
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSignal transduction and amplification through enzyme-triggered ligand release and accelerated catalysis†
Sean
Goggins
a,
Barrie J.
Marsh
b,
Anneke T.
Lubben
a and
Christopher G.
Frost
*a
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: C.G.Frost@bath.ac.uk; Fax: +44 (0)1225 386231; Tel: +44 (0)1225 386142
bAtlas Genetics, Derby Court, Epsom Square, White Horse Business Park, Trowbridge, Wiltshire, BA14 0XG, UK. E-mail: barrie.marsh@atlasgenetics.com; Tel: +44 (0)1225 717932
First published on 15th June 2015
Abstract
Signal transduction and signal amplification are both important mechanisms used within biological signalling pathways. Inspired by this process, we have developed a signal amplification methodology that utilises the selectivity and high activity of enzymes in combination with the robustness and generality of an organometallic catalyst, achieving a hybrid biological and synthetic catalyst cascade. A proligand enzyme substrate was designed to selectively self-immolate in the presence of the enzyme to release a ligand that can bind to a metal pre-catalyst and accelerate the rate of a transfer hydrogenation reaction. Enzyme-triggered catalytic signal amplification was then applied to a range of catalyst substrates demonstrating that signal amplification and signal transduction can both be achieved through this methodology.
Introduction
In nature, living organisms utilise signal transduction and signal amplification for selective and amplified chemical responses towards particular stimuli.1 These powerful systems are used for a number of purposes, such as visual excitation,2 blood clotting,3 and biosignalling.4 Typically, these biological responses are realised through enzyme cascade mechanisms.5 Naturally, the high signal amplification and rapid response times of these systems have inspired the development of a range of versatile, synthetic signal amplification systems for stimuli-responsive materials,6 supramolecular signalling,7 and sensing.8 With regard to the latter, approaches towards signal amplification for sensing vary tremendously with nanomaterials,9 conjugated polymers,10 and supramolecular aggregates,11 all being successfully incorporated into analyte detection methodologies and achieving low limits of detection. Recently, the introduction of self-immolative dendrimers,12 has propelled the development of small molecule-based signal amplification protocols.13 In particular, the dendritic chain reaction (DCR) developed by Sella and Shabat,14 and the two-component system developed by Baker and Phillips,15 have both demonstrated autoinductive and autocatalytic exponential signal amplification for the detection of a number of analytes.16 Despite the high amplification achieved and the thermal stability of the reagents, these methods suffer from high background reactions due to reagent hydrolysis and long reaction times.17 As such, there is still a need for stable and selective reagents that can provide rapid signal amplification.The ability of catalysts to generate multiple product molecules without themselves being chemically altered has seen them integrated as the amplifier component within multiple signal amplification methodologies.18 Signal amplification by supramolecular catalysis (SAAC),19 has by far become the most popular catalytic approach to signal amplification due to the plethora of supramolecular analyte receptors and synthetic organometallic catalysts available that can be combined to create an effective amplified detection assay.20 However, turnover numbers are inadequate and the assays often require the use of organic solvents under an inert atmosphere. Contrastingly, a range of iridium-based, water-soluble organometallic complexes have been synthesised and shown to perform a number of organic transformations “in- or on-water”.21 Iridium-catalysed transfer hydrogenation in particular, is of significant interest due to its low catalytic loading, fast reaction times and high substrate conversion.22 To the best of our knowledge, these outstanding catalyst attributes are yet to be harnessed for signal amplification within a sensing methodology. Herein, we describe an enzyme-triggered iridium-catalysed transfer hydrogenation for signal transduction and amplification.
Results and discussion
Concept
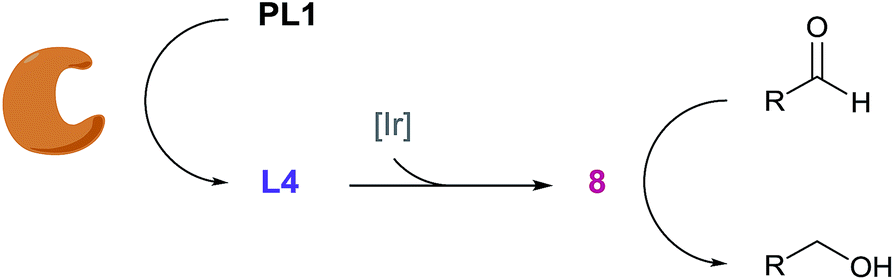
Inspired by biosignalling enzyme cascades, we designed an enzyme-activated synthetic catalyst cascade that could be used for signal transduction and signal amplification purposes (Fig. 1). In principle, this would combine the selectivity and high activity of an enzyme plus add the control and scope of a synthetic catalyst. To gain proof of concept, we initially looked into identifying an efficient catalyst that we could employ as both our signal transducer and signal amplifier. Also, we required a catalyst that could be selectively switched on in the presence of an enzyme. One catalytic system that we believed fulfilled these criteria was an iridium-based transfer hydrogenation catalyst, which has been shown to efficiently reduce aldehydes in aqueous media.22 Not only this, but the addition of a ligand was shown to increase the turnover frequency (TOF) of the catalyst 600-fold, which we could potentially manipulate for catalyst activation.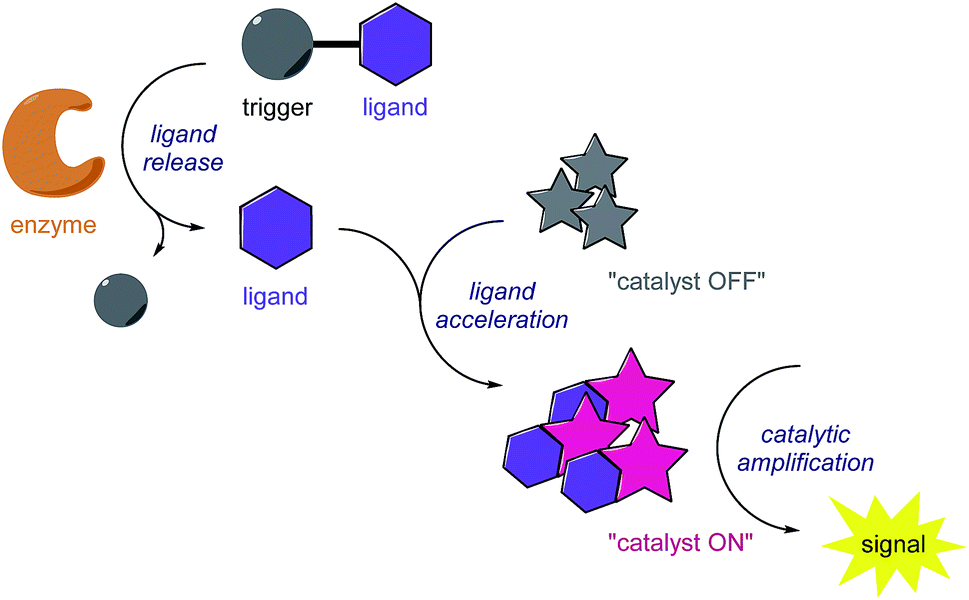 | ||
| Fig. 1 Signal amplification concept through enzyme-triggered ligand release and subsequent ligand-accelerated organometallic catalysis. | ||
To begin our investigation, we looked to use an electroactive aldehyde as the catalyst substrate as this would allow reaction progress to be monitored ‘real-time’ through simple electrochemical analysis without the need for compound extraction or isolation. The stability of ferrocene in aqueous media, its compatibility with biological components and its favourable electrochemical properties made ferrocenecarboxaldehyde 1 an obvious initial choice.23 Electrochemical analysis of 1, and its corresponding reduced product, ferrocenemethanol 2, using differential pulse voltammetry (DPV), revealed that aldehyde 1 and alcohol 2 have considerably different oxidation potentials (ΔEox ≈ 300 mV) (Fig. 2). As no peak overlap was observed, peak integration could be used to calculate reaction conversion allowing reaction progress to be monitored using ratiometric electrochemical analysis. Ratiometric sensing has the benefit of obtaining reaction conversions without internal or external standards, and also minimises errors associated with sampling and electrode or instrument variations. Thus, the reaction conversion of the transfer hydrogenation reaction can be calculated using eqn (1).
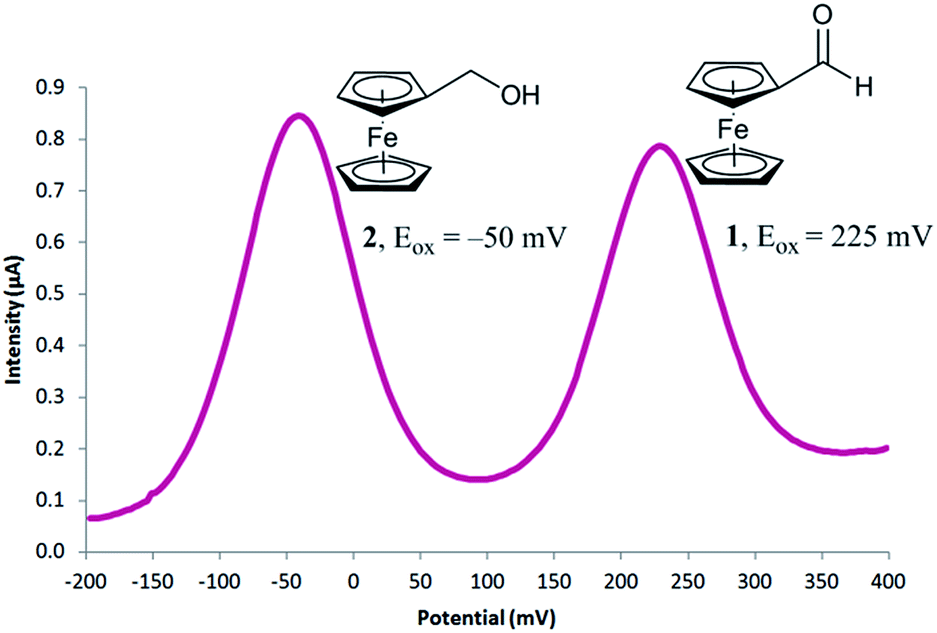 | ||
| Fig. 2 Differential pulse voltammogram obtained for a mixture of 0.25 mM ferrocenecarboxaldehyde 1 and 0.25 mM ferrocenemethanol 2. | ||
Pleasingly, when the literature conditions were applied to ferrocenecarboxaldehyde 1, the product, ferrocenemethanol 2, was isolated in 71% yield. By increasing the catalytic loading and introducing an enzyme-compatible, water-miscible co-solvent to improve the solubility of the substrate, quantitative yield was achieved in just 30 minutes at room temperature. In order to further improve catalytic activity under the optimised conditions we synthesised and tested a range of ligands with varying aryl substituents (ESI†). Only mesityl-derivative L4 provided any improvement over the previously-used tosyl-derivative and was subsequently used in all further studies. Importantly, in the absence of any ligand, minimal conversion was observed, highlighting the importance of a ligand in order to achieve high catalytic activity (Table S1†). Thus, we hypothesised that a programmed, selective release of the ligand would accelerate the transfer hydrogenation reaction in the presence of an enzyme.
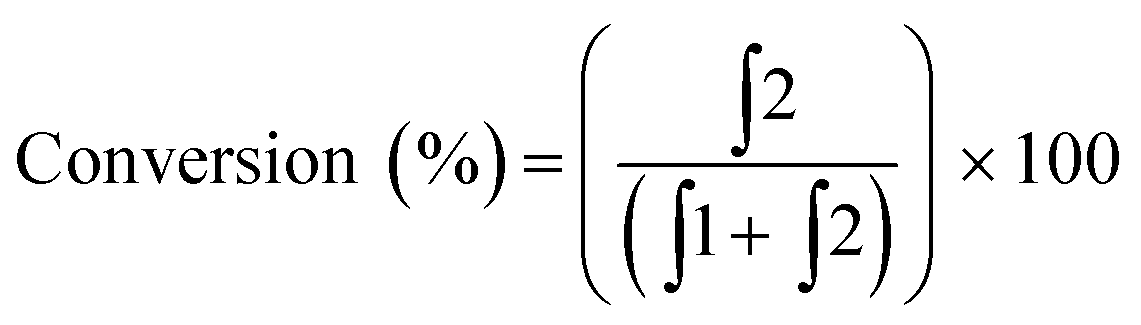 | (1) |
Ligand functionalisation studies towards proligand design
Enzyme-triggered compound release is typically associated with prodrug methodologies, where active pharmaceutical ingredients are manipulated with enzyme-responsive functional groups to improve their physical properties as well as their potency.24 We therefore reasoned that we could combine an enzyme-cleavable trigger moiety with a self-immolative linker to design a proligand which would selectively release our optimised ligand in the presence of an enzyme. To achieve a successful proligand, we first required further information regarding the essential components and the limitations of the ligand. Therefore, we synthesised (ESI†) another set of ligand derivatives and tested them under the optimised reaction conditions (Table 1).
Ethylenediamine, along with its mono- and di-methylated derivatives (Table 1, entries 1–4) were all found to be ineffective ligands under these conditions. Also, tetramethylethylenediamine (Table 1, entry 5) was shown to completely inhibit the reaction. Expectedly, the best-performing ligand deduced from the optimisation, mono-mesityl sulfonamide L4 (Table 1, entry 6), delivered quantitative conversion in 30 minutes, confirming that a sulfonamide is required for a high reaction rate. Methylation at the primary amine position of L4 (Table 1, entry 7), leads to a drop-off in conversion but interestingly, does not inhibit the reaction completely. Since a proton is still available at this position to activate the carbonyl towards attack of the hydride, this result corresponds with a typical transfer hydrogenation reaction mechanism.25 Methylation of the sulfonamide of L4 (Table 1, entry 8) however, leads to a significantly greater drop-off in conversion as functionalisation at this position removes the acidic sulfonamide proton required for iridium insertion to form the active catalyst. Unsurprisingly therefore, both dimethyl derivatives of L4 (Table 1, entries 9 and 10) completely inhibit the transfer hydrogenation reaction with no conversion at all observed. Clearly, as functionalisation at the primary amine position of L4 was enough to prevent ligand acceleration of the catalyst, we next looked at ligands with alternative functionality at this position. Protecting the primary amine position as a carbamate (Table 1, entries 11–13), was indeed found to have a detrimental impact on reaction conversion. Interestingly, benzyl carbamate derivatives L21 and L22 exhibit minimal conversion in the transfer hydrogenation reaction, leading us to believe that we could release ligand L4 through a quinone-methide elimination mechanism.26
Proligand synthesis and mechanistic studies
Consequently, proligand PL1 was designed (Scheme 1) containing a benzyl carbamate linkage along with a phosphate functional group as the enzyme-cleavable trigger. A phosphate trigger enables PL1 to be an appropriate enzyme substrate for alkaline phosphatase (ALP, EC 3.1.3.1), chosen for its high catalytic activity, robustness and inexpensive cost.27 ALP is also a target of interest since abnormal serum levels can be used to preliminarily diagnose diseases such as bone disease,28 liver dysfunction,29 breast and prostatic cancer,30 and diabetes.31 Moreover, ALP is commonly used as an enzyme label within enzyme-linked immunosorbent assays (ELISA) due to its ease of antibody conjugation allowing for this signal amplification methodology to be applied to sensitive protein detection.32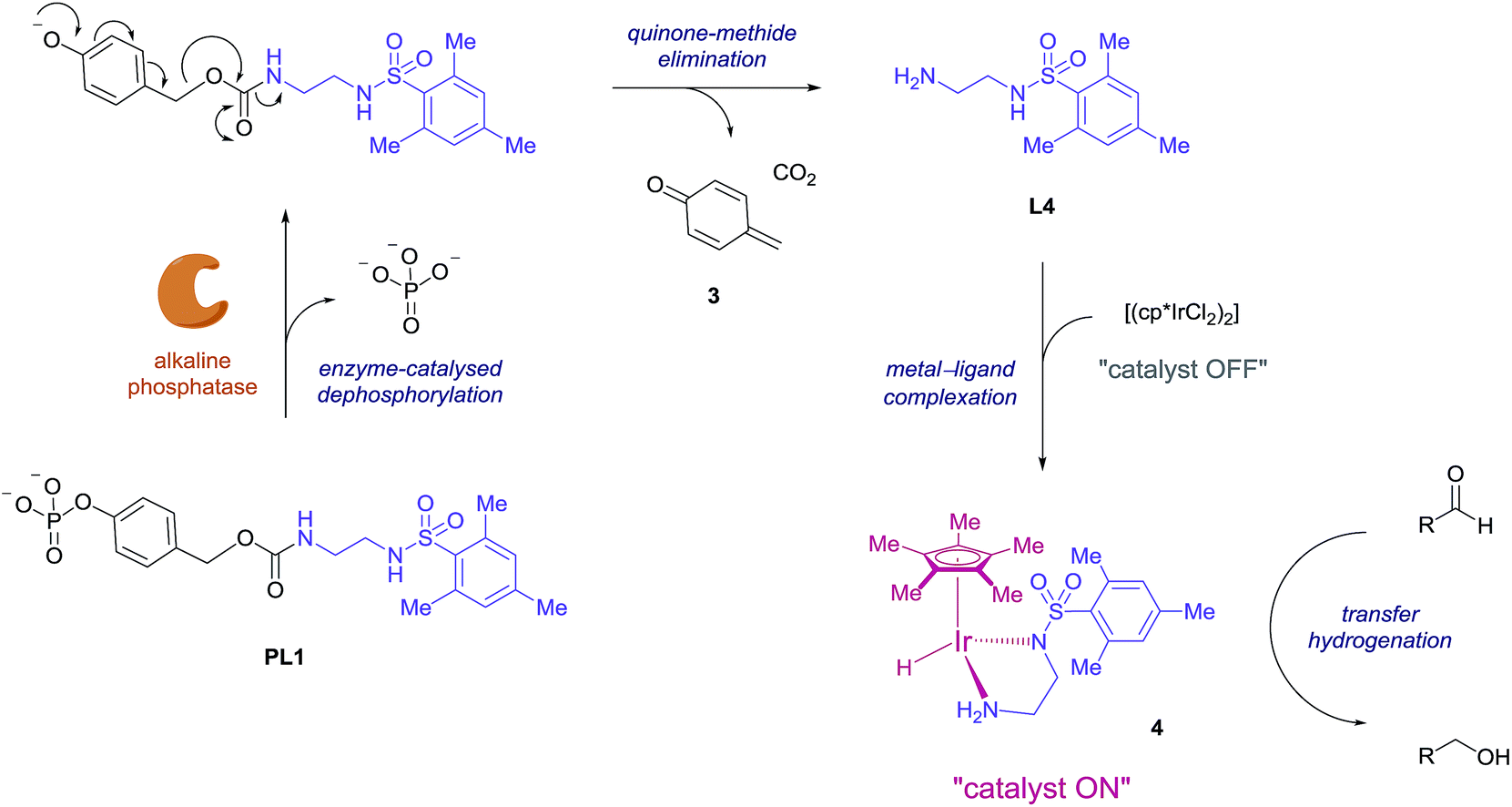 | ||
| Scheme 1 Proposed mechanism for enzyme-triggered ligand release and subsequent ligand-accelerated transfer hydrogenation. | ||
The mechanism we propose for this signal amplification methodology is illustrated in Scheme 1. Initially, selective dephosphorylation of PL1 by the enzyme ALP should occur and the resulting phenol intermediate would be unstable in the alkaline buffered conditions preferred by the enzyme. Subsequent 1,6-elimination should then take place releasing carbon dioxide, quinone methide 3 and ligand L4. Complexation of the newly released ligand with the iridium precatalyst would generate the active transfer hydrogenation catalyst 4 and an accelerated conversion of aldehyde to alcohol should then be observed.
The synthesis of PL1 (Scheme 2) was achieved through 1,1′-carbonyldiimidazole (CDI) coupling of previously reported benzyl alcohol 5,33 and L4 to afford diallyl-protected carbamate 6. Allyl deprotection was then performed through the use of a polymer-bound palladium catalyst under reductive conditions and subsequent sodium salt formation furnished proligand PL1 in 64% overall yield. Once isolated, PL1 was found to be an air-stable solid which can be stored in a vial at room temperature for several months with any sign of degradation.
 | ||
| Scheme 2 Synthetic route to PL1. | ||
To determine whether proligand PL1 is an appropriate substrate for ALP, PL1 was subjected to a buffered solution containing ALP and the interaction monitored by 31P NMR (Fig. 3). Over time, PL1 was shown to be completely consumed by the enzyme and simultaneously, the appearance of another peak corresponding to the phosphate anion, indicated that enzyme-catalysed hydrolysis of the reagent was indeed occurring. In another experiment, this time in the absence of the enzyme, the phosphate peak was not observed showing that PL1 is stable towards background hydrolysis and that phosphate cleavage occurs selectively in the presence of the enzyme. Due to the need for a proton-saturated buffer solution and the poor solubility of the ligand L4 in an aqueous solution, we were unable to detect the free ligand, or any elimination products by 1H NMR.
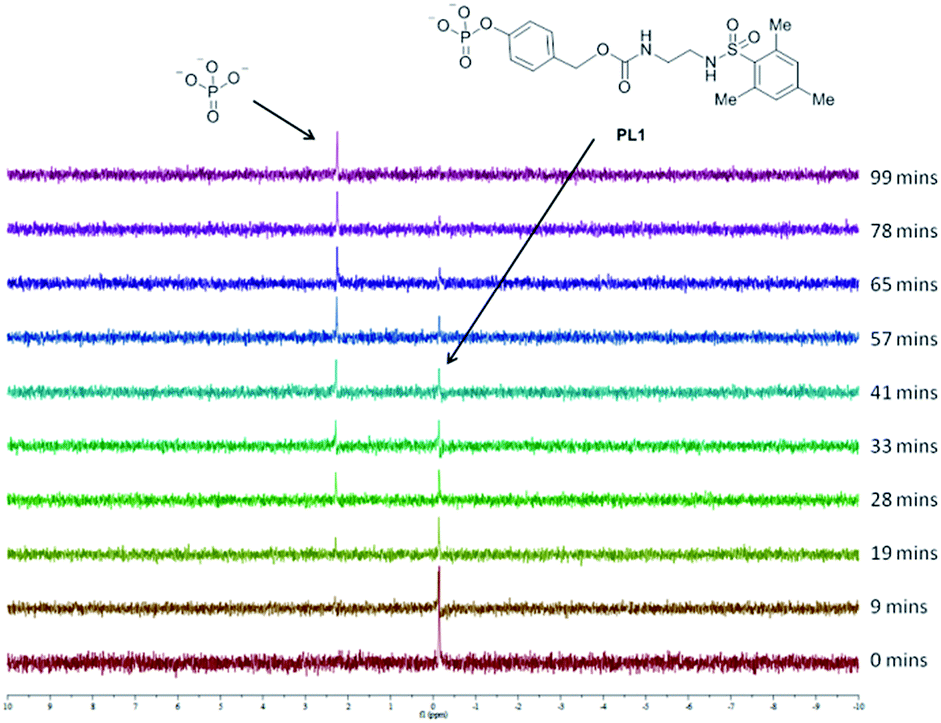 | ||
| Fig. 3 Stacked 31P NMR plots of PL1 after addition of ALP. | ||
In order to detect the elimination products, we studied the reaction using direct infusion Electrospray Time-of-Flight mass spectrometry (ESI-UHR-TOF MS, negative ion mode). Initially, an aqueous solution of PL1 was monitored over a ten minute time-frame and in this period, only the ion corresponding to the [M − H]− of PL1 was observed, and no ion corresponding to ligand L4 was seen (ESI – Fig. S2†). This confirms the stability of PL1 to hydrolysis. Then, immediately after the addition of a solution of ALP, the appearance of the [M − H]− ion corresponding to L4 is seen and this ion is shown to increase with intensity over time (Fig. 4). Concomitantly, the enzyme substrate ion decreases in intensity, indicating that enzyme-catalysed consumption of PL1 and simultaneous production of L4 is occurring. Additionally, [M − H]− ions relating to eliminated linker 3 and 4-hydroxybenzyl alcohol, a result of quinone methide 3 being trapped by water, are also observed and intensities for these ions increase slowly over time (ESI – Fig. S3†). Over the course of the experiment, the [M − H]− ion for the phenol intermediate was never observed. This suggests that elimination to L4 and 3 occurs rapidly under the reaction conditions. Together, this data confirms that both dephosphorylation of PL1 and subsequent elimination to release L4 occurs selectively in the presence of enzyme, which supports the enzyme-triggered catalyst activation mechanism we propose.
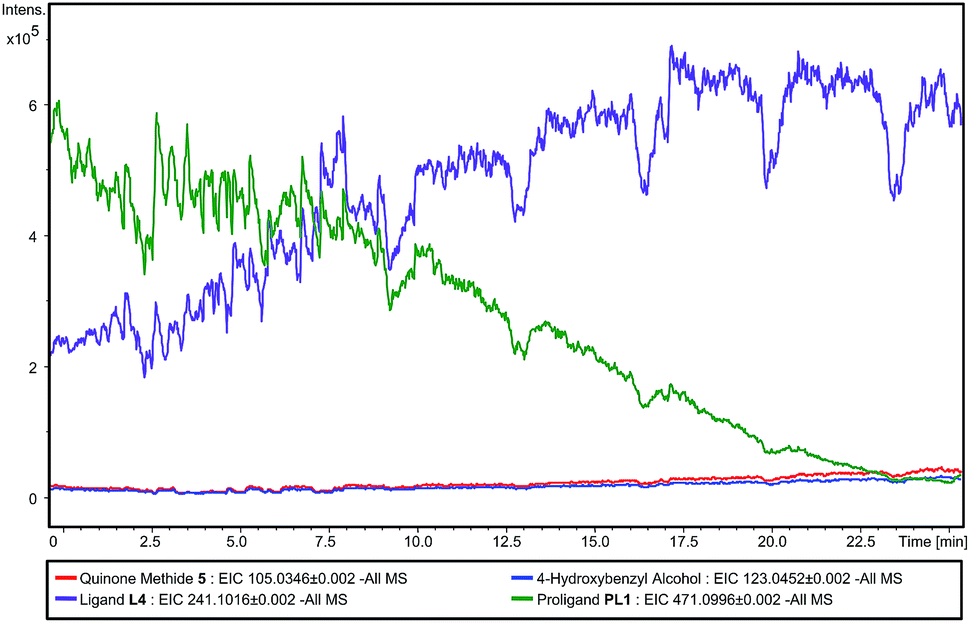 | ||
| Fig. 4 Reaction profile of PL1 after addition of ALP monitored by mass spectrometry. | ||
Enzyme-triggered catalysis
Having established that ligand release occurs selectively as a direct result of enzyme activity, a number of background reactions were performed to determine if catalyst activation is attributed to the combination of the presence of the enzyme and the enzyme substrate (Table 2). In the presence of just the enzyme and no PL1 (Table 2, entry 1), no conversion of 1 to 2 was observed showing that any ligand-like amino acid side-chains on the enzyme itself or any enzyme additives such as amino alcohol-derived stabilisers do not contribute to any undesired reduction. In the absence of the enzyme, a satisfactorily low yield of ferrocenemethanol 2 was obtained in the presence of just the proligand (Table 2, entry 2). Gratifyingly, in the presence of both the enzyme and the proligand (Table 2, entry 3), a near quantitative yield was obtained which we attribute to enzyme-triggered ligand release and ligand-accelerated catalytic transfer hydrogenation.
With catalyst activity shown only to be activated by both the enzyme and the enzyme substrate, we next wanted to optimise the signal amplification cascade using ratiometric electrochemical analysis to monitor the cascade. Prior to this however, electrochemically-derived reaction profiles were needed for the active catalytic species in order to determine the lowest catalytic loading required to obtain full conversion in the shortest amount of time. As such, ferrocenecarboxaldehyde 1 was exposed to different catalytic loadings and the reaction was sampled every 3 minutes (Fig. 5). At a catalyst loading of 0.5 mol%, only ≈50% conversion was observed after 30 minutes. By doubling the catalyst loading to 1 mol%, quantitative conversion was able to be obtained within the same timeframe. Doubling the catalytic loading further to 2 mol% enabled quantitative conversion to be achieved in less than 6 minutes.
 | ||
| Fig. 5 Electrochemically-determined reaction profiles under different catalytic loadings of active catalyst 4. | ||
Encouraged by the efficiency of the transfer hydrogenation of 1 at the higher catalyst loading, we set to electrochemically monitor enzyme activity over time (Fig. 6). Pleasingly, in the presence of high concentrations of ALP, conversion of 1 to 2 was indeed observed. The sigmoidal shape of the conversion curves indicate a slow catalyst-activation induction period is apparent until a sufficient number of iridium complexes have been activated which leads to a pseudo-exponential increase in conversion. A number of signal amplification methodologies previously reported exhibit sigmoidal reaction profiles that are indicative of an exponential-like mechanism being in effect.14a,15a,34 Importantly, in the absence of the enzyme, a minimal background reaction was observed showing that activation of the catalytic cascade occurs selectively. This further enhances the potential of this methodology since no protection, separation or manipulation of the reagents is required to achieve successful activation of the catalytic cascade.
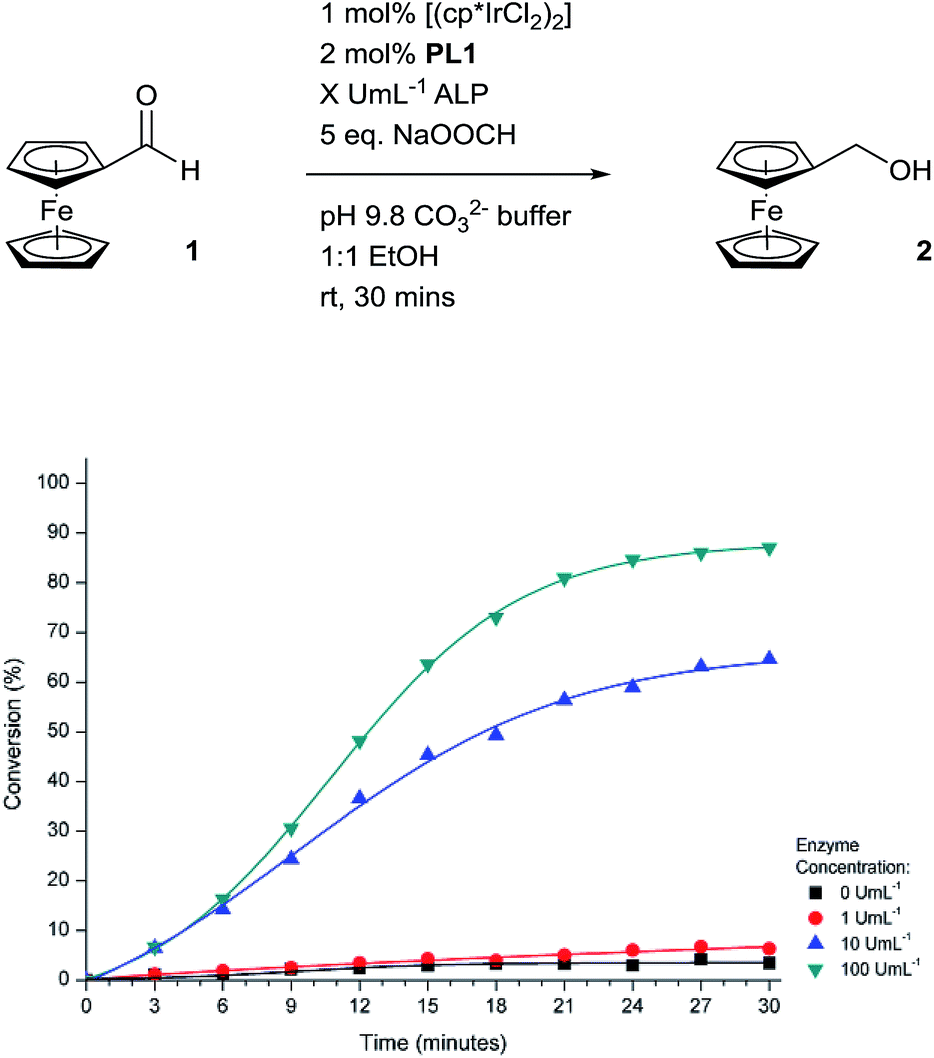 | ||
| Fig. 6 Electrochemically-determined reaction profiles under different concentrations of ALP at room temperature. | ||
To improve the efficiency of the signal amplification cascade further, we increased the temperature of the system to coincide with the optimum working temperature of the enzyme.32 This would not only increase the rate of PL1 consumption but also increase the rate of the transfer hydrogenation reaction, thus leading to a much improved signal-amplified response. Indeed, at these elevated temperatures, much greater conversions were observed in comparison to the conversions obtained for the same enzyme concentration at room temperature (Fig. 7). A high concentration of ALP at 37 °C allows for quantitative conversion of the transfer hydrogenation reaction to be obtained within 15 minutes. Good conversions are also seen at lower enzyme concentrations indicating both the excellent sensitivity and good dynamic range of the catalytic cascade. However, the increase in temperature also leads to an increase in the background rate, which is expected since ligand-less iridium pre-catalysts are known to catalyse the transfer hydrogenation reaction.22a Using the conversions obtained under these conditions after 3 minutes, a calibration curve was plotted (ESI†). From this, a limit of detection for this methodology was calculated to be 7.6 pM after a reaction time of 3 minutes which is comparable with that of redox cycling-based amplification methodologies for rapid electrochemical ALP detection.35
 | ||
| Fig. 7 Electrochemically-determined reaction profiles under different concentrations of ALP at 37 °C. | ||
Substrate scope
We next wanted to determine if the catalyst cascade could be applied to the enzyme-triggered reduction of aldehydes other than ferrocenecarboxaldehyde. Therefore, the optimised conditions were then directly applied to a range of different aldehydes (Table 3). Initially, we looked at catalyst substrates that could also be used to monitor reaction progress, and therefore enzyme activity, by means other than electrochemistry. First, we evaluated fluorophore-containing aldehydes (Table 3, entries 1–3) as this would allow signal transduction from enzyme activity to fluorescence to be obtained. Pleasingly, all fluorescent aldehydes tested delivered good to quantitative conversions in the presence of ALP, whilst maintaining a satisfactorily low background in its absence. Next, we investigated fluorine-containing aromatic aldehydes (Table 3, entries 4–6), which would enable 19F NMR to be used to monitor reaction progress. Again, good to quantitative conversions were obtained using these benzaldehydes. Electron-withdrawing substituents upon the aromatic ring (Table 3, entries 5–6) were found to over-activate the aldehyde towards hydride attack and a subsequent increase in background reaction is observed. In contrast, electron-donating groups (Table 3, entries 7–8) were able to suppress this background reaction. Interestingly, when a di-aldehyde was used as the catalyst substrate (Table 3, entry 10), the mono-hydrogenated product was found to be the major product over the di-hydrogenated product in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Aliphatic aldehydes (Table 3, entry 11) are also tolerated, but an undesirably high background was observed which we attribute to the high solubility of the substrate in the reaction medium. Cinnamaldehydes are also reduced efficiently (Table 3, entry 12) in the presence of ALP with no alkene reduction being observed.
:1 ratio. Aliphatic aldehydes (Table 3, entry 11) are also tolerated, but an undesirably high background was observed which we attribute to the high solubility of the substrate in the reaction medium. Cinnamaldehydes are also reduced efficiently (Table 3, entry 12) in the presence of ALP with no alkene reduction being observed.
|
|
|||
|---|---|---|---|
| Entry | Product | Conversiona | |
| ALP | |||
| 0 U mL−1 | 25 U mL−1 | ||
| a Conversion determined by 1H NMR. b Reaction conditions: 0.25 mol% [Ir(cp*Cl2)2], 0.5 mol% PL1, 25 U mL−1 ALP, 0.5 mmol R–CHO, 5 eq. NaOOCH, 1 mL EtOH, 1 mL pH 9.8 CO32− buffer, 37 °C, 30 min. c Reaction conditions: 0.25 mol% [Ir(cp*Cl2)2], 0.5 mol% PL1, 25 U mL−1 ALP, 1 mmol R–CHO, 5 eq. NaOOCH, 2 mL EtOH, 2 mL pH 9.8 CO32− buffer, 37 °C, 30 min. d Combined conversion of 20 (75%) and 1,4-benzenedimethanol (25%). | |||
| 1b |
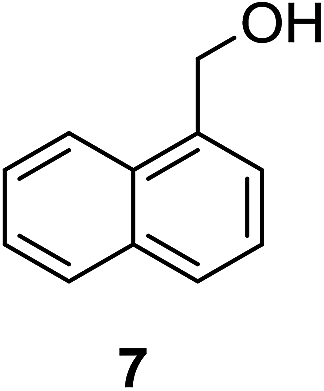
|
10% | >99% |
| 2b |

|
2% | 77% |
| 3b |
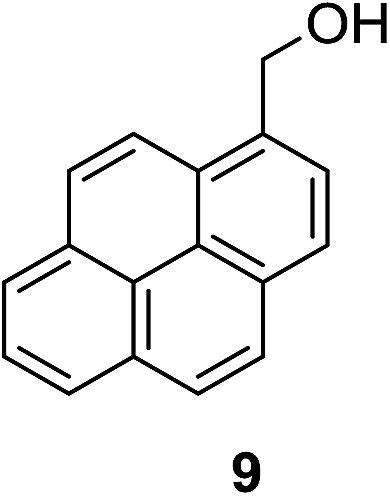
|
2% | 95% |
| 4c |
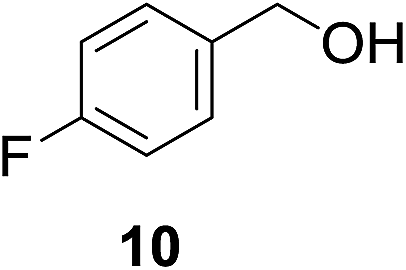
|
4% | >99% |
| 5c |
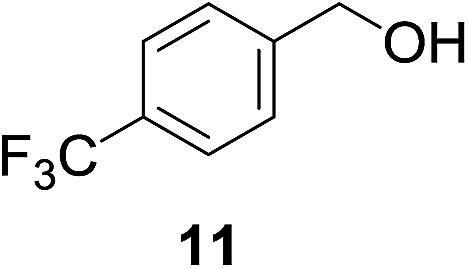
|
20% | >99% |
| 6c |
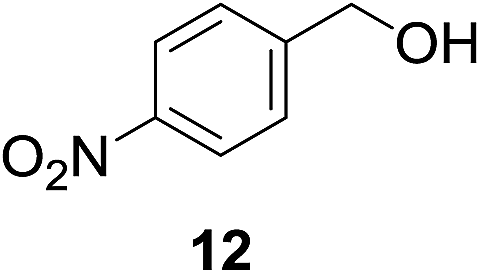
|
21% | >99% |
| 7b |
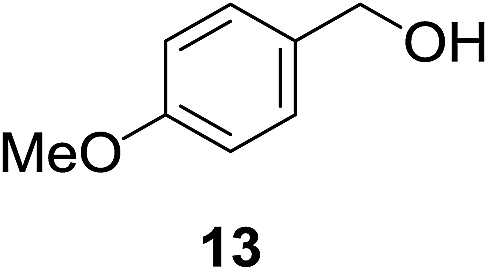
|
11% | >99% |
| 8b |
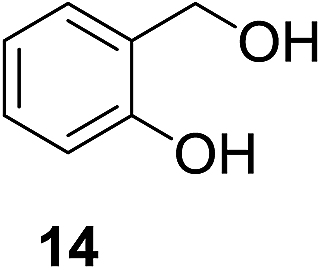
|
13% | >99% |
| 9b |
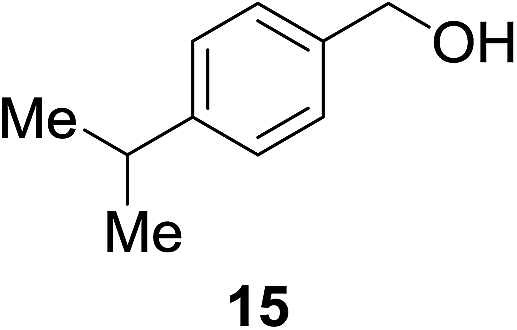
|
14% | >99% |
| 10b |
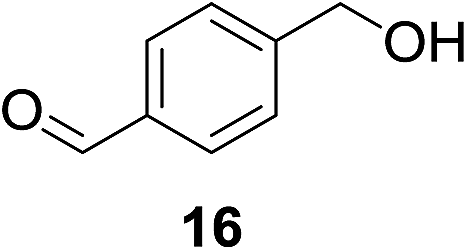
|
26% | >99%d |
| 11b |
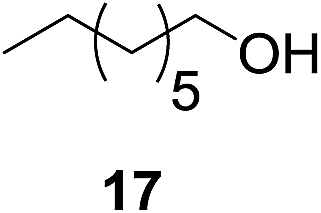
|
24% | >99% |
| 12b |
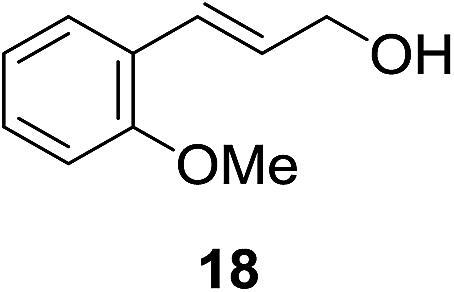
|
8% | 75% |
On the whole, signal transduction from enzyme activity to a range of output signals was successfully performed whilst maintaining sufficiently low signals in the absence of ALP. Despite this, we are currently investigating methods to nullify the background rate to improve the signal-to-noise ratio. Also, the application of this catalyst cascade to the detection of other enzymes, as well as proteins via ELISA techniques, is currently underway.
Conclusions
In summary, we have successfully developed a biosynthetic signal transduction and signal amplification methodology by utilising the selectivity and high activity of an enzyme, in combination with a water-tolerant organometallic catalyst. Through simple manipulation of the proligand trigger, this methodology can be applied towards the detection of a number of different enzymes as well as other analytes of interest. We have also shown that selective catalyst activation by an enzyme can be used to reduce a range of aldehydes allowing the amplification cascade to be tailored to the desired signal output. In principle, this technology could easily be used as a general signal amplification strategy for improving the sensitivity of enzyme detection assays and, by extension, protein detection assays through enzyme-linked immunosorbent assay (ELISA) protocols. Additionally, the ability for the system to process a simple input signal and convert it into an amplified signal output lends itself towards molecular computing applications as well as stimuli-responsive materials.Acknowledgements
We would like to thank Atlas Genetics Ltd. for studentship funding (S.G.) and for the kind donation of a potentiostat. We would also like to acknowledge the extensive use of NMR and MS facilities within the Chemical Characterisation and Analysis Facility (CCAF) at the University of Bath for compound characterisation.Notes and references
- A. L. Lehninger, D. L. Nelson and M. M. Cox, Lehninger Principles of Biochemistry, W. H. Freeman, New York, 4th edn, 2004, ch. 12, pp. 421–439 Search PubMed.
- R. G. Macfarlane, Nature, 1964, 202, 498 CrossRef CAS PubMed.
- G. Wald, Science, 1965, 150, 1028 CAS.
- J. M. Bowness, Science, 1966, 152, 1370 CrossRef CAS.
- G. Krauss, Biochemistry of Signal Transduction and Regulation, Wiley–VCH, Weinheim, 3rd edn, 2008, ch. 3, pp. 115–149 Search PubMed.
- For reviews: (a) R. Yerushalmi, A. Scherz, M. E. van der Boom and H.-B. Kraatz, J. Mater. Chem., 2005, 15, 4480 RSC; (b) P. Theato, B. S. Sumerlin, R. K. O'Reilly and T. H. Epps III, Chem. Soc. Rev., 2013, 42, 7055 RSC; (c) J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276 CrossRef CAS PubMed.
- J. G. Millán and L. J. Prins, in Supramolecular Systems in Biomedical Fields, ed. H.-J. Schneider, The Royal Society of Chemistry, Cambridge, 2013, ch. 2, pp. 7–34 Search PubMed.
- P. Scrimin and L. J. Prins, Chem. Soc. Rev., 2011, 40, 4488 RSC.
- (a) N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS PubMed; (b) X. Cao, Y. Ye and S. Liu, Anal. Biochem., 2011, 417, 1 CrossRef CAS PubMed; (c) K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739 CrossRef CAS PubMed; (d) J. Lei and H. Ju, Chem. Soc. Rev., 2012, 41, 2122 RSC.
- S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef PubMed.
- (a) J. Liu and Y. Lu, J. Am. Chem. Soc., 2003, 125, 6642 CrossRef CAS PubMed; (b) T. Takeuchi and S. Matile, J. Am. Chem. Soc., 2009, 131, 18048 CrossRef CAS PubMed; (c) R. de la Rica and M. M. Stevens, Nat. Nanotechnol., 2012, 7, 821 CrossRef CAS PubMed; (d) Z. Gao, L. Hou, M. Xu and D. Tang, Sci. Rep., 2014, 4, 3966 Search PubMed.
- (a) F. M. H. de Groot, C. Albrecht, R. Koekkoek, P. H. Beusker and H. W. Scheeren, Angew. Chem., Int. Ed., 2003, 42, 4490 CrossRef CAS PubMed; (b) M. L. Szalai, R. M. Kevwitch and D. V. McGrath, J. Am. Chem. Soc., 2003, 125, 15688 CrossRef CAS PubMed; (c) R. J. Amir, N. Pessah, M. Shamis and D. Shabat, Angew. Chem., Int. Ed., 2003, 42, 4494 CrossRef CAS PubMed.
- M. Avital-Schmilovici and D. Shabat, Soft Matter, 2010, 6, 1073 RSC.
- (a) E. Sella and D. Shabat, J. Am. Chem. Soc., 2009, 131, 9934 CrossRef CAS PubMed; (b) E. Sella, A. Lubelski, J. Klafter and D. Shabat, J. Am. Chem. Soc., 2010, 132, 3945 CrossRef CAS PubMed.
- (a) M. S. Baker and S. T. Phillips, J. Am. Chem. Soc., 2011, 133, 5170 CrossRef CAS PubMed; (b) M. S. Baker and S. T. Phillips, Org. Biomol. Chem., 2012, 10, 3595 RSC.
- (a) N. Karton-Lifshin and D. Shabat, New J. Chem., 2012, 36, 386 RSC; (b) K. Yeung, K. M. Schmid and S. T. Phillips, Chem. Commun., 2013, 49, 394 RSC; (c) M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511 CrossRef CAS PubMed; (d) T. Yoshii, S. Onogi, H. Shigemitsu and I. Hamachi, J. Am. Chem. Soc., 2015, 137, 3360 CrossRef CAS PubMed; (e) H. Kim, M. S. Baker and S. T. Phillips, Chem. Sci., 2015, 6, 3388 RSC.
- M. A. Swiderska and J.-L. Reymond, Nat. Chem., 2009, 1, 527 CrossRef CAS PubMed.
- O. V. Makhlynets and I. V. Korendovych, Biomolecules, 2014, 4, 402 CrossRef PubMed.
- (a) L. Zhu and E. V. Anslyn, Angew. Chem., Int. Ed., 2006, 45, 1190 CrossRef CAS PubMed; (b) H. J. Yoon and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 11590 CrossRef CAS PubMed; (c) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114 CrossRef CAS PubMed.
- L. Kovbasyuk and R. Krämer, Chem. Rev., 2004, 104, 3161 CrossRef CAS PubMed.
- (a) T. Abura, S. Ogo, Y. Watanabe and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 4149 CrossRef CAS PubMed; (b) J. M. McFarland and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 13490 CrossRef CAS PubMed; (c) N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210 CrossRef CAS PubMed; (d) R. Kawahara, K. Fujita and R. Yamaguchi, J. Am. Chem. Soc., 2010, 132, 15108 CrossRef CAS PubMed; (e) R. Kawahara, K. Fujita and R. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 3643 CrossRef CAS PubMed; (f) J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383 CrossRef CAS PubMed.
- (a) X. Wu, J. Liu, X. Li, A. Zanotti-Gerosa, F. Hancock, D. Vinci, J. Ruan and J. Xiao, Angew. Chem., Int. Ed., 2006, 45, 6718 CrossRef CAS PubMed; (b) X. Wu, X. Li, A. Zanotti-Gerosa, A. Pettman, J. Liu, A. J. Mills and J. Xiao, Chem.–Eur. J., 2008, 14, 2209 CrossRef CAS PubMed; (c) J. Tan, W. Tang, Y. Sun, Z. Jiang, F. Chen, L. Xu, Q. Fan and J. Xiao, Tetrahedron, 2011, 67, 6206 CrossRef CAS PubMed; (d) Y. Wei, D. Xue, Q. Lei, C. Wang and J. Xiao, Green Chem., 2013, 15, 629 RSC.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931 CrossRef CAS PubMed.
- (a) L. N. Jungheim and T. A. Shepherd, Chem. Rev., 1994, 94, 1553 CrossRef CAS; (b) J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255 CrossRef CAS PubMed.
- R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931 CrossRef CAS.
- S. Gnaim and D. Shabat, Acc. Chem. Res., 2014, 47, 2970 CrossRef CAS PubMed.
- R. B. McComb, G. N. Bowers Jr and S. Posen, Alkaline Phosphatase, Plenum Press, New York, 1979 Search PubMed.
- D. Goltzman and D. Miao, Encycl. Endocr. Dis., 2004, 1, 164 CAS.
- J. Ooi, K. Shiraki, Y. Morishita and T. Nobori, J. Clin. Lab. Anal., 2007, 21, 133 CrossRef PubMed.
- G. Ramaswamy, V. R. Rao, L. Krishnamoorthy, G. Ramesh, R. Gomathy and D. Renukadevi, Indian J. Clin. Biochem., 2000, 15, 110 CrossRef CAS PubMed.
- G. M. M. Rao and L. O. Morghom, Enzyme, 1986, 35, 57 CAS.
- D. M. Kemeny and S. Chantler in ELISA and Other Solid Phase Immunoassays, ed. D. M. Kemeny and S. J. Challacombe, John Wiley & Sons, Chichester, 1988 Search PubMed.
- S. Goggins, C. Naz, B. J. Marsh and C. G. Frost, Chem. Commun., 2015, 51, 561 RSC.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2009, 48, 386 CrossRef CAS PubMed.
- (a) C. G. Bauer, A. V. Eremenko, E. Ehrentreich-Förster, F. F. Bier, A. Makower, H. B. Halsall, W. R. Heineman and F. W. Scheller, Anal. Chem., 1996, 68, 2453 CrossRef CAS; (b) C. Ruan and Y. Li, Talanta, 2001, 54, 1095 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterisation data and copies of NMR spectra for novel compounds. See DOI: 10.1039/c5sc01588j |
| This journal is © The Royal Society of Chemistry 2015 |