
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomimetic versus enzymatic high-potential electrocatalytic reduction of hydrogen peroxide on a functionalized carbon nanotube electrode†
Bertrand
Reuillard
,
Solène
Gentil
,
Marie
Carrière
,
Alan
Le Goff
 * and
Serge
Cosnier
* and
Serge
Cosnier
Univ. Grenoble Alpes, DCM UMR 5250, F-38000 Grenoble, France. and CNRS, DCM UMR 5250, F-38000 Grenoble, France. E-mail: alan.le-goff@ujf-grenoble.fr
First published on 22nd May 2015
Abstract
We report the non-covalent functionalization of a multi-walled carbon nanotube (MWCNT) electrode with a biomimetic model of the horseradish peroxidase (HRP) active site. By modifying the MWCNT electrode surface with imidazole-modified polypyrrole, a new biomimetic complex of HRP was synthesized on the MWCNT sidewalls via the coordination of imidazole (Im) to the metal centre of iron protoporphyrin IX, affording (Im)(PP)FeIII. Compared to the pi-stacking of non-coordinated (PP)FeIII on a MWCNT electrode, the (Im)(PP)FeIII-modified MWCNT electrode exhibits higher electrocatalytic activity with an Imax = 0.52 mA cm−2 for the reduction of H2O2, accompanied by a high onset potential of 0.43 V vs. Ag/AgCl. The performances of these novel surface-confined HRP mimics were compared to those of a MWCNT electrode modified by HRP. Although the enzyme electrode displays a higher electrocatalytic activity towards H2O2 reduction, the (Im)(PP)FeIII-modified MWCNT electrode exhibits a markedly higher operational stability, retaining 63% of its initial activity after one month.
Introduction
Horseradish peroxidase (HRP) is the classical enzyme usually used to detect and reduce H2O2 as it is its natural substrate to detoxify media through oxidation of different natural organic substrates.1 The crystal structure of this protein, along with studies of the mechanism of the catalytic cycle, have shown that the catalytic activity is mainly due to the stabilization of an iron oxo bond (FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)2,3 within its cofactor, iron protoporphyrin IX or hemin. This stabilization is due to the presence of specific amino acids surrounding the active site, two histidine groups (His170 and His42) and an arginine residue (Arg38) stabilize the high oxidation state of the iron metal centre and also act as proton relays. The instability of this high oxidation state compound makes it difficult to study. Nevertheless, the redox potential of this reactive species has been evaluated to be around 0.7 V vs. SCE at pH 7 with minor variations according to the protein type.4 HRP has been among the most studied enzymes for H2O2 biosensing applications. Recently, we and others have been able to achieve efficient electron transfer between an electrode and an enzyme for electrocatalytic or biosensing means, leading to high electrocatalytic current densities accompanied by low overpotentials.5–7 The reduction of H2O2 at redox potentials as high as 0.5 V at pH 7, has led to the design of oxygen-reducing biocathodes through the combination of glucose oxidase (producing H2O2 while oxidizing glucose) and HRP, which performs the reduction of H2O2 to H2O.8–11 These oxygen-reducing biocathodes achieve the global 4H+/4e− reduction of oxygen with onset potentials of 0.42 V vs. SCE at pH 7, which make this bienzymatic system a good candidate to replace high potential multicopper oxidase12 in biofuel cell biocathodes.
O)2,3 within its cofactor, iron protoporphyrin IX or hemin. This stabilization is due to the presence of specific amino acids surrounding the active site, two histidine groups (His170 and His42) and an arginine residue (Arg38) stabilize the high oxidation state of the iron metal centre and also act as proton relays. The instability of this high oxidation state compound makes it difficult to study. Nevertheless, the redox potential of this reactive species has been evaluated to be around 0.7 V vs. SCE at pH 7 with minor variations according to the protein type.4 HRP has been among the most studied enzymes for H2O2 biosensing applications. Recently, we and others have been able to achieve efficient electron transfer between an electrode and an enzyme for electrocatalytic or biosensing means, leading to high electrocatalytic current densities accompanied by low overpotentials.5–7 The reduction of H2O2 at redox potentials as high as 0.5 V at pH 7, has led to the design of oxygen-reducing biocathodes through the combination of glucose oxidase (producing H2O2 while oxidizing glucose) and HRP, which performs the reduction of H2O2 to H2O.8–11 These oxygen-reducing biocathodes achieve the global 4H+/4e− reduction of oxygen with onset potentials of 0.42 V vs. SCE at pH 7, which make this bienzymatic system a good candidate to replace high potential multicopper oxidase12 in biofuel cell biocathodes.
Interesting approaches have also focused on using inorganic complexes mimicking the HRP active site. For more than 30 years now, porphyrin or phthalocyanin iron(III) have been used to electrochemically reduce O2 and H2O2 on different types of electrodes.13–17 The group of Fukuzumi has recently published several functional H2O2 fuel cells based on iron porphyrin peroxidase mimics.18,19 The possibility of oxidizing H2O2 at low potential and reducing it at high potential gives the possibility of generating a sufficient voltage, using only H2O2 as the fuel for both the anode and cathode. However, all these iron porphyrin complexes perform H2O2 electrocatalytic reduction at lower turnover frequencies and lower potential compared to HRP. This enzyme has been proven to reduce H2O2 at an overvoltage of 0.55 V compared to the thermodynamic H2O2/H2O redox potential.5,10 The difference in catalytic activity is most likely due to the lack of assistance from the surrounding ligands in these complexes compared to the heme enzyme active site. For years now, several groups have also tried to mimic more closely the active site pocket of hemoproteins by modifying the iron porphyrin skeleton or adding an external ligand.20–23 For instance, Collman's group has described elegant examples of the reconstitution of the active site of Cytochrome c Oxidase (CcO), where the iron porphyrin is usually directly modified by an imidazole ligand that also helps the complexation of an additional copper centre, forming a dinuclear biomimetic Fe–Cu complex. Similar studies have also focused on synthesized HRP models, either through the direct modification of the hemin core with imidazole residues or by adding another chelating reagent, to bind to the iron cation.24,25 All of these contributions clearly show that the nature of the axial ligand coordinating to the iron center is crucial in the stabilization of the high oxidation state species. Axial imidazole ligands mimic the histidine amino acid of the protein in the stabilization of the iron peroxo, hydroperoxo and oxo species.26–29
Carbon nanotubes (CNTs) have been widely used for biomimetic catalyst immobilization30–34 and especially for electrocatalytic applications. Their ability to immobilize large amounts of redox catalysts and improve heterogeneous electron transfer rates make them a suitable platform to elaborate efficient molecular electrocatalytic materials. CNTs can be functionalized by many different covalent and non-covalent methods increasing the scope of their use, especially for the activation of small molecules. In particular, metal porphyrins display strong pi-stacking interactions with CNTs, which allow their stable and intimate binding33 and hence their use in the oxygen reduction reaction.35,36 Furthermore, recent studies have reported the covalent functionalization of CNTs by chelating units like pyridine and imidazole groups, enabling the coordination of iron(III) porphyrin derivatives.37,38 These heme enzyme biomimetic models exhibited excellent catalytic properties toward the oxygen reduction reaction, but at high overvoltages of 0.8 V for H2O2 reduction.37
This work describes the synthesis of an original pyrrole-imidazole monomer that enables non-covalent modification of multi-walled carbon nanotubes (MWCNTs) via oxidative electropolymerization of the pyrrole subunit. Once the electrode is fully covered by the poly[imidazole-pyrrole] (p-Im) film, the addition of iron protoporphyrin IX, (PP)FeIII, enables the formation of a biomimetic species through the coordination of the imidazole ligand to the iron(III) centre. The “on-CNT” synthesis of (imidazole)(protoporphyrinato) iron(III) ((Im)(PP)FeIII) HRP biomimetic complex, forming a poly-[(imidazole)(protoporphyrinato) iron(III)] polymer p-[(Im)(PP)FeIII], was then compared with the pi-stacking of free (PP)FeIII on pristine MWCNT sidewalls and with the direct wiring of HRP immobilized on a MWCNT electrode. We then investigated the performance of these different electrodes towards the electrocatalytic reduction of hydrogen peroxide.
Results and discussion
Functionalization of MWCNT electrodes with free (PP)FeIII and p-[(Im)(PP)FeIII]
The functionalized MWCNT electrodes were prepared in several steps. For the preparation of the poly[imidazole-pyrrole]-functionalized MWCNT electrode (Fig. 1A), p-Im-MWCNT, the modified electrode was first functionalized with the imidazole-pyrrole monomer via oxidative electropolymerization performed in acetonitrile (MeCN) + 0.1 M TBAP. Fig. 1B shows the electropolymerization process. 80 CV scans were performed from 0 to 0.9 V vs. Ag/AgNO3 to ensure full coverage of MWCNT sidewalls with the poly-[imidazole-pyrrole] polymer.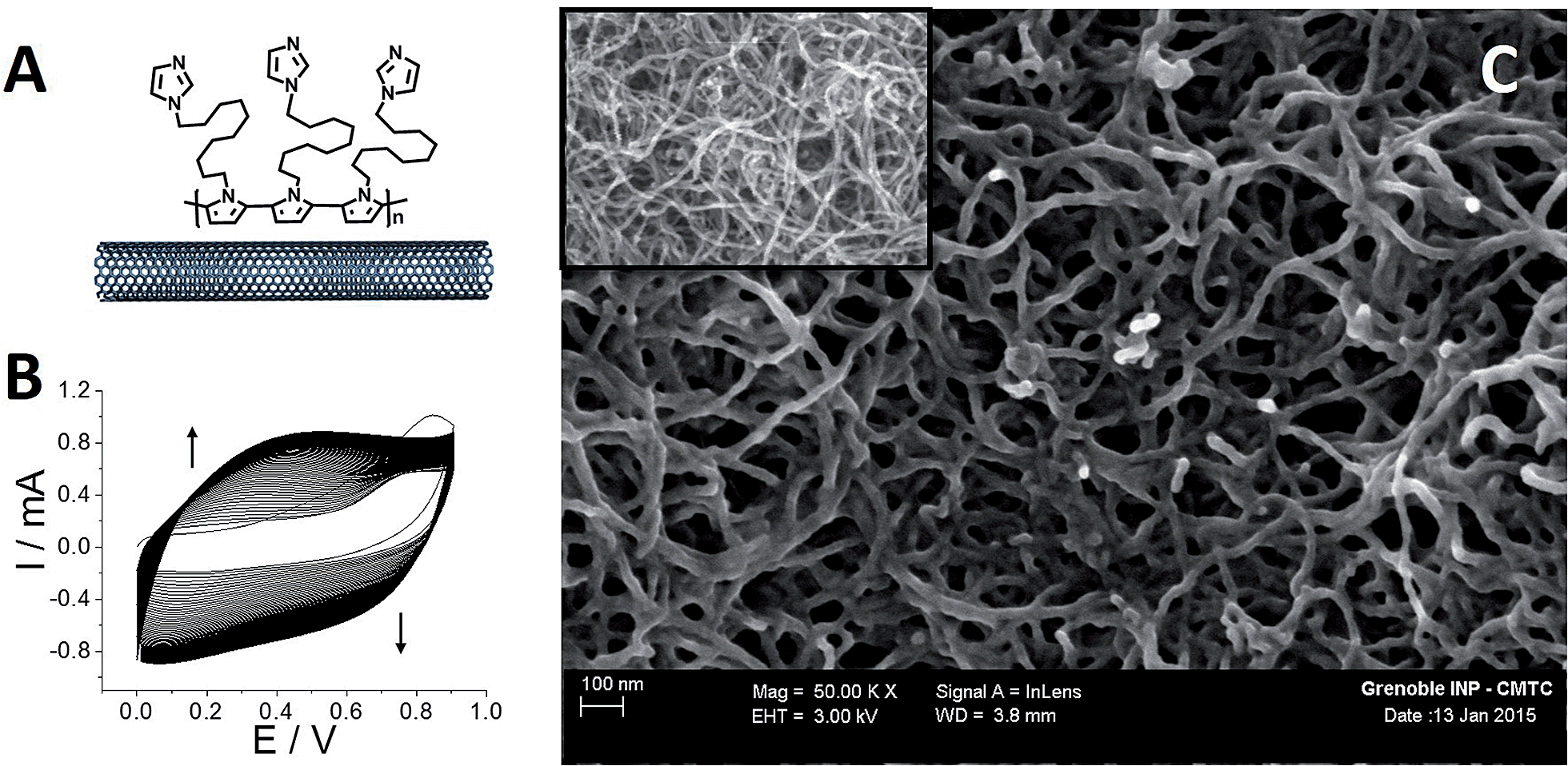 | ||
| Fig. 1 (A) Schematic representation of [poly-imidazole]-pyrrole; (B) oxidative electropolymerization of 1-(11-(1H-pyrrol-1-yl)undecyl)-1H-imidazole (2 mM) by repeated potential scanning (80 scans) over the range 0–0.9 V in MeCN + 0.1 M TBAP (v = 100 mV s−1); (C) SEM image of a MWCNT electrode after electrodeposition of [poly-imidazole]-pyrrole, (inset) SEM image of a pristine MWCNT electrode. | ||
The evolution of the thickness of the poly-[imidazole-pyrrole] film is correlated to the increase of the redox signal at around 0.4 V, corresponding to the electroactivity of the polypyrrole backbone. Fig. 1C displays SEM images of the pristine MWCNT film and the resulting p-Im-MWCNT electrodes. These images confirm the homogenous deposition of a few-nanometer-thin layer of poly[imidazole-pyrrole] on the 10 nm-diameter MWCNTs. It is noteworthy that the highly porous MWCNT nanostructure is preserved during the electropolymerization process.
Then, both pristine and p-Im-MWCNT electrodes were incubated in a 10 mM (PP)FeIII DMF solution. Cyclic voltammetry was performed in 0.1 M phosphate buffer to investigate the redox response corresponding to the Fe(III)/Fe(II) redox couple (Fig. 2). In both cases, one reversible peak system was obtained with E1/2 = −0.34 V for (PP)FeIII and −0.24 V for p-[(Im)(PP)FeIII]. The higher capacitive current for p-[(Im)(PP)FeIII] arises from the presence of the conjugated polypyrrole film. In addition, these redox systems have a linear dependence of both anodic and cathodic current intensity on scan rate, confirming the immobilization of (PP)FeIII on the electrodes (Fig. 3).
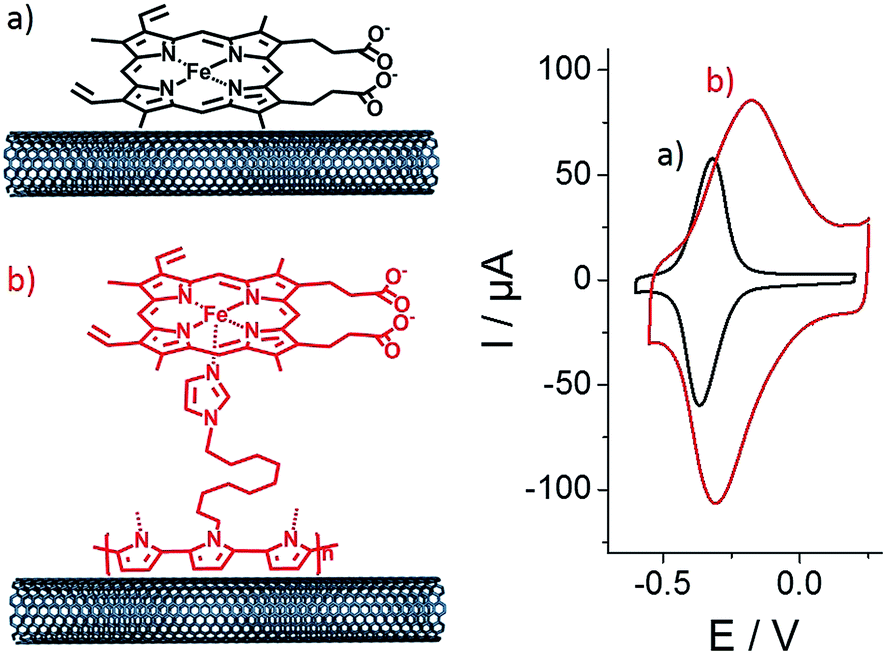 | ||
| Fig. 2 Schematic representation and associated cyclic voltammetry for (a) (PP)FeIII-functionalized MWCNT electrodes and (b) p-[(Im)(PP)FeIII]-functionalized MWCNT electrodes in phosphate buffer at pH 7. Electrodes were functionalized by incubation in DMF containing (PP)FeIII (10 mM), v = 10 mV s−1. | ||
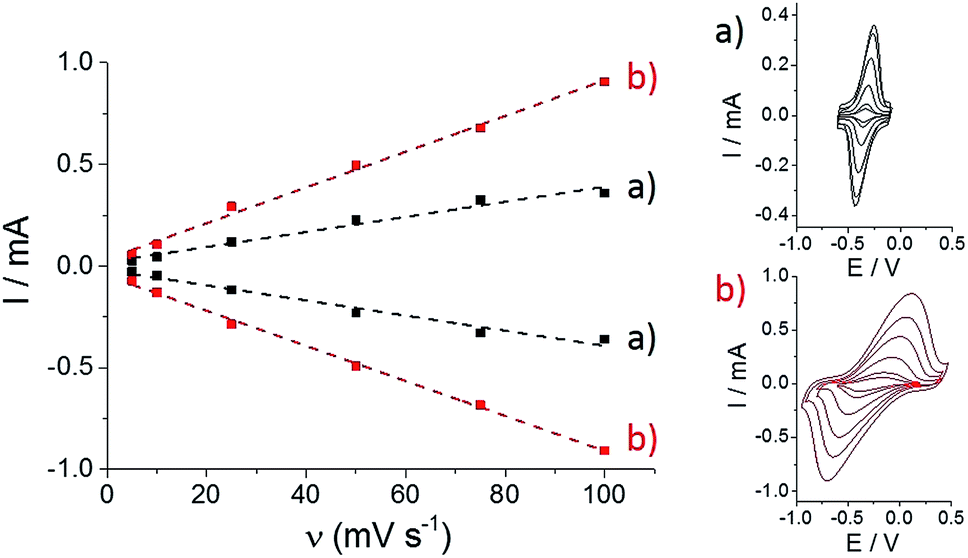 | ||
| Fig. 3 Evolution of the increase in anodic and cathodic peak currents with scan rate for (a) (PP)FeIII-functionalized MWCNT electrodes and (b) p-[(Im)(PP)FeIII]-functionalized MWCNT electrodes in phosphate buffer (pH 7). | ||
As observed by CV in Fig. 3, ΔE for the Fe(III)/Fe(II) redox couple also increases with scan rate. According to the Laviron equation for interfacial electron transfer in adsorbed redox systems,39 ΔE variations correspond to a ks of 0.35 (±0.5) for (PP)FeIII directly adsorbed on MWCNT sidewalls and 0.12 (±0.5) s−1 for p-[(Im)(PP)FeIII]. The latter exhibits a slower electron transfer rate, which likely arises from the presence of the partially-insulating poly-[imidazole-pyrrole] layer. While (PP)FeIII is immobilized by taking advantage of the pi–pi interactions between the porphyrin ring and MWCNT sidewalls, coordination of imidazole to the iron centre also allows immobilization of the (PP)FeIII through the formation of the [(Im)(PP)FeIII] complex on functionalized MWCNTs. A 100 mV shift towards a positive potential for the p-[(Im)(PP)FeIII] redox potential is in good agreement with the bottom-up synthesis of an (imidazole)(protoporphyrinato) iron(III) complex on the electrode.
We further investigated the association behavior of (PP)FeIII by pi–pi interactions and by imidazole ligand coordination on MWCNT surfaces. Fig. 4 shows the influence of the concentration of the (PP)FeIII incubation solution on the final porphyrin iron(III) apparent surface coverage. The surface concentrations were estimated by integrating the charge under the anodic or cathodic peak for both of the functionalized-MWCNT electrodes (inset, Fig. 4). For both MWCNT electrodes, the apparent surface coverage of the iron(III) complex increases with the increasing (PP)FeIII concentration of the incubation solution. In the case of p-(Im)(PP)FeIII, a shoulder corresponding to a small redox system around −0.35 V indicates that the pi-stacking of a small amount of (PP)FeIII on MWCNTs cannot be avoided, despite the presence of the polymer layer.
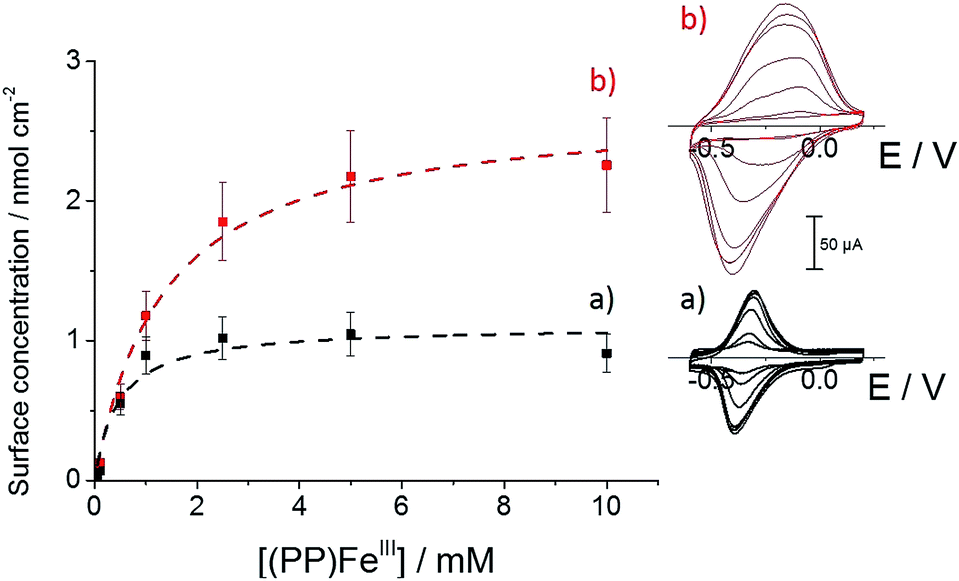 | ||
| Fig. 4 Variation of (a) (PP)FeIII and (b) (Im)(PP)FeIII surface coverage with (PP)FeIII incubation solution concentration in DMF, accompanied by fitting binding isotherms (dashed lines); (inset) associated cyclic voltammetry for (a) (PP)FeIII-functionalized MWCNT electrodes and (b) p-[(Im)(PP)FeIII]-functionalized MWCNT electrodes obtained after incubation in a DMF solution of (PP)FeIII (0.05, 0.1, 0.5, 1, 2.5, 5 and 10 mM) (v = 10 mV s−1, 0.1 M phosphate buffer, pH 7). | ||
For both types of electrode, this increase follows a simple Langmuir binding isotherm, according to the equation:
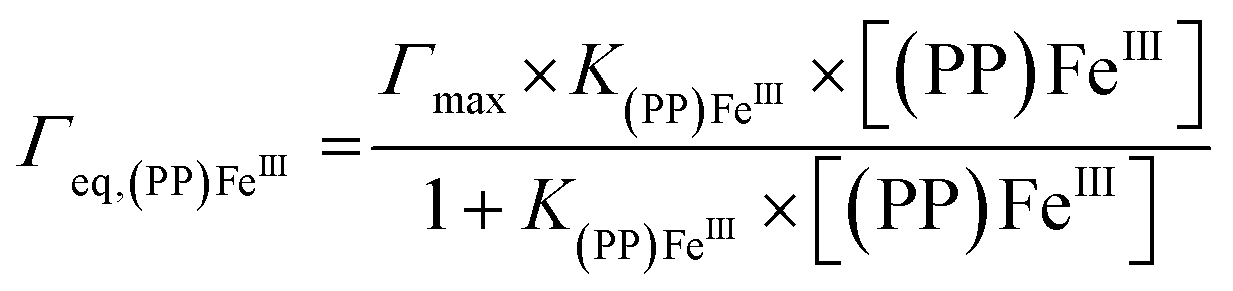
Electrocatalytic H2O2 reduction: biomimetic vs. enzymatic electrocatalysis
The electrocatalytic properties of these modified MWCNT electrodes were investigated towards H2O2 reduction. The performances of these biomimetic MWCNT electrodes were compared with those of MWCNT electrodes functionalized with HRP. This enzyme was immobilized through the formation of a boronic ester covalent linkage with a pyrene-boronic-acid derivative.10Fig. 5 displays the CV data for the three different MWCNT electrodes in the presence of 10 mM H2O2. | ||
| Fig. 5 Cyclic voltammetry for (a) (PP)FeIII-functionalized MWCNT electrodes, (b) p-[(Im)(PP)FeIII]-functionalized MWCNT electrodes and (c) HRP-functionalized MWCNT electrodes in the absence and presence of 10 mM H2O2 (v = 10 mV s−1, phosphate buffer, pH 7). | ||
All three electrodes exhibited electrocatalysis of both H2O2 reduction and oxidation. Onset potentials of +0.30 (±0.01), +0.43 (±0.02) and +0.59 (±0.02) V were measured for (PP)FeIII, p-[(Im)(PP)FeIII] and HRP respectively (see Fig. 4 and Table 1). Chronoamperometric measurements were performed at a fixed potential of 0.30 V in the presence of different concentrations of H2O2 (Fig. 6A). As expected, the HRP-based electrode was able to reduce H2O2 at low overpotentials with an excellent maximum current density of 2.1 mA cm−2. The p-[(Im)(PP)FeIII]-based electrode exhibited an onset potential of 0.43 (±0.02) V, representing an intermediate value between the (PP)FeIII-based electrode and the HRP-based electrode and confirming that coordination of imidazole results in the increase in the redox potential of the Fe(IV)O intermediate.
| MWCNT electrode | E 1/2(FeIII/FeII) (V) | E onset (V) | I max at 0.3 V (mA cm−2) |
|---|---|---|---|
| (PP)FeIII | −0.34 (±0.01) | +0.30 (±0.01) | 0.050 (±0.003) |
| p-[(Im)(PP)FeIII] | −0.24 (±0.01) | +0.43 (±0.02) | 0.52 (±0.06) |
| HRP | −0.10 | +0.59 (±0.02) | 2.1 (±0.1) |
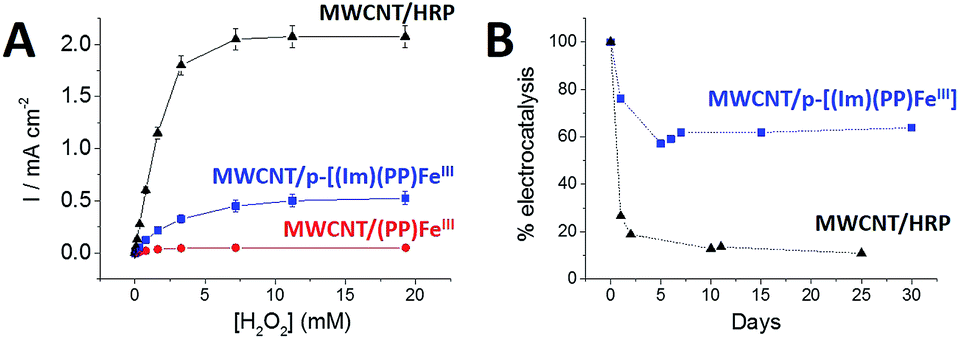 | ||
| Fig. 6 (A) Cathodic catalytic current densities as a function of H2O2 concentration recorded at the (●) (PP)FeIII-functionalized MWCNT electrode, (■) p-[(Im)(PP)FeIII]-functionalized MWCNT electrode and (▲) HRP-functionalized MWCNT electrode. Chronoamperometric experiments were performed at a fixed potential of 0.3 V in 0.1 M phosphate buffer pH 7. (B) Evolution of the maximum catalytic current density for the (■) p-[(Im)(PP)FeIII]-functionalized MWCNT electrode and the (▲) HRP-functionalized MWCNT electrode as a function of time. Error bars were estimated from experiments performed on three electrodes. | ||
Furthermore, p-[(Im)(PP)FeIII] exhibited an excellent current density for H2O2 reduction of 0.52 mA cm−2 accompanied by excellent stability (Fig. 6B). An apparent turnover frequency (TOF) of 4.8 s−1 was estimated from the (Im)(PP)FeIII surface coverage. Although the p-[(Im)(PP)FeIII]-based electrode exhibits a lower maximum current density (0.52 mA cm−2) for H2O2 reduction than the HRP-based electrode, the biomimetic electrode displays a markedly better operational stability. It appears that the HRP-based electrode retains only 11% of its initial electroactivity after 25 days whereas the p-[(Im)(PP)FeIII]-functionalized MWCNT electrode still retains 63% of its initial activity after one month (Fig. 6B).
Conclusions
These functionalized MWCNT electrodes, from the pi-stacked free (PP)FeIII cofactor to the immobilized directly-wired HRP, all have in common the surface-confined (PP)FeIII core. The functionalization of MWCNTs with an imidazole-modified polymer allows both the immobilization of (PP)FeIII and the modification of the iron(III) coordination sphere. The electrochemistry of these functionalized MWCNTs gets closer to the electrochemistry of HRP-based electrodes in terms of the redox potential of the Fe(III)/Fe(II) couple and the redox potentials of the Fe(IV)O intermediate, as confirmed by the onset potentials of the cathodic electrocatalytic wave in the presence of H2O2. Thanks to the high conductivity and high electroactive surfaces, these biomimetic electrodes constitute an efficient tool for the electrocatalytic reduction of H2O2. Furthermore, the improved stability of the biomimetic catalyst over weeks represents an important advantage in the design of a new generation of biocathodes and hence biofuel cells. However, there is still room for improvement to approach the true redox potential of the HRP Fe(IV)O catalytic intermediate. This could especially be achieved by providing a proton source in the vicinity of the iron centre. Thanks to the flexibility of these functionalization techniques, this work is underway, especially in the design of more sophisticated pyrrole monomers and porphyrin ligands.
Acknowledgements
The Région Rhones-Alpes is acknowledged for the PhD funding of B. Reuillard. The authors also thank the ANR-12-JS08-0006-01 PHOBIOS for financial support. The authors wish to acknowledge the support from the platform Chimie NanoBio ICMG FR 2607 (PCN-ICMG) and from the LabEx ARCANE (ANR-11-LABX-0003-01).Notes and references
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szöke, A. Henriksen and J. Hajdu, Nature, 2002, 417, 463–468 CrossRef CAS PubMed.
- J. N. Rodríguez-López, D. J. Lowe, J. Hernández-Ruiz, A. N. P. Hiner, F. García-Cánovas and R. N. F. Thorneley, J. Am. Chem. Soc., 2001, 123, 11838–11847 CrossRef PubMed.
- G. Battistuzzi, M. Bellei, C. A. Bortolotti and M. Sola, Arch. Biochem. Biophys., 2010, 500, 21–36 CrossRef CAS PubMed.
- W. Jia, S. Schwamborn, C. Jin, W. Xia, M. Muhler, W. Schuhmann and L. Stoica, Phys. Chem. Chem. Phys., 2010, 12, 10088–10092 RSC.
- M. Varničić, K. Bettenbrock, D. Hermsdorf, T. Vidaković-Koch and K. Sundmacher, RSC Adv., 2014, 4, 36471–36479 RSC.
- E. Suraniti, S. Ben-Amor, P. Landry, M. Rigoulet, E. Fontaine, S. Bottari, A. Devin, N. Sojic, N. Mano and S. Arbault, Angew. Chem., Int. Ed., 2014, 53, 6655–6658 CrossRef CAS PubMed.
- W. Jia, C. Jin, W. Xia, M. Muhler, W. Schuhmann and L. Stoica, Chem.–Eur. J., 2012, 18, 2783–2786 CrossRef CAS PubMed.
- C. Agnès, B. Reuillard, A. Le Goff, M. Holzinger and S. Cosnier, Electrochem. Commun., 2013, 34, 105–108 CrossRef PubMed.
- B. Reuillard, A. Le Goff, M. Holzinger and S. Cosnier, J. Mater. Chem. B, 2014, 2, 2228–2232 RSC.
- V. Krikstolaityte, Y. Oztekin, J. Kuliesius, A. Ramanaviciene, Z. Yazicigil, M. Ersoz, A. Okumus, A. Kausaite-Minkstimiene, Z. Kilic, A. O. Solak, A. Makaraviciute and A. Ramanavicius, Electroanalysis, 2013, 25, 2677–2683 CrossRef CAS PubMed.
- A. Le Goff, M. Holzinger and S. Cosnier, Cell. Mol. Life Sci., 2015, 72, 941–952 CrossRef CAS PubMed.
- T. Lötzbeyer, W. Schuhmann and H.-L. Schmidt, J. Electroanal. Chem., 1995, 395, 341–344 CrossRef.
- R. K. Raman and A. K. Shukla, J. Appl. Electrochem., 2005, 35, 1157–1161 CrossRef CAS.
- G. L. Turdean, I. C. Popescu, A. Curulli and G. Palleschi, Electrochim. Acta, 2006, 51, 6435–6441 CrossRef CAS PubMed.
- J. Chen, L. Zhao, H. Bai and G. Shi, J. Electroanal. Chem., 2011, 657, 34–38 CrossRef CAS PubMed.
- Y. Guo, J. Li and S. Dong, Sens. Actuators, B, 2011, 160, 295–300 CrossRef CAS PubMed.
- Y. Yamada, S. Yoshida, T. Honda and S. Fukuzumi, Energy Environ. Sci., 2011, 4, 2822–2825 CAS.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Inorg. Chem., 2014, 53, 1272–1274 CrossRef CAS PubMed.
- J. P. Collman, Inorg. Chem., 1997, 36, 5145–5155 CrossRef CAS.
- M. C. Feiters, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2000, 29, 375–384 RSC.
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS PubMed.
- R. Ricoux, Q. Raffy and J.-P. Mahy, C. R. Chim., 2007, 10, 684–702 CrossRef CAS PubMed.
- G. Ashkenasy, D. Margulies, C. E. Felder, A. Shanzer and L. S. Powers, Chem.–Eur. J., 2002, 8, 4017–4026 CrossRef CAS.
- Q. Wang, Z. Yang, M. Ma, C. K. Chang and B. Xu, Chem.–Eur. J., 2008, 14, 5073–5078 CrossRef CAS PubMed.
- J.-G. Liu, T. Ohta, S. Yamaguchi, T. Ogura, S. Sakamoto, Y. Maeda and Y. Naruta, Angew. Chem., Int. Ed., 2009, 48, 9262–9267 CrossRef CAS PubMed.
- J.-G. Liu, Y. Shimizu, T. Ohta and Y. Naruta, J. Am. Chem. Soc., 2010, 132, 3672–3673 CrossRef CAS PubMed.
- S. P. de Visser, J. S. Valentine and W. Nam, Angew. Chem., Int. Ed., 2010, 49, 2099–2101 CrossRef CAS PubMed.
- M. T. Kieber-Emmons, M. F. Qayyum, Y. Li, Z. Halime, K. O. Hodgson, B. Hedman, K. D. Karlin and E. I. Solomon, Angew. Chem., Int. Ed., 2012, 51, 168–172 CrossRef CAS PubMed.
- J. D. Blakemore, A. Gupta, J. J. Warren, B. S. Brunschwig and H. B. Gray, J. Am. Chem. Soc., 2013, 135, 18288–18291 CrossRef CAS PubMed.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371–1374 CrossRef CAS PubMed.
- B. Reuillard, A. L. Goff and S. Cosnier, Chem. Commun., 2014, 50, 11731–11734 RSC.
- K. Elouarzaki, A. Le Goff, M. Holzinger, J. Thery and S. Cosnier, J. Am. Chem. Soc., 2012, 14078–14085 CrossRef CAS PubMed.
- P. Kang, S. Zhang, T. J. Meyer and M. Brookhart, Angew. Chem., Int. Ed., 2014, 53, 8709–8713 CrossRef CAS PubMed.
- I. Hijazi, T. Bourgeteau, R. Cornut, A. Morozan, A. Filoramo, J. Leroy, V. Derycke, B. Jousselme and S. Campidelli, J. Am. Chem. Soc., 2014, 136, 6348–6354 CrossRef CAS PubMed.
- A. Morozan, S. Campidelli, A. Filoramo, B. Jousselme and S. Palacin, Carbon, 2011, 49, 4839–4847 CrossRef CAS PubMed.
- P.-J. Wei, G.-Q. Yu, Y. Naruta and J.-G. Liu, Angew. Chem., Int. Ed., 2014, 53, 6659–6663 CrossRef CAS PubMed.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. Gyu Kim, Q. Li, N. Park, M. Liu and J. Cho, Nat. Commun., 2013, 4, 2076 Search PubMed.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1979, 101, 19–28 CrossRef CAS.
- F. A. Walker, M.-W. Lo and M. T. Ree, J. Am. Chem. Soc., 1976, 98, 5552–5560 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc01473e |
| This journal is © The Royal Society of Chemistry 2015 |