also true of the delocalisation indices, reinforcing the NBO conclusion of greater covalency in 6-Uvs.6. This is also the case for 7vs.7-U; the M–S and M–N QTAIM metrics are all larger in an absolute sense in the uranium system.
| M–O | O–K | M–N (av) | M–S | S–K | M–N (av) | |||
|---|---|---|---|---|---|---|---|---|
| 6 | ρ | 0.175 | 0.024 | 0.075 | 7 | 0.090 | 0.018 | 0.084 |
| H | 0.094 | 0.003 | −0.016 | 0.031 | 0.002 | −0.022 | ||
| ε | 0.000 | 0.000 | 0.166 | 0.000 | 0.000 | 0.192 | ||
| δ(A,B) | 1.387 | 0.163 | 0.625 | 1.184 | 0.157 | 0.705 | ||
| 6-U | ρ | 0.199 | 0.023 | 0.083 | 7-U | 0.101 | 0.018 | 0.098 |
| H | −0.119 | 0.003 | −0.020 | −0.037 | 0.002 | −0.029 | ||
| ε | 0.062 | 0.002 | 0.207 | 0.000 | 0.000 | 0.155 | ||
| δ(A,B) | 1.575 | 0.147 | 0.702 | 1.372 | 0.152 | 0.829 |
The M–E ρ and H and, to a lesser extent, δ(A,B) are significantly smaller in the sulphur compounds than the oxygen. We have previously cautioned, however, in the context of Th/U–S/Se bonding,70 against the interpretation of such reductions in terms of reduced covalency. The QTAIM covalency metrics show very strong dependence on bond length, and we believe that the very significant (>0.5 Å) difference between M–S and M–O is the dominant factor here.
Conclusions
We have demonstrated the synthesis of oxo and sulphide complexes of thorium via reductive removal of the trityl protecting group. This work further demonstrates the generality of the reduction deprotection methodology, suggesting that this method will be broadly applicable towards the synthesis of multiple bonds in other metal systems, including lanthanides and transition metals, and we are currently exploring this possibility. Quantum chemical analysis (NBO and QTAIM) of the bonding in the thorium systems, and analogous uranium oxo and sulphido molecules, indicates that the M–E interactions are σ + 2π triple bonds that are strongly polarised toward the chalcogen, while the M–N bonds (also largely ligand-based) have double bond character. For both the M–E and M–N bonds, there is greater metal–ligand orbital mixing (which, in the early part of the actinide series, we are comfortable describing as covalency) in the sulphur than the oxygen compounds. The Th–ligand bonds are found to be more ionic than the uranium analogues. Finally, the 6d orbitals play a larger role in the Th–E and Th–N bonds than do the 5f, while the latter are more involved in the uranium–ligand bonding.Experimental
General
All reactions and subsequent manipulations were performed under anaerobic and anhydrous conditions under an atmosphere of nitrogen. Hexanes, Et2O, THF, and toluene were dried using a Vacuum Atmospheres DRI-SOLV Solvent Purification system and stored over 3 Å sieves for 24 h prior to use. Benzene-d6 was dried over 3 Å molecular sieves for 24 h prior to use. ThCl4(DME)2 was synthesized according to the previously reported procedure.71 All other reagents were purchased from commercial suppliers and used as received.NMR spectra were recorded on a Varian UNITY INOVA 400, a Varian UNITY INOVA 500 spectrometer, a Varian UNITY INOVA 600 MHz spectrometer, or an Agilent Technologies 400-MR DD2 400 MHz Spectrometer. 1H and 13C{1H} NMR spectra were referenced to external SiMe4 using the residual protio solvent peaks as internal standards. IR spectra were recorded on a Nicolet 6700 FT-IR spectrometer. Elemental analyses were performed by the Micro-Analytical Facility at the University of California, Berkeley.
[Th(Cl)(NR2)3] (1)
To a colourless, cold (−25 °C), solution of ThCl4(DME)2 (385.7 mg, 0.70 mmol), in THF (4 mL) was added a cold (−25 °C) solution of NaN(SiMe3)2 (381.6 mg, 2.08 mmol) in THF (4 mL). This mixture was allowed to stir for 18 h, whereupon the solvent was removed in vacuo to afford a colourless solid. This solid was triturated with hexanes (3 × 4 mL) to yield a colourless powder. The resulting powder was extracted with diethyl ether (10 mL) and filtered through a Celite column supported on glass wool (0.5 cm × 3 cm). The cloudy filtrate was again filtered through a Celite column supported on glass wool (0.5 cm × 3 cm) to give a clear colourless filtrate. The volume of this filtrate was reduced in vacuo to 4 mL and layered with hexanes (5 mL). Storage of this mixture at −25 °C for 24 h resulted in the deposition of colourless crystals, which were isolated by decanting off the supernatant (167 mg, 32%). The supernatant was then dried in vacuo to afford a colourless solid. This solid was then extracted with diethyl ether (5 mL) and filtered through a Celite column supported on glass wool (0.5 cm × 3 cm) to afford a colourless filtrate. The volume of this filtrate was reduced to 2 mL in vacuo and layered with hexanes (4 mL). Storage of this mixture at −25 °C for 24 h resulted in the deposition of an additional batch of colourless crystals, which were isolated by decanting off the supernatant. Total yield: 294.2 mg, 56%. Crystals suitable for X-ray crystallographic analysis were grown from a concentrated Et2O solution stored at −25 °C for 24 h. Melting point: 208–210 °C (lit. value = 210–212 °C).521H NMR (400 MHz, 25 °C, benzene-d6): δ 0.41 (s, 54H, NSiCH3). 13C{1H} NMR (100 MHz, 25 °C, benzene-d6): δ 4.26 (NSiCH3). IR (KBr pellet, cm−1): 611 (s), 657 (m), 678 (m), 771 (s), 830 (s), 850 (s), 923 (s), 1073 (m), 1182 (w), 1248 (s), 1406 (m).[Th(I)(NR2)3] (2)
To a stirring suspension of [Th(Cl)(NR2)3] (1) (852.3 mg, 1.14 mmol) in hexanes (8 mL) was added TMSI (2 mL, 14.05 mmol). This mixture was allowed to stir for 96 h, whereupon the solvent was removed in vacuo to afford a white solid. The solid was triturated with pentane (2 × 3 mL) to yield a white powder (908.2 mg, 95%). Crystals suitable for X-ray crystallographic analysis were grown from a concentrated CH2Cl2 solution stored at −25 °C for 24 h. Anal. calcd for C18H54IN3Si6Th: C, 25.73; H, 6.48; N, 5.00. Found: C, 25.34; H, 6.32; N, 5.24. 1H NMR (400 MHz, 25 °C, benzene-d6): δ 0.45 (s, 54H, NSiCH3). 13C{1H} NMR (100 MHz, 25 °C, benzene-d6): δ 5.13 (NSiCH3). IR (KBr pellet, cm−1): 612 (m), 657 (m), 676 (m), 772 (m), 830 (s), 850 (s), 909 (s), 1073 (m), 1249 (s), 1408 (w).[Th(OCPh3)(NR2)3] (3)
To a colourless, stirring suspension of 2 (231.4 mg, 0.28 mmol) in toluene (4 mL) was added a cold (−25 °C) solution of KOCPh3 (84.7 mg, 0.28 mmol) in toluene (4 mL), in two portions over the course of 1 h. This mixture was allowed to stir for another hour, resulting in the deposition of a fine white powder. An aliquot (0.25 mL) of the reaction mixture was taken, the solvent was removed in vacuo, and a 1H NMR spectrum in benzene-d6 was recorded. This spectrum indicated the presence of starting material, complex 3, and a small amount complex 5. The amount of remaining starting material was estimated from relative area of its silylamide resonance, whereupon an additional portion of KOCPh3 (13.4 mg, 0.045 mmol) was added to the reaction mixture. After 1 h of stirring, this mixture was filtered through a Celite column supported on glass wool (0.5 cm × 3 cm) to afford a colourless filtrate. The solvent was then removed in vacuo to yield a colourless oil. Storage of this oil at −25 °C for 24 h resulted in the formation of crystals within the matrix of the oil. The crystalline material was isolated by decanting off the remaining oil and then washed with cold (−25 °C) pentane (2 mL). This material consisted mostly of complex 5 and was discarded. The oil and the pentane washings were combined and the solvent was removed in vacuo to yield a colourless oil. Storage of this oil at −25 °C for 24 h resulted in the deposition of colourless crystals, which were isolated by decanting off the remaining oil. 88.0 mg, 33%. Anal. calcd for C37H69N3OSi6Th: C, 45.70; H, 7.15; N, 4.32. Found: C, 45.55; H, 7.24; N, 4.09. 1H NMR (400 MHz, 25 °C, benzene-d6): δ 0.39 (s, 54H, NSiCH3), 7.09 (t, 3H, JHH = 7.2 Hz, p-CH), 7.18 (t, 6H, JHH = 7.6 Hz, m-CH), 7.39 (d, 6H, JHH = 7.6 Hz, o-CH). 13C{1H} NMR (100 MHz, 25 °C, benzene-d6): δ 5.53 (NSiCH3), 96.13 (C(C6H5)3), 127.56 (p-C), 127.88 (o-C), 129.90 (m-C), 148.16 (Cipso). IR (KBr pellet, cm−1): 475 (w), 610 (m), 639 (w), 662 (m), 700 (m), 759 (m), 773 (m), 849 (s), 882 (w), 901 (s), 1012 (m), 1035 (m), 1051 (m), 1090 (w), 1151 (w), 1159 (w), 1184 (w), 1201 (w), 1252 (s), 1445 (w), 1491 (w).[Th(SCPh3)(NR2)3] (4)
To a stirring suspension of KSCPh3 (51.4 mg, 0.16 mmol) in toluene (5 mL) was added 2 (137.4 mg, 0.16 mmol). This solution was allowed to stir for 1 h, whereupon the solvent was removed in vacuo. The resulting white solid was extracted with hexanes (10 mL) and filtered through a Celite column supported on glass wool (0.5 cm × 3 cm), to provide a colourless filtrate. The volume of the filtrate was reduced to 3 mL in vacuo. Storage of this solution for 48 h resulted in the deposition of colourless crystals, which were isolated by decanting off the supernatant (92.3 mg, 57%). Anal. calcd for C37H69N3SSi6Th: C, 44.95; H, 7.04; N, 4.25. Found: C, 44.83; H, 6.90; N, 4.15. 1H NMR (400 MHz, 25 °C, benzene-d6): δ 0.42 (s, 54H, NSiCH3), 7.02 (t, 3H, JHH = 7.4 Hz, p-CH), 7.16 (t, 6H, JHH = 7.6 Hz, m-CH), 7.66 (d, 6H, JHH = 7.6 Hz, o-CH). 13C{1H} NMR (100 MHz, 25 °C, benzene-d6): δ 5.21 (NSiCH3), 80.70 (C(C6H5)3), 126.78 (p-C), 130.97 (m-C), 149.57 (Cipso). The resonance assignable to the o-C was not observed due to overlap with the benzene-d6 resonance. IR (KBr pellet, cm−1): 614 (m), 662 (m), 700 (m), 742 (m), 759 (m), 773 (m), 834 (s), 844 (s), 852 (s), 898 (s), 1034 (w), 1184 (w), 1254 (s), 1443 (w), 1484 (w).[K(18-crown-6)][Th(O)(NR2)3] (6)
To a colourless, cold (−25 °C), stirring solution of 3 (189.9 mg, 0.20 mmol) in THF (3 mL) was added KC8 (56.1 mg, 0.42 mmol), which immediately yielded a dark red mixture. After 2 min, a cold (−25 °C), colourless solution of 18-crown-6 (104.3 mg, 0.39 mmol) in THF (3 mL) was added to this mixture. The solution was allowed to stir for 30 min, whereupon it was filtered through a Celite column supported on glass wool (0.5 cm × 3 cm) to provide a vibrant red filtrate. The filtrate was dried in vacuo to provide a red solid that was triturated with diethyl ether (3 × 3 mL). The resulting red powder was extracted with diethyl ether (5 mL) and filtered through a Celite column supported on glass wool (0.5 cm × 3 cm) to afford a large plug of bright red solid and a pale orange-red filtrate. The volume of the filtrate was reduced to 1 mL in vacuo. Storage of this solution at −25 °C for 24 h resulted in the deposition of colourless crystals, which were isolated by decanting off the supernatant (47.0 mg, 23%). Anal. calcd for C30H78KN3O7Si6Th·0.5C4H10O: C, 35.93; H, 7.82; N, 3.93. Found: C, 36.53; H, 7.82; N, 3.89. 1H NMR (400 MHz, 25 °C, benzene-d6): δ 0.64 (s, 54H, NSiCH3), 3.09 (s, 24H, 18-crown-6). 13C{1H} NMR (100 MHz, 25 °C, benzene-d6): 5.47 (NSiCH3), 70.30 (18-crown-6). IR (KBr pellet, cm−1): 599 (m), 665 (m), 677 (m), 724 (w), 755 (m), 770 (m), 832 (s), 867 (s), 966 (s), 986 (s), 1116 (s), 1182 (w), 1243 (s), 1285 (w), 1353 (m), 1455 (w), 1474 (w). Raman (neat solid, cm−1): 389 (w), 615 (s), 678 (m).[K(18-crown-6)][Th(S)(NR2)3] (7)
To a colourless, cold (−25 °C), stirring solution of 4 (144.7 mg, 0.15 mmol) in THF (3 mL) was added KC8 (41.2 mg, 0.30 mmol), which immediately yielded a dark red mixture. After 2 min, a cold (−25 °C), colourless solution of 18-crown-6 (76.5 mg, 0.29 mmol) in THF (3 mL) was added to this mixture. This solution was allowed to stir for 15 min, whereupon it was filtered through a Celite column supported on glass wool (0.5 cm × 3 cm) to provide a vibrant red filtrate. The filtrate was dried in vacuo to provide a red solid that was triturated with diethyl ether (8 mL). The resulting red powder was extracted with diethyl ether (8 mL) and filtered through a Celite column supported on glass wool (0.5 cm × 3 cm) to afford a large plug of bright red solid and a pale orange-red filtrate. The volume of the filtrate was reduced to 2 mL in vacuo. Storage of this solution at −25 °C for 24 h resulted in the deposition of colourless crystals, which were isolated by decanting off the supernatant (48.7 mg, 32%). Subsequent concentration of the mother liquor and storage at −25 °C for 24 h resulted in the deposition of additional crystals. Total yield: 95.6 mg, 62%. Anal. calcd for C30H78KN3O6SSi6Th: C, 34.36; H, 7.50; N, 4.01. Found: C, 34.85; H, 7.94; N, 3.64. 1H NMR (400 MHz, 25 °C, benzene-d6): δ 0.74 (s, 54H, NSiCH3), 3.17 (s, 24H, 18-crown-6). 13C{1H} NMR (100 MHz, 25 °C, benzene-d6): 5.49 (NSiCH3), 70.12 (18-crown-6). IR (KBr pellet, cm−1): 605 (m), 664 (m), 685 (w), 699 (w), 785 (sh), 771 (m), 842 (s), 882 (sh), 936 (s), 963 (s), 1108 (s), 1182 (m), 1252 (s), 1285 (w), 1352 (m), 1455 (w), 1474 (w). Raman (neat solid, cm−1): 385 (w), 578 (s), 630 (s), 682 (s), 843 (m), 883 (m), 1014 (s).X-ray crystallography
Data for 1, [Na(THF)4.5][Th(Cl)2(NR2)3], 2, 4–7 were collected on a Bruker KAPPA APEX II diffractometer equipped with an APEX II CCD detector using a TRIUMPH monochromator with a Mo Kα X-ray source (α = 0.71073 Å). The crystals were mounted on a cryoloop under Paratone-N oil, and all data were collected at 100(2) K using an Oxford nitrogen gas cryostream. Data were collected using ω scans with 0.5° frame widths. Frame exposures of 2 s were used for 1 and [Na(THF)4.5][Th(Cl)2(NR2)3]. Frame exposures of 5 s were used for 2. Frame exposures of 10 s were used for 4 and 7. Frame exposures of 5 s (low angle) and 10 s (high angle) were used for 5 and 6. Data collection and cell parameter determination were conducted using the SMART program.72 Integration of the data frames and final cell parameter refinement were performed using SAINT software.73 Absorption correction of the data was carried out using the multi-scan method SADABS.74 Subsequent calculations were carried out using SHELXTL.75 Structure determination was done using direct or Patterson methods and difference Fourier techniques. All hydrogen atom positions were idealized, and rode on the atom of attachment. Structure solution, refinement, graphics, and creation of publication materials were performed using SHELXTL.75 Further crystallographic details can be found in Tables S1 and S2.†For [Na(THF)4.5][Th(Cl)2(NR2)3], one sodium atom and its coordinated THF molecules exhibited positional disorder and were modelled over two positions in a 50:50 ratio. The C–C and C–O bond were constrained to 1.5 and 1.4 Å, respectively, using the DFIX command. In addition, the diethyl ether solvate of 6 exhibited positional disorder; one of the carbon atoms of this molecule was modelled over two positions in a 50:50 ratio. The anisotropic parameters of the disordered carbon atoms were constrained using the EADP command. Hydrogen atoms were not added to disordered carbon atoms.
Computational details
Density functional theory calculations were carried out using the PBE functional,76,77 as implemented in the Gaussian 09 Rev. D.01 quantum chemistry code.78 Dispersion corrections (D3) due to Grimme et al.79 were included, as discussed in the main text. (14s 13p 10d 8f)/[10s 9p 5d 4f] segmented valence basis sets with Stuttgart–Bonn variety relativistic pseudopotentials were used for Th and U.80 For the geometry optimisations, the 6-31G** basis sets were used for all other atoms. The ultrafine integration grid was employed in all calculations, as were the SCF convergence criteria. The default RMS force geometry convergence criterion was relaxed to 0.000667 au using IOP 1/7; the maximum force at each converged geometry is given in the ESI.† The electronic structures at the PBE + D3 geometries were recalculated using improved basis sets for the ligands; 6-311G** for O, S, N, K; 6-31G** for C and H. Natural bond orbital calculations were performed using the NBO6 code, interfaced with Gaussian.81 QTAIM analyses were performed using the AIMAll program package,82 with wfx files generated in Gaussian used as input.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Chemical Sciences, Biosciences, and Geosciences Division under Contract no. DE-FG02-09ER16067. N. K. thanks University College London for computing resources via the Research Computing “Legion” cluster Legion@UCL and associated services, and is also grateful for computational resources from the EPSRC's National Service for Computational Chemistry Software, http://www.nsccs.ac.uk. We thank Tracy Chuong for assistance with the Raman spectroscopy and Jeff Carmichael for preliminary investigations in this area.Notes and references
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- N. Kaltsoyannis, Inorg. Chem., 2013, 52, 3407–3413 CrossRef CAS PubMed.
- S. G. Minasian, J. L. Krinsky and J. Arnold, Chem.–Eur. J., 2011, 17, 12234–12245 CrossRef CAS PubMed.
- I. Kirker and N. Kaltsoyannis, Dalton Trans., 2011, 40, 124–131 RSC.
- M. P. Jensen and A. H. Bond, J. Am. Chem. Soc., 2002, 124, 9870–9877 CrossRef CAS PubMed.
- K. I. M. Ingram, M. J. Tassell, A. J. Gaunt and N. Kaltsoyannis, Inorg. Chem., 2008, 47, 7824–7833 CrossRef CAS PubMed.
- T. W. Hayton, Dalton Trans., 2010, 39, 1145–1158 RSC.
- T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC.
- M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed.
- S. Fortier, J. L. Brown, N. Kaltsoyannis, G. Wu and T. W. Hayton, Inorg. Chem., 2012, 51, 1625–1633 CrossRef CAS PubMed.
- S. Fortier, N. Kaltsoyannis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 14224–14227 CrossRef CAS PubMed.
- G. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS.
- S. C. Bart, C. Anthon, F. W. Heinemann, E. Bill, N. M. Edelstein and K. Meyer, J. Am. Chem. Soc., 2008, 130, 12536–12546 CrossRef CAS PubMed.
- A.-C. Schmidt, F. W. Heinemann, L. Maron and K. Meyer, Inorg. Chem., 2014, 53, 13142–13153 CrossRef CAS PubMed.
- K. C. Mullane, A. J. Lewis, H. Yin, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2014, 53, 9129–9139 CrossRef CAS PubMed.
- N. H. Anderson, S. O. Odoh, Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919–926 CrossRef CAS PubMed.
- D. M. King, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2014, 136, 5619–5622 CrossRef CAS PubMed.
- J. G. Brennan and R. A. Andersen, J. Am. Chem. Soc., 1985, 107, 514–516 CrossRef CAS.
- T. W. Hayton, J. M. Boncella, B. L. Scott, P. D. Palmer, E. R. Batista and P. J. Hay, Science, 2005, 310, 1941–1943 CrossRef CAS PubMed.
- O. P. Lam, S. M. Franke, H. Nakai, F. W. Heinemann, W. Hieringer and K. Meyer, Inorg. Chem., 2012, 51, 6190–6199 CrossRef CAS PubMed.
- W. Ren, G. Zi, D.-C. Fang and M. D. Walter, Chem.–Eur. J., 2011, 17, 12669–12682 CrossRef CAS PubMed.
- E. M. Matson, M. G. Crestani, P. E. Fanwick and S. C. Bart, Dalton Trans., 2012, 41, 7952–7958 RSC.
- J.-C. Tourneux, J.-C. Berthet, T. Cantat, P. Thuéry, N. Mézailles and M. Ephritikhine, J. Am. Chem. Soc., 2011, 133, 6162–6165 CrossRef CAS PubMed.
- D. P. Mills, F. Moro, J. McMaster, J. van Slageren, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2011, 3, 454–460 CrossRef CAS PubMed.
- E. Lu, O. J. Cooper, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 6696–6700 CrossRef CAS PubMed.
- R. E. Cramer, R. B. Maynard, J. C. Paw and J. W. Gilje, J. Am. Chem. Soc., 1981, 103, 3589–3590 CrossRef CAS.
- D. P. Mills, O. J. Cooper, F. Tuna, E. J. L. McInnes, E. S. Davies, J. McMaster, F. Moro, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2012, 134, 10047–10054 CrossRef CAS PubMed.
- S. Fortier, J. R. Walensky, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 6894–6897 CrossRef CAS PubMed.
- O. J. Cooper, D. P. Mills, J. McMaster, F. Moro, E. S. Davies, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 2383–2386 CrossRef CAS PubMed.
- D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266–267, 2–15 CrossRef CAS.
- A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Science, 2005, 309, 1835–1838 CrossRef CAS PubMed.
- S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482–488 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed.
- D. S. J. Arney, R. C. Schnabel, B. C. Scott and C. J. Burns, J. Am. Chem. Soc., 1996, 118, 6780–6781 CrossRef CAS.
- B. M. Gardner, G. Balázs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 4484–4488 CrossRef CAS PubMed.
- L. Ventelon, C. Lescop, T. Arliguie, P. C. Leverd, M. Lance, M. Nierlich and M. Ephritikhine, Chem. Commun., 1999, 659–660 RSC.
- J. L. Brown, S. Fortier, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 15468–15475 CrossRef CAS PubMed.
- J. L. Brown, S. Fortier, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352–5355 CrossRef CAS PubMed.
- D. E. Smiles, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2014, 136, 96–99 CrossRef CAS PubMed.
- D. E. Smiles, G. Wu and T. W. Hayton, Inorg. Chem., 2014, 53, 10240–10247 CrossRef CAS PubMed.
- G. Zi, Sci. China: Chem., 2014, 57, 1064–1072 CrossRef CAS.
- W. Ren, G. Zi, D.-C. Fang and M. D. Walter, J. Am. Chem. Soc., 2011, 133, 13183–13196 CrossRef CAS PubMed.
- W. Ren, G. Zi and M. D. Walter, Organometallics, 2012, 31, 672–679 CrossRef CAS.
- A. Haskel, T. Straub and M. S. Eisen, Organometallics, 1996, 15, 3773–3775 CrossRef CAS.
- G. Ma, M. J. Ferguson, R. McDonald and R. G. Cavell, Inorg. Chem., 2011, 50, 6500–6508 CrossRef CAS PubMed.
- W. Ren, X. Deng, G. Zi and D.-C. Fang, Dalton Trans., 2011, 40, 9662–9664 RSC.
- E. Lu, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 9356–9359 CrossRef CAS PubMed.
- B. E. Bursten and R. J. Strittmatter, Angew. Chem., Int. Ed., 1991, 30, 1069–1085 CrossRef.
- D. C. Bradley, J. S. Ghotra and F. A. Hart, Inorg. Nucl. Chem. Lett., 1974, 10, 209–211 CrossRef CAS.
- H. W. Turner, R. A. Andersen, A. Zalkin and D. H. Templeton, Inorg. Chem., 1979, 18, 1221–1224 CrossRef CAS.
- U. J. Williams, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2014, 53, 6338–6345 CrossRef CAS PubMed.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- Z. Lin, C. P. Brock and T. J. Marks, Inorg. Chim. Acta, 1988, 141, 145–149 CrossRef CAS.
- W. J. Evans, K. A. Miller, S. A. Kozimor, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2007, 26, 3568–3576 CrossRef CAS.
- The formation of [K(18-crown-6)(THF)2][CPh3] during the reaction was further confirmed by comparison of its 1H NMR spectrum with that previously reported for this species.
- D. M. Barnhart, D. L. Clark, J. C. Gordon, J. C. Huffman and J. G. Watkin, Inorg. Chem., 1994, 33, 3939–3944 CrossRef CAS.
- I. Korobkov, A. Arunachalampillai and S. Gambarotta, Organometallics, 2004, 23, 6248–6252 CrossRef CAS.
- S. M. Beshouri, P. E. Fanwick, I. P. Rothwell and J. C. Huffman, Organometallics, 1987, 6, 2498–2502 CrossRef CAS.
- D. M. Barnhart, D. L. Clark, J. C. Gordon, J. C. Huffman, J. G. Watkin and B. D. Zwick, Inorg. Chem., 1995, 34, 5416–5423 CrossRef CAS.
- D. L. Clark and J. G. Watkin, Inorg. Chem., 1993, 32, 1766–1772 CrossRef CAS.
- J. M. Berg, D. L. Clark, J. C. Huffman, D. E. Morris, A. P. Sattelberger, W. E. Streib, W. G. Van Der Sluys and J. G. Watkin, J. Am. Chem. Soc., 1992, 114, 10811–10821 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- W. Ren, W. W. Lukens, G. Zi, L. Maron and M. D. Walter, Chem. Sci., 2013, 4, 1168–1174 RSC.
- A. R. E. Mountain and N. Kaltsoyannis, Dalton Trans., 2013, 42, 13477–13486 RSC.
- Q.-R. Huang, J. R. Kingham and N. Kaltsoyannis, Dalton Trans., 2015, 44, 2554–2566 RSC.
- C. F. Matta and R. J. Boyd, in The quantum theory of atoms in molecules, ed. C. F. Matta and R. J. Boyd, Wiley-VCH, Weinheim, Germany, 2007, pp. 1–34 Search PubMed.
- S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed.
- A. C. Behrle, C. L. Barnes, N. Kaltsoyannis and J. R. Walensky, Inorg. Chem., 2013, 52, 10623–10631 CrossRef CAS PubMed.
- T. Cantat, B. L. Scott and J. L. Kiplinger, Chem. Commun., 2010, 46, 919–921 RSC.
- SMART Apex II, Version 2.1, Bruker AXS Inc., Madison, WI, 2005 Search PubMed.
- SAINT Software User's Guide, Version 7.34a, Bruker AXS Inc., Madison, WI, 2005 Search PubMed.
- SADABS, University of Gottingen, Germany, 2005 Search PubMed.
- SHELXTL PC, Version 6.12, Bruker AXS Inc., Madison, WI, 2005 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Gaussian 09, Revision D.01, Gaussian, Inc., Wallington, CT, 2009 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- X. Cao and M. Dolg, J. Mol. Struct.: THEOCHEM, 2004, 673, 203–209 CrossRef CAS.
- Theoretical Chemistry Institute, NBO 6.0, University of Wisconsin, Madison WI, 2013 Search PubMed.
- AIMAll, 14.11.23, TK Gristmill Software, Overland Park KS, USA, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental details, figures, spectral data, converged Cartesian coordinates, total energies, maximum force at converged geometries for 6, 6-U, 7 and 7-U, and crystallographic data for 1, [Na(THF)4.5][Th(Cl)2(NR2)3], 2, 4–7. CCDC 1057750–1057756. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01248a |
| This journal is © The Royal Society of Chemistry 2015 |