
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of 5-methylcytosine and 5-hydroxymethylcytosine in DNA via host–guest interactions inside α-hemolysin nanopores†
Tao
Zeng‡
ab,
Lei
Liu‡
a,
Ting
Li
a,
Yuru
Li
a,
Juan
Gao
a,
Yuliang
Zhao
*b and
Hai-Chen
Wu
 *a
*a
aKey Laboratory for Biomedical Effects of Nanomaterials & Nanosafety, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China. E-mail: haichenwu@ihep.ac.cn; Fax: +86-10-88235745; Tel: +86-10-88235745
bNational Center for Nanoscience and Technology of China, Beijing 100190, China. E-mail: zhaoyl@nanoctr.cn
First published on 24th June 2015
Abstract
Cytosine methylation and hydroxymethylation are both important epigenetic modifications of DNA in mammalian cells. Therefore, profiling DNA (hydroxy)methylation across the genome is vital for understanding their roles in gene regulation. Here, we report a nanopore-based approach for quick and reliable detection of 5-methylcytosine and 5-hydroxymethylcytosine in DNA at the single-molecule level. The single-stranded DNA containing 5-methylcytosine or 5-hydroxymethylcytosine was first selectively modified on the epigenetic base to attach a host–guest complex. Threading of the modified DNA molecules through α-hemolysin nanopores causes unbinding of the host–guest complex and generates highly characteristic current signatures. Statistical analysis of the signature events affords quantitative information about 5-methylcytosine and 5-hydroxymethylcytosine in DNA. Our results suggest that other DNA modifications could also be detected with the developed method. Furthermore, we anticipate our nanopore sensing strategy to be generally useful in biochemical analysis and to find applications in the early diagnosis of diseases.
Introduction
DNA methylation has been one of the most intensively studied epigenetic events in mammalian cells owing to its essential roles in genomic imprinting,1 regulation of gene expression,2 mammalian development,3,4etc. In humans, DNA methylation occurs exclusively at the 5-position of cytosine in the context of the CpG dinucleotide. These CpG sites tend to be clustered into regions, called CpG islands, that are characterised by a high frequency of CpG sites and a high (G + C) content. However, in cancer cells many CpG islands in the promoter regions of tumour suppressor genes are aberrantly methylated. Hypermethylation of the CpG islands may silence tumour suppressor gene expression and result in tumour formation and progression.5 Therefore, DNA methylation has been regarded as a potential biomarker for cancer risk assessment, early diagnosis, prognosis and epigenetic therapy.6,7 Recently, it was discovered that 5-hydroxymethylcytosine (5hmC) is abundant in embryonic stem cells and the brain.8 Although it has been accepted that 5hmC is an intermediate of 5mC on its way to demethylation,9 5hmC is now regarded as the sixth base10 and has been found to play important roles in epigenetic reprogramming and regulation of tissue-specific gene expression.11 Currently, its full potential as a true epigenetic mark is being actively explored.Because of methylation's important implications for normal biology and disease, DNA methylation detection is vital to understanding the influence of epigenetics. Numerous methods have been developed for the detection of DNA methylation, especially in the last 10 years.12 Currently available techniques for DNA methylation detection can be roughly categorised into four types including bisulfite sequencing,13,14 endonuclease digestion,15,16 affinity enrichment,17 and emerging single-molecule techniques.18–20 Each type of strategy has its own distinct advantages and disadvantages for specific purposes. Detection of 5hmC has also attracted much attention after it was identified as a nucleobase in mammalian genomic DNA.8 Oxidative bisulfite sequencing can yield quantitative analysis of 5hmC at the single-base resolution.21 Other faster and cheaper methods are under active development.
Nanopore sensing is a powerful single-molecule technique based on detecting a modulation in the ionic current due to the partial blockade of nanopores caused by analytes. It has shown promise for rapid and low-cost DNA sequencing,22–26 and many other sensing applications.27–29 Recently, both protein nanopores and solid-state nanopores have been used to detect 5mC and 5hmC.30–36 A more recent report demonstrated the detection of 5hmC with a new type of nanopore sensor comprised of single-walled carbon nanotubes inserted into a lipid bilayer.37 Although interesting results have been obtained in these studies, a quantitative method that can be used for genomic DNA analysis remains elusive. In this report, we employed the combination of selective chemical labelling of the (hydroxy)methylated cytosines and host–guest interactions inside α-hemolysin (αHL) nanopores to quantitatively detect 5mC and 5hmC in short single-stranded DNA (ssDNA). The DNA strand containing 5mC or 5hmC is first modified with a ferrocene⊂cucurbit[7]uril (Fc⊂CB[7]) complex. Translocation of the DNA–Fc⊂CB[7] hybrid through αHL produces highly characteristic signature events which can be correlated with the existence of 5mC or 5hmC. Therefore, it is a fast and low-cost approach suitable for high-throughput screening of epigenetic biomarkers in genomic DNAs, enabling early diagnosis of (hydroxy)methylation-related diseases.
Results and discussion
Selective modification of 5mC/5hmC in ssDNA with a host–guest complex
Selective chemical modification of the (hydroxy)methylated sites is probably the most straightforward way of making the epigenetic bases easy to differentiate by nanopores. However, finding such an appropriate modification group remains a challenging task. So far, there have only been two literature reports in which the modified moiety on DNA successfully produced characteristic signals.38,39 During the course of our studies on 5mC/5hmC detection, we envisioned that attaching a host–guest complex to a specific site of the DNA strand might generate characteristic current events when it passes through αHL (Fig. 1a). Thus, the first key step of our strategy is the selective modification of 5mC/5hmC with a “guest” molecule. Successful examples of the specific modification of 5mC are rare in the literature. Although it was reported that oxidation with potassium osmate40 or sodium periodates41 could discriminate between 5mC and C/T, we found that either C or T could interfere when we adopted their conditions for the conversion of 5mC in model DNAs (for DNA sequences, see Table S1†). Eventually, we achieved the selective modification of 5mC by combining bisulfite treatment and the reaction of hydroxylamine with cytosine derivatives.42 The alkyne moiety attached during the latter step enabled the following “click” chemistry that successfully linked a ferrocene or adamantine derivative to the DNA strand under very mild conditions (Fig. 1b and c, S1 and S2 and ESI†). Upon incubation with CB[7], the noncovalent hybrid DNA1–Fc⊂CB[7] was formed for translocation analysis. This 5mC modification procedure can effectively avoid the interference of C and T in the DNA strand, but 5hmC seems to be indistinguishable since it converts to cytosine 5-methylenesulfonate during the bisulfite treatment and then reacts with hydroxylamine derivatives in the following step (Fig. S3†). As a result, quantitative analysis of both 5mC- and 5hmC-containing DNAs needs to be combined with 5hmC-containing DNA detection, as will be discussed vide infra (Fig. 2).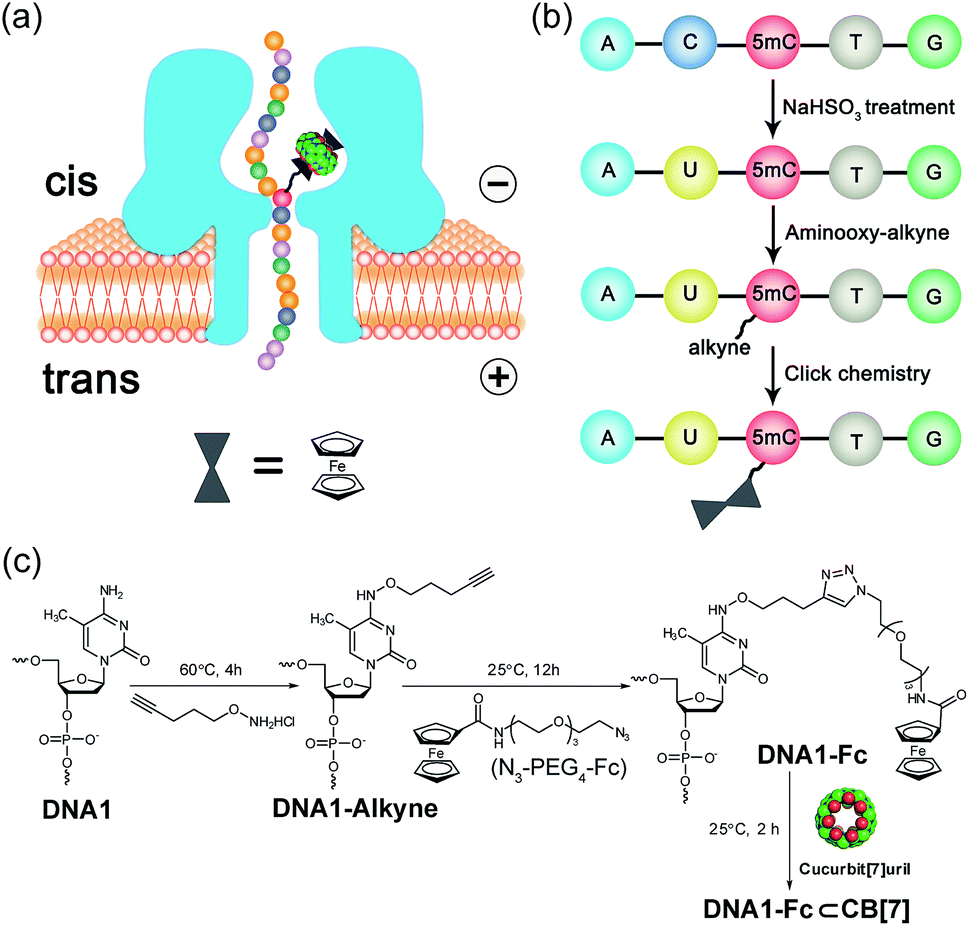 | ||
| Fig. 1 Schematic illustration of the detection strategy and chemical labeling of 5mC-containing DNA. (a) Translocation of a 5mC/5hmC-containing DNA molecule modified with the Fc⊂CB[7] complex through an αHL nanopore. (b) Selective modification steps of 5mC-containing DNA to afford the covalent DNA–Fc complex. The aminooxy-alkyne is O-(pent-4-yn-1-yl)hydroxylamine. (c) Chemical reactions of the modification of 5mC-containing DNA in (b). | ||
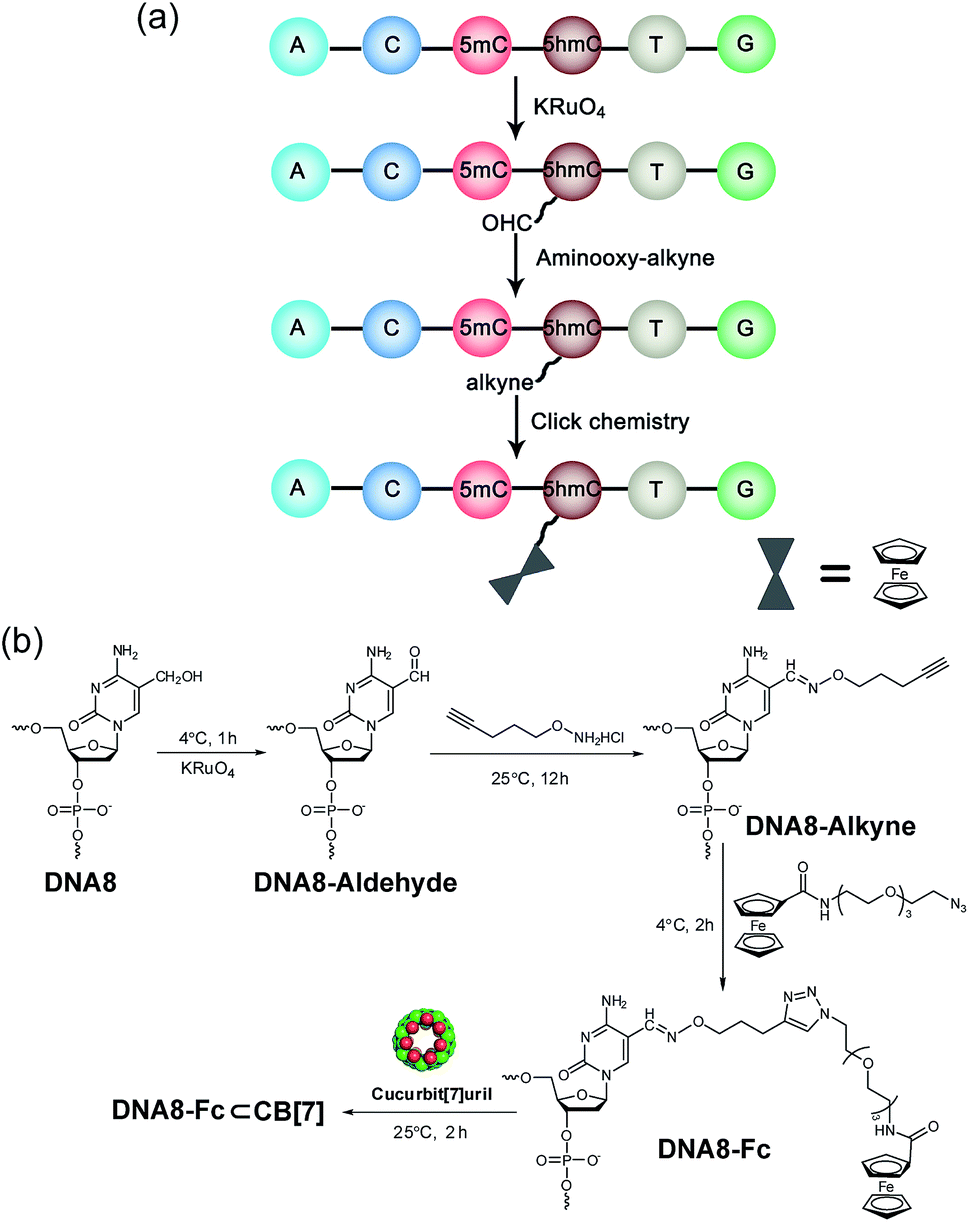 | ||
| Fig. 2 Selective modification of 5hmC-containing DNA. (a) Selective modification steps of 5hmC-containing DNA8 to afford the covalent DNA8–Fc complex. (b) Chemical reactions of the modification of DNA8. | ||
The selective modification of 5hmC was facilitated by oxidation with KRuO4 which afforded a versatile aldehyde group.21 Subsequent coupling with O-(pent-4-yn-1-yl)hydroxylamine followed by a “click” reaction attached an Fc moiety to the DNA strand. This modification procedure is highly selective for 5hmC, leaving C and 5mC in the strand intact (Fig. S4†). After incubation with CB[7], the DNA8–Fc⊂CB[7] hybrid was generated for single-channel recording studies.
Generation of signature events during the translocation of DNA–Fc⊂CB[7] through αHL
Translocation of modified 5mC-containing ssDNA through αHL was conducted in a buffer of 3 M KCl and 10 mM Tris (pH 8.0) with the transmembrane potential held at +160 mV. When DNA1–Fc⊂CB[7] was placed in the cis compartment, we observed highly characteristic current events at a frequency of around 8.2 min−1 (Fig. 3a). These events are composed of two consecutive parts, with Level 1 featuring long and deep blockades and Level 2–2′ featuring transient current oscillations (Fig. 3b). The scatter plots of residual current versus duration of Levels 1 and 2 are shown in Fig. 3c.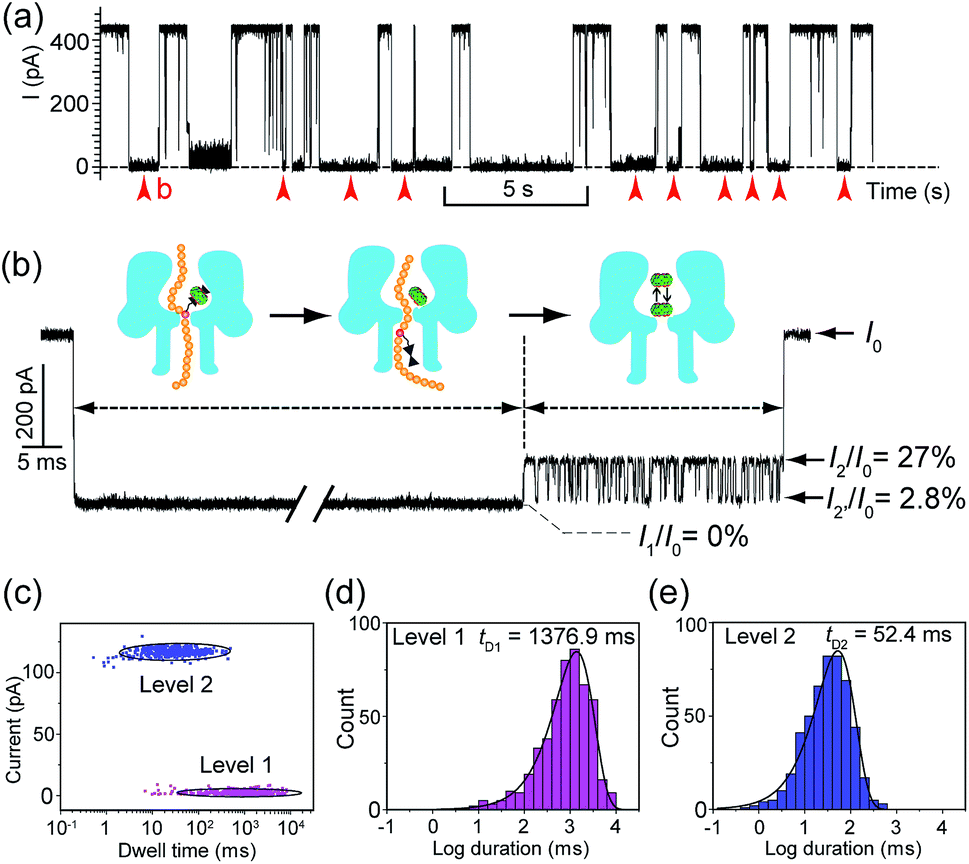 | ||
Fig. 3 Translocation of the DNA1–Fc⊂CB[7] hybrid through αHL. (a) A representative current trace of the translocation of DNA1–Fc⊂CB[7] (final concentration 0.25 μM) through αHL. Data were acquired in a buffer of 3 M KCl and 10 mM Tris (pH 8.0) with the transmembrane potential held at +160 mV. Red arrows indicate the multi-level current events. (b) A typical multi-level signature event generated by DNA1–Fc⊂CB[7] translocation (marked in (a)). Diagrams above the trace show the molecular mechanism of hybrid dissociation and translocation. Level 0: the open pore current level. Level 1: trapping of DNA1–Fc⊂CB[7] in the pore, dissociation of the DNA1–Fc⊂CB[7] and translocation of the DNA1–Fc through the pore. Level 2 and 2′: oscillation of CB[7] in the cis vestibule of αHL. (c) Scatter plots showing current blockage versus event durations of Level 1 and Level 2 in the signature events. (d) The dwell-time histogram for Level 1. The solid line in the histogram is a single exponential fit to the data. For Level 1, the mean duration tD1 = 1376.9 ms. (e) The dwell-time histogram for Level 2. The solid line in the histogram is a single exponential fit to the data. For Level 2, the mean duration tD2 = 52.4 ms. The sequence of DNA1: 5′-AAAAAAAAAA(![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif) ![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) )AAAAAAAAAA-3′. )AAAAAAAAAA-3′. | ||
Since none of the control experiments showed any long and deep current blockades (for control experimental traces, see Fig. S5†), we attributed the Level 1 state to the translocation of DNA1–Fc⊂CB[7] and concomitant dissociation of the Fc⊂CB[7] complex at the constriction of αHL. There are two features that support this assignment. First, the frequency of the occurrence of signature events is highly dependent on the applied voltage (Fig. S6†). As the applied potential was increased from 160 to 240 mV, the frequency of multi-level signature events rose exponentially. This clearly indicates that a higher transmembrane potential facilitates the unbinding of the Fc⊂CB[7] complex and also promotes DNA translocation from cis to trans. Second, the DNA1–Fc⊂CB[7] hybrid also produced single-level long blockades with no residual current that resembles Level 1 of the multi-level signature events (Fig. 3a and b), and the frequency ratio of multi-level to single-level events monotonically increased from 3.30 at 160 mV to 5.26 at 240 mV (Fig. S7†). The single-level blockade was formed by the trapped hybrid in a vestibule that returned to the cis solution without unbinding and translocation.43 It is conceivable that a higher voltage promotes unbinding of the Fc⊂CB[7] complexes and decreases the probability of DNA escaping from the cis side.
The current oscillation between Levels 2 and 2′ is reminiscent of trapping an analyte in the nanocavity of a protein pore (Fig. 3b).44 Thus, we speculated that the Level 2–2′ alternation was due to the trapping and oscillation of CB[7] inside the vestibule of αHL. To confirm the association of the signature events with CB[7], we carried out a competition experiment by adding 1-aminoadamantane (final concentration 1.0 μM) to the solution of DNA1–Fc⊂CB[7] (final concentration 0.25 μM) during the translocation studies. 1-Aminoadamantane is known to bind with CB[7] as tightly as ferrocene derivatives, and it may replace the ferrocene moiety when present in higher concentrations. After 30 minutes of incubation, we found that all the signature events produced by DNA1–Fc⊂CB[7] completely disappeared, and only ssDNA translocation events were observed, whose duration and conductance matched well with that of DNA1–Fc (Fig. S8†).
The translocation of CB[7] (outer diameter ∼1.6 nm) to the trans side would be prevented by the narrow constriction (∼1.4 nm) of αHL; however, the cis opening of the vestibule is wide enough (∼2.6 nm) for CB[7] to escape to the cis solution. So there must be a mechanism that could trap CB[7] inside the vestibule for tens of milliseconds (the mean duration of Level 2: tD2 = 52.4 ms, Fig. 3e). It is well known that cucurbiturils have a strong affinity with positively charged organic guests.45 We presumed that the side chains of some basic amino acids inside the vestibule might have interactions with CB[7] which provide the driving force for the current oscillation. By examining the crystal structure of αHL in the vestibule region, we found that the two lysine (K) residues at positions 8 and 147 might be crucial for the temporal trapping of CB[7] in the vestibule (Fig. S9†). To validate this assumption, we carried out translocation of DNA1–Fc⊂CB[7] through the αHL mutants K147N and K8L. The mutant pores are homoheptamers, so the mutations appear in all of the 7 subunits. In the K147N mutant, we found that the multi-level signature events completely vanished and the frequency of events dropped to only ∼1 s−1 even though the transmembrane potential was held at +200 mV (Fig. S10†). It was reported by Bayley and coworkers that elimination of one positively charged residue near the constriction of αHL might significantly reduce the DNA capture.46 Interestingly, we observed multi-level events in the K8L mutant (Fig. 4a). Although elimination of positively charged residues at the cis entrance of αHL could also affect the DNA capture, the frequency of the multi-level events was only reduced by ∼25% in the K8L mutant compared with the wildtype αHL. However, the feature of the “signature” event has drastically changed. Level 1 remains about the same, but the duration of Level 2 was shortened to ∼10 ms and Level 2′ completely disappeared (Fig. 4b). This result provided compelling evidence that the lysines at positions 8 and 147 play important roles in the trapping and oscillation of CB[7] in the vestibule. We also examined the translocation of the hybrid DNA1–adamantane⊂β-cyclodextrin (Ad⊂βCD) through αHL and found that the events generated did not have Levels 2 and 2′ (Fig. S11†). This is a further proof that the current oscillation between Levels 2 and 2′ is caused by “host–guest” interactions inside the vestibule of αHL.
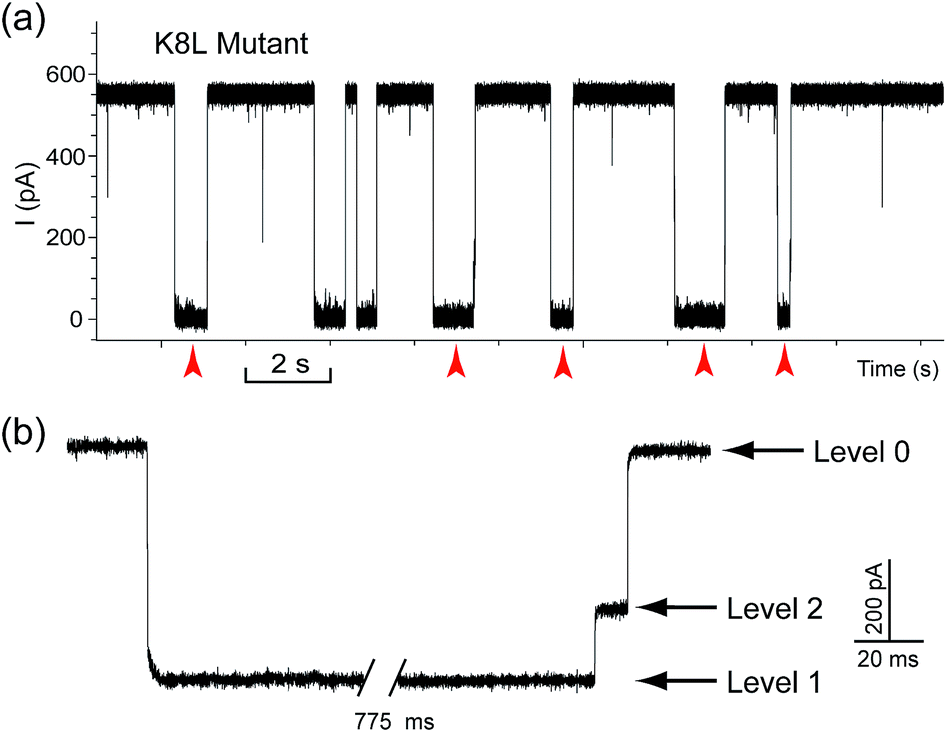 | ||
| Fig. 4 Translocation of DNA1–Fc⊂CB[7] through the engineered αHL mutant K8L. (a) Representative current trace of the translocation of DNA1–Fc⊂CB[7] through mutant K8L. Red arrows indicate the multi-level current events. (b) Expanded view of a multi-level signature event. Traces were recorded in 3 M KCl buffered with 10 mM Tris (pH 8.0). DNA1–Fc (final concentration 0.25 μM) was incubated with CB[7] (final concentration 50 μM) at room temperature for 2 hours before measurement. The transmembrane potential was held at +200 mV. | ||
In addition to the singly methylated ssDNA, we also investigated the detection of two 5mC sites in one DNA strand with the above strategy. We observed that the current signature generated by the doubly methylated DNA10 was similar to that of the singly methylated control DNA11 except for a slightly longer Level 1 state (Fig. S12†). Apparently, the current pattern of the first methylation site was affected by the tandem translocation of the second site. This could be a minor disadvantage when densely methylated DNA samples are being examined. One possible solution to circumvent this problem is to anchor the “host” molecule inside the nanopore, and then thread DNA strands modified with multiple “guest” molecules through the pore. Another problem we encountered was that when longer methylated DNAs (>40 bases) were tested, the Level 1 state in the current signatures became prolonged and sometimes led to long-lived blockades (>10 s). By elevating the temperature to around 46 °C, we nicely solved this clogging problem and obtained a high frequency of current signature around 35.5 min−1 (Fig. S13†).
Systematic studies of the factors affecting characteristic signature events
We performed systematic studies of the influence of different experimental conditions on the generation of characteristic signature events. First, we investigated the effects of the transmembrane potential on each current level of the signature events. When the transmembrane potential was relatively low (≤140 mV), only a few characteristic events could be observed. Thus we set the voltage range to between 160 and 240 mV, and found that the duration of the Level 1 state decreased monotonically as the applied voltage was increased (Fig. 5a). This trend is often given as strong evidence to prove the translocation of an analyte through a nanopore.23 It supports our assumption that Level 1 was caused by the translocation of the DNA1–Fc⊂CB[7] hybrid, and the unbinding of the Fc⊂CB[7] complex should be mainly responsible for the long duration of the level.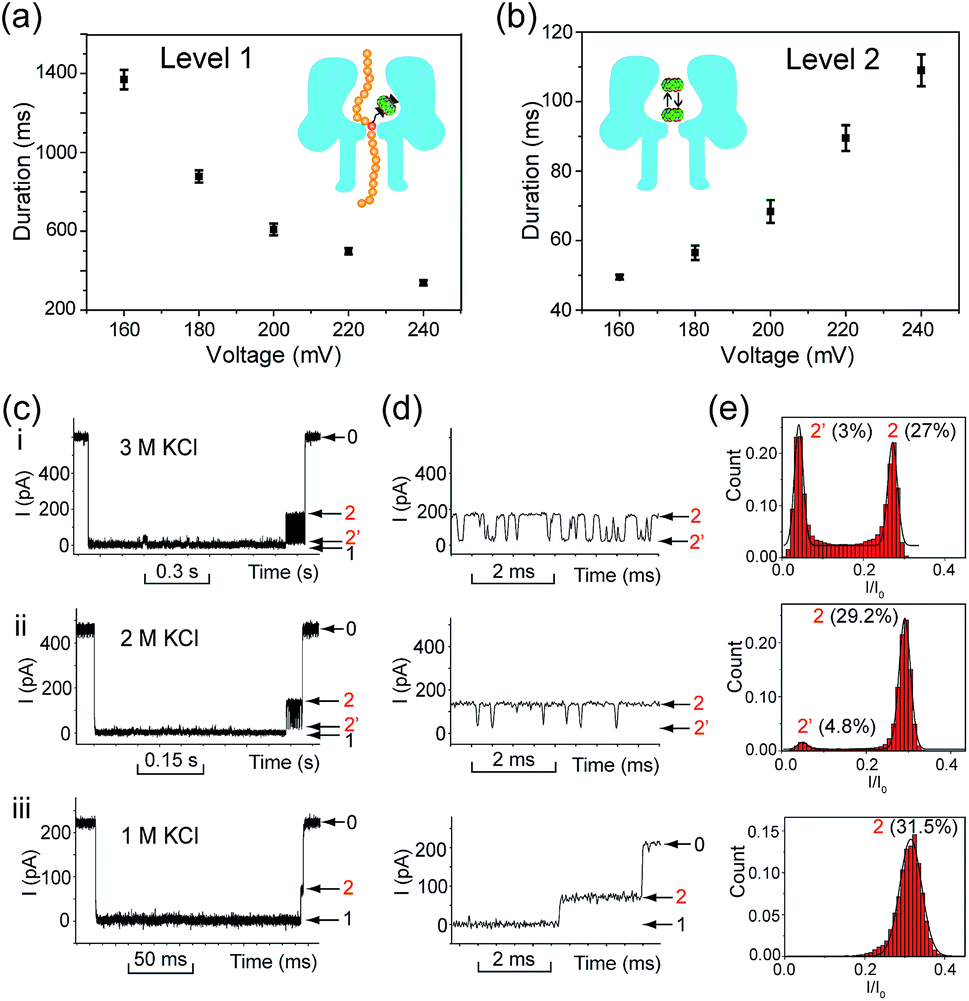 | ||
| Fig. 5 Investigation of the influence of the applied potentials and ionic strength on the translocation of DNA1–Fc⊂CB[7]. (a) Voltage dependence of the duration of Level 1. All the data points were obtained from a single exponential fit to the corresponding dwell time histograms. Recordings were conducted in a buffer of 3 M KCl and 10 mM Tris (pH 8.0) in the presence of 0.25 μM DNA1–Fc⊂CB[7]. The number of individual experiments n = 6. (b) Voltage dependence of the duration of Level 2 as a whole. The recording conditions are the same as in (a). The number of individual experiments n = 6. (c) Representative signature events of the translocation of DNA1–Fc⊂CB[7] (0.25 μM) in a buffer of 3 M KCl (i), 2 M KCl (ii), and 1 M KCl (iii) with 10 mM Tris (pH 8.0). The transmembrane potential was held at +200 mV. (d) Expanded views of Levels 2 and 2′ in (c). (e) Normalized all-point current amplitude histograms of Levels 2 and 2′. The current blockage levels are shown in parenthesis. The solid lines are Gaussian fit to the histograms. | ||
The duration of the Level 2 state as a whole increased as the transmembrane potential became more positive (Fig. 5b). This phenomenon is very similar to the interaction of βCD with anion-selective mutant αHL pores from the cis side, where the electroosmotic flow accounted for the enhanced binding of βCD to the sites within αHL.47 In the current study, we used wildtype αHL which is also weakly anion-selective at pH 7.5 (PK+/PCl− = 0.79). Therefore, electroosmotic flow from cis to trans should play a crucial role in the voltage-dependent oscillation of CB[7] in the vestibule. In addition, unlike βCD which is neutral under the aforementioned conditions, CB[7] is in equilibrium with CB[7]·K+ and CB[7]·K+·K+ in the electrolyte solutions (K1 600 M−1, K2, 53 M−1).48 The driving force exerted by the electric field on the CB[7]–metal complexes might be in good balance with the electroosmotic force.
In order to clarify the role of electroosmosis in the generation of characteristic signature events, we studied the translocation of DNA1–Fc⊂CB[7] in different concentrations of electrolyte. According to ref. 47, the net water flux generated by the electroosmotic flow (Jw) should conform to Jw ∝ VcKCl, where V is the transmembrane potential and cKCl is the KCl concentration in moles l−1 (ESI† text). Generally, different concentrations of KCl would produce varied electroosmotic force on the DNA1–Fc⊂CB[7] hybrid, while the electric field force remains about the same. When the translocation was conducted in 1.0, 2.0 and 3.0 M KCl respectively, we observed prominent changes in the pattern of signature events (Fig. 5c). The Level 1 state only showed minor changes in the duration when cKCl was lowered from 3.0 M to 1.0 M; however, the Level 2 and 2′ states underwent significant changes along with the decrease of cKCl. When cKCl dropped from 3.0 M to 2.0 M, the current signals switched from oscillating between Levels 2 and 2′ to residing mainly on Level 2 with occasional bumps to Level 2′. When cKCl was further lowered down to 1.0 M, we found that Level 2′ completely disappeared (Fig. 5d and e). Besides, the duration of Level 2 as a whole decreased drastically from 68.4 ms in 3.0 M KCl to 22.9 ms in 2.0 M KCl, and further to 0.81 ms in 1.0 M KCl (Table S2†). These results clearly indicated that lowering the KCl concentration while maintaining the transmembrane potential would greatly reduce the electroosmotic flow from cis to trans, and thus result in shortened retainment of CB[7] in the vestibule.
Quantitative determination of (hydroxy)methylation levels in DNA samples
The occurrence of characteristic signature events during the translocation of the modified 5mC/5hmC-containing DNA–Fc⊂CB[7] complex could be unambiguously correlated with the presence of 5mC/5hmC in ssDNA. Thus, the frequency of signature events (fsig) can be used to quantify 5mC/5hmC-containing ssDNA in a mixture. However, the presence of unmodified ssDNA in the solution might complicate the situation. In order to establish the quantitative determination of 5mC/5hmC in ssDNA, we constructed the standard working curves of fsigversus different percentages of (hydroxy)methylated DNA in the mixture (Fig. 6). Here, we used DNA7 and DNA8 with sequences predicted to have minimal secondary structures to conduct DNA modification and translocation experiments. First, we selectively modified the 5mC/5hmC in DNA7/DNA8 following the aforementioned procedures. Next, we mixed the modified DNA7/DNA8 with DNA9 in different ratios. DNA9 was used as the internal control DNA in the mixture and was also subject to the modification conditions. The plots of fsig–(5mC-containing DNA)% and fsig–(5hmC-containing DNA)% are shown in Fig. 6a and b, respectively.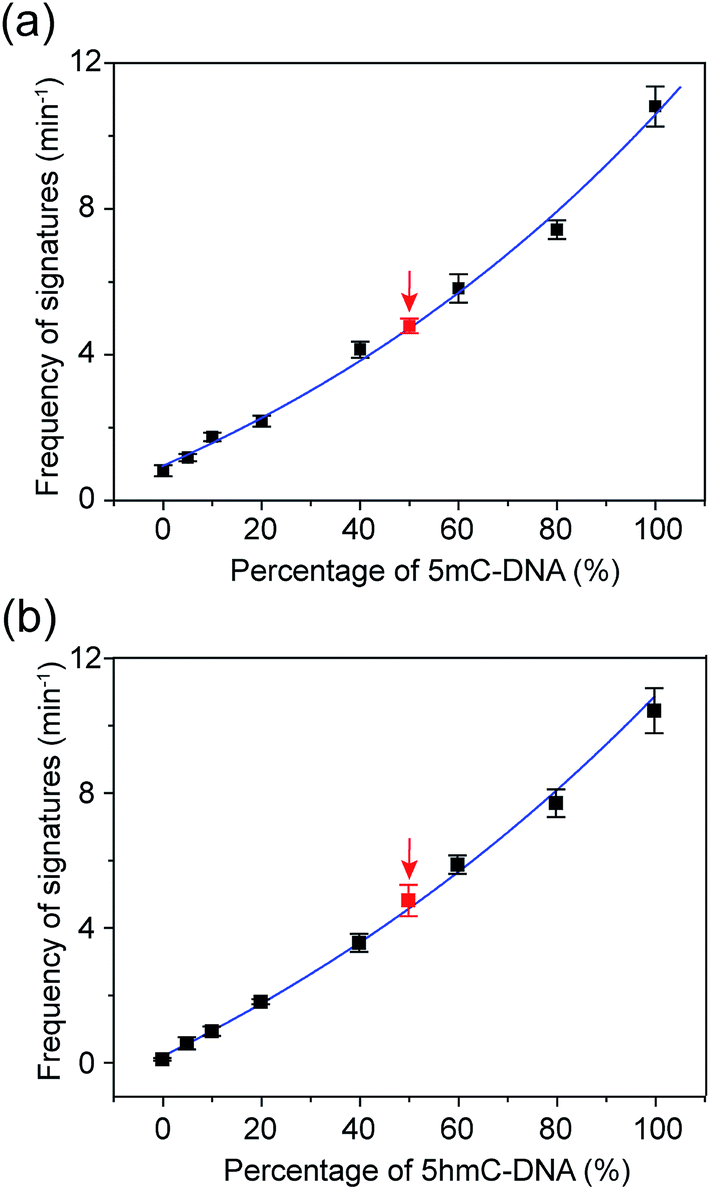 | ||
Fig. 6 Quantitative determination of 5mC- and 5hmC-containing DNAs with translocation recordings of DNA7–Fc⊂CB[7] and DNA8–Fc⊂CB[7]. (a) Correlation of the frequency of signature events (min−1) with the percentage of 5mC-containing DNA. The blue line is a single exponential fit to the data. The black data points were obtained by recording the events of premixed DNA7–Fc⊂CB[7] and DNA9 with known ratios. The red data point was the result of a mimicked real DNA sample which was composed of DNA7 and DNA9 for a blind test. The total DNA concentration is 0.20 μM. The number of individual experiments n = 3. (b) Correlation of the frequency of the signature events (min−1) with the percentage of 5hmC-containing DNA. The definition of black and red data points is the same as in (a) except for the use of DNA8–Fc⊂CB[7]. The total DNA concentration is 0.25 μM. All data was acquired in a buffer of 3 M KCl and 10 mM Tris (pH 8.0) with the transmembrane potential held at +200 mV. The number of individual experiments n = 3. The sequences of DNA7: 5′-TATGACCTGA()TAGATACGCT-3′; DNA8: 5′-TATGACCTGA(![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif) )TAGATACGCT-3′. )TAGATACGCT-3′. | ||
To verify the practical feasibility of the quantitative approach, we premixed DNA7/DNA8 and DNA9 with different ratios to mimic “real” DNA samples and subjected the mixture to modification and translocation studies. Interestingly, the results of fsig of the premixed samples nicely matched with the values interpolated from the standard working curves in Fig. 6 (red data points). This clearly indicates that the approach is suitable for “blind” sample testing. It should be noted that the 5mC detection strategy presented in this work does not discriminate between 5mC and 5hmC. Therefore, if the sample contains both 5mC and 5hmC, the real value of percentage of methylated DNA should be (5mC-containing DNA)% minus (5hmC-containing DNA)%.
Conclusion
We have developed a nanopore-based approach for quick and efficient detection of 5mC and 5hmC in ssDNA by generating characteristic current signatures during the translocation of modified DNA through αHL. The host–guest (Fc⊂CB[7]) complex attached on the (hydroxyl)methylated sites of DNA is crucial for the generation of characteristic events. This feature endows the detection with very high confidence at the single-molecule level. The high selectivity of the approach relies on specific chemical modifications of 5mC and 5hmC in the DNA strand. All the chemical treatments are carried out under very mild conditions and independent of DNA sequence context. Compared with the existing nanopore-based methods for the detection of 5mC and 5hmC,30–36 the current approach neither involves any restriction enzyme nor requires sequence information about the (hydroxyl)methylated loci. Thus, it can detect any (hydroxyl)methylation cytosines in ssDNA without knowing the sequences of the testing samples. The features of quick execution, ease of implementation and low cost makes it highly suitable for high-throughput screening of DNA fragments that contain (hydroxy)methylated cytosines. In particular, in combination with enzymatic digestion, this approach should find applications in screening (hydroxy)methylated DNAs in massive genomic DNA samples.The integration of host–guest interactions in the nanopore system is a novel and useful strategy, complementary to existing nanopore sensing techniques.38,39,43,49,50 Apart from 5mC and 5hmC, other modified bases such as 8-oxo-guanine,51 N6-methyladenine,19 and the recently identified 5-carboxylcytosine and 5-formylcytosine52 could also be selectively modified and detected using the same approach. Overall, we expect the sensing strategy presented in this work to be practically useful in nanopore sensing, particularly in the detection of DNA modifications and early diagnosis of diseases.
Acknowledgements
We thank Q. S. Liu (IHEP) for providing the αHL mutants used in this work. This project was funded by National Basic Research Program of China (973 program, numbers 2013CB932800), the National Natural Science Foundation of China (numbers 21175135, 21375130, 21205119, 21475132), and the CAS Hundred Talents Program.Notes and references
- W. Reik and J. Walter, Nat. Rev. Genet., 2001, 2, 21–32 CrossRef CAS PubMed.
- K. D. Robertson and A. P. Wolffe, Nat. Rev. Genet., 2000, 1, 11–19 CrossRef CAS PubMed.
- B. E. Bernstein, A. Meissner and E. S. Lander, Cell, 2007, 128, 669–681 CrossRef CAS PubMed.
- S. H. Feng, S. E. Jacobsen and W. Reik, Science, 2010, 330, 622–627 CrossRef CAS PubMed.
- M. Esteller, Oncogene, 2002, 21, 5427–5440 CrossRef CAS PubMed.
- E. N. Gal-Yam, Y. Saito, G. Egger and P. A. Jones, Annu. Rev. Med., 2008, 59, 267–280 CrossRef CAS PubMed.
- H. Heyn and M. Esteller, Nat. Rev. Genet., 2012, 13, 679–692 CrossRef CAS PubMed.
- S. Kriaucionis and N. Heintz, Science, 2009, 324, 929–930 CrossRef CAS PubMed.
- M. Tahiliani, K. P. Koh, Y. H. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 2009, 324, 930–935 CrossRef CAS PubMed.
- N. Rusk, Nat. Methods, 2012, 9, 646 CrossRef PubMed.
- M. R. Branco, G. Ficz and W. Reik, Nat. Rev. Genet., 2012, 13, 7–13 CAS.
- P. W. Laird, Nat. Rev. Genet., 2010, 11, 191–203 CrossRef CAS PubMed.
- M. Frommer, L. E. Mcdonald, D. S. Millar, C. M. Collis, F. Watt, G. W. Grigg, P. L. Molloy and C. L. Paul, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 1827–1831 CrossRef CAS.
- C. Grunau, S. J. Clark and A. Rosenthal, Nucleic Acids Res., 2001, 29, E65 CrossRef CAS PubMed.
- B. Khulan, R. F. Thompson, K. Ye, M. J. Fazzari, M. Suzuki, E. Stasiek, M. E. Figueroa, J. L. Glass, Q. Chen, C. Montagna, E. Hatchwell, R. R. Selzer, T. A. Richmond, R. D. Green, A. Melnick and J. M. Greally, Genome Res., 2006, 16, 1046–1055 CrossRef CAS PubMed.
- R. A. Irizarry, C. Ladd-Acosta, B. Carvalho, H. Wu, S. A. Brandenburg, J. A. Jeddeloh, B. Wen and A. P. Feinberg, Genome Res., 2008, 18, 780–790 CrossRef CAS PubMed.
- D. Serre, B. H. Lee and A. H. Ting, Nucleic Acids Res., 2010, 38, 391–399 CrossRef CAS PubMed.
- J. Eid, A. Fehr, J. Gray, K. Luong, J. Lyle, G. Otto, P. Peluso, D. Rank, P. Baybayan, B. Bettman, A. Bibillo, K. Bjornson, B. Chaudhuri, F. Christians, R. Cicero, S. Clark, R. Dalal, A. Dewinter, J. Dixon, M. Foquet, A. Gaertner, P. Hardenbol, C. Heiner, K. Hester, D. Holden, G. Kearns, X. Kong, R. Kuse, Y. Lacroix, S. Lin, P. Lundquist, C. Ma, P. Marks, M. Maxham, D. Murphy, I. Park, T. Pham, M. Phillips, J. Roy, R. Sebra, G. Shen, J. Sorenson, A. Tomaney, K. Travers, M. Trulson, J. Vieceli, J. Wegener, D. Wu, A. Yang, D. Zaccarin, P. Zhao, F. Zhong, J. Korlach and S. Turner, Science, 2009, 323, 133–138 CrossRef CAS PubMed.
- B. A. Flusberg, D. R. Webster, J. H. Lee, K. J. Travers, E. C. Olivares, T. A. Clark, J. Korlach and S. W. Turner, Nat. Methods, 2010, 7, 461–465 CrossRef CAS PubMed.
- C. X. Song, T. A. Clark, X. Y. Lu, A. Kislyuk, Q. Dai, S. W. Turner, C. He and J. Korlach, Nat. Methods, 2012, 9, 75–77 CrossRef CAS PubMed.
- M. J. Booth, M. R. Branco, G. Ficz, D. Oxley, F. Krueger, W. Reik and S. Balasubramanian, Science, 2012, 336, 934–937 CrossRef CAS PubMed.
- J. J. Kasianowicz, E. Brandin, D. Branton and D. W. Deamer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13770–13773 CrossRef CAS.
- J. Clarke, H. C. Wu, L. Jayasinghe, A. Patel, S. Reid and H. Bayley, Nat. Nanotechnol., 2009, 4, 265–270 CrossRef CAS PubMed.
- G. M. Cherf, K. R. Lieberman, H. Rashid, C. E. Lam, K. Karplus and M. Akeson, Nat. Biotechnol., 2012, 30, 344–348 CrossRef CAS PubMed.
- E. A. Manrao, I. M. Derrington, A. H. Laszlo, K. W. Langford, M. K. Hopper, N. Gillgren, M. Pavlenok, M. Niederweis and J. H. Gundlach, Nat. Biotechnol., 2012, 30, 349–353 CrossRef CAS PubMed.
- D. Wendell, P. Jing, J. Geng, V. Subramaniam, T. J. Lee, C. Montemagno and P. Guo, Nat. Nanotechnol., 2009, 4, 765–772 CrossRef CAS PubMed.
- S. Howorka and Z. Siwy, Chem. Soc. Rev., 2009, 38, 2360–2384 RSC.
- B. M. Venkatesan and R. Bashir, Nat. Nanotechnol., 2011, 6, 615–624 CrossRef CAS PubMed.
- F. Haque, J. Li, H.-C. Wu, X.-J. Liang and P. Guo, Nano Today, 2013, 8, 56–74 CrossRef CAS PubMed.
- E. V. B. Wallace, D. Stoddart, A. J. Heron, E. Mikhailova, G. Maglia, T. J. Donohoe and H. Bayley, Chem. Commun., 2010, 46, 8195–8197 RSC.
- M. Wanunu, D. Cohen-Karni, R. R. Johnson, L. Fields, J. Benner, N. Peterman, Y. Zheng, M. L. Klein and M. Drndic, J. Am. Chem. Soc., 2011, 133, 486–492 CrossRef CAS PubMed.
- W. W. Li, L. Z. Gong and H. Bayley, Angew. Chem., Int. Ed., 2013, 52, 4350–4355 CrossRef CAS PubMed.
- J. Shim, G. I. Humphreys, B. M. Venkatesan, J. M. Munz, X. Q. Zou, C. Sathe, K. Schulten, F. Kosari, A. M. Nardulli, G. Vasmatzis and R. Bashir, Sci. Rep., 2013, 3, 1389 Search PubMed.
- I. Kang, Y. Wang, C. Reagan, Y. M. Fu, M. X. Wang and L. Q. Gu, Sci. Rep., 2013, 3, 2381 Search PubMed.
- A. H. Laszlo, I. M. Derrington, H. Brinkerhoff, K. W. Langford, I. C. Nova, J. M. Samson, J. J. Bartlett, M. Pavlenok and J. H. Gundlach, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18904–18909 CrossRef CAS PubMed.
- Z. L. Wescoe, J. Schreiber and M. Akeson, J. Am. Chem. Soc., 2014, 136, 16582–16587 CrossRef CAS PubMed.
- L. Liu, C. Yang, K. Zhao, J. Y. Li and H. C. Wu, Nat. Commun., 2013, 4, 2989 Search PubMed.
- N. Mitchell and S. Howorka, Angew. Chem., Int. Ed., 2008, 47, 5565–5568 CrossRef CAS PubMed.
- N. An, A. M. Fleming, H. S. White and C. J. Burrows, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11504–11509 CrossRef CAS PubMed.
- K. Tanaka, K. Tainaka, T. Kamei and A. Okamoto, J. Am. Chem. Soc., 2007, 129, 5612–5620 CrossRef CAS PubMed.
- S. Bareyt and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 181–184 CrossRef CAS PubMed.
- M. Munzel, L. Lercher, M. Muller and T. Carell, Nucleic Acids Res., 2010, 38, e192 CrossRef PubMed.
- Y. Wang, D. L. Zheng, Q. L. Tan, M. X. Wang and L. Q. Gu, Nat. Nanotechnol., 2011, 6, 668–674 CrossRef CAS PubMed.
- M. Soskine, A. Biesemans, M. De Maeyer and G. Maglia, J. Am. Chem. Soc., 2013, 135, 13456–13463 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- G. Maglia, M. R. Restrepo, E. Mikhailova and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 19720–19725 CrossRef CAS PubMed.
- L. Q. Gu, S. Cheley and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15498–15503 CrossRef CAS PubMed.
- E. Mezzina, F. Cruciani, G. F. Pedulli and M. Lucarini, Chem.–Eur. J., 2007, 13, 7223–7233 CrossRef CAS PubMed.
- F. Haque, J. Lunn, H. Fang, D. Smithrud and P. Guo, ACS Nano, 2012, 6, 3251–3261 CrossRef CAS PubMed.
- S. Wang, F. Haque, P. G. Rychahou, B. M. Evers and P. Guo, ACS Nano, 2013, 7, 9814–9822 CrossRef CAS PubMed.
- A. E. P. Schibel, N. An, Q. A. Jin, A. M. Fleming, C. J. Burrows and H. S. White, J. Am. Chem. Soc., 2010, 132, 17992–17995 CrossRef CAS PubMed.
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Science, 2011, 333, 1300–1303 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc01436k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |