DOI:
10.1039/C5SC01722J
(Edge Article)
Chem. Sci., 2015,
6, 4828-4836
Synthesis of triazolium-based mono- and tris-branched [1]rotaxanes using a molecular transporter of dibenzo-24-crown-8†
Received
11th May 2015
, Accepted 28th May 2015
First published on 2nd June 2015
Abstract
We report a diverted route to [1]rotaxane and tris-branched [1]rotaxane that are devoid of any efficient template and which could not be obtained by classical straightforward strategies. The described chemical route relies on the utilization of a “macrocycle transporter”, which is able first to bind a macrocycle, second to link temporarily a triazolium-containing molecular axle, and third to deliver the macrocycle around the new docked axle through molecular machinery in a [1]rotaxane structure. The extended encircled thread is eventually cleaved by an amine or a triamine to afford the triazolium-containing [1]rotaxanes, releasing at the same time, the macrocycle transporter as a recyclable species.
Introduction
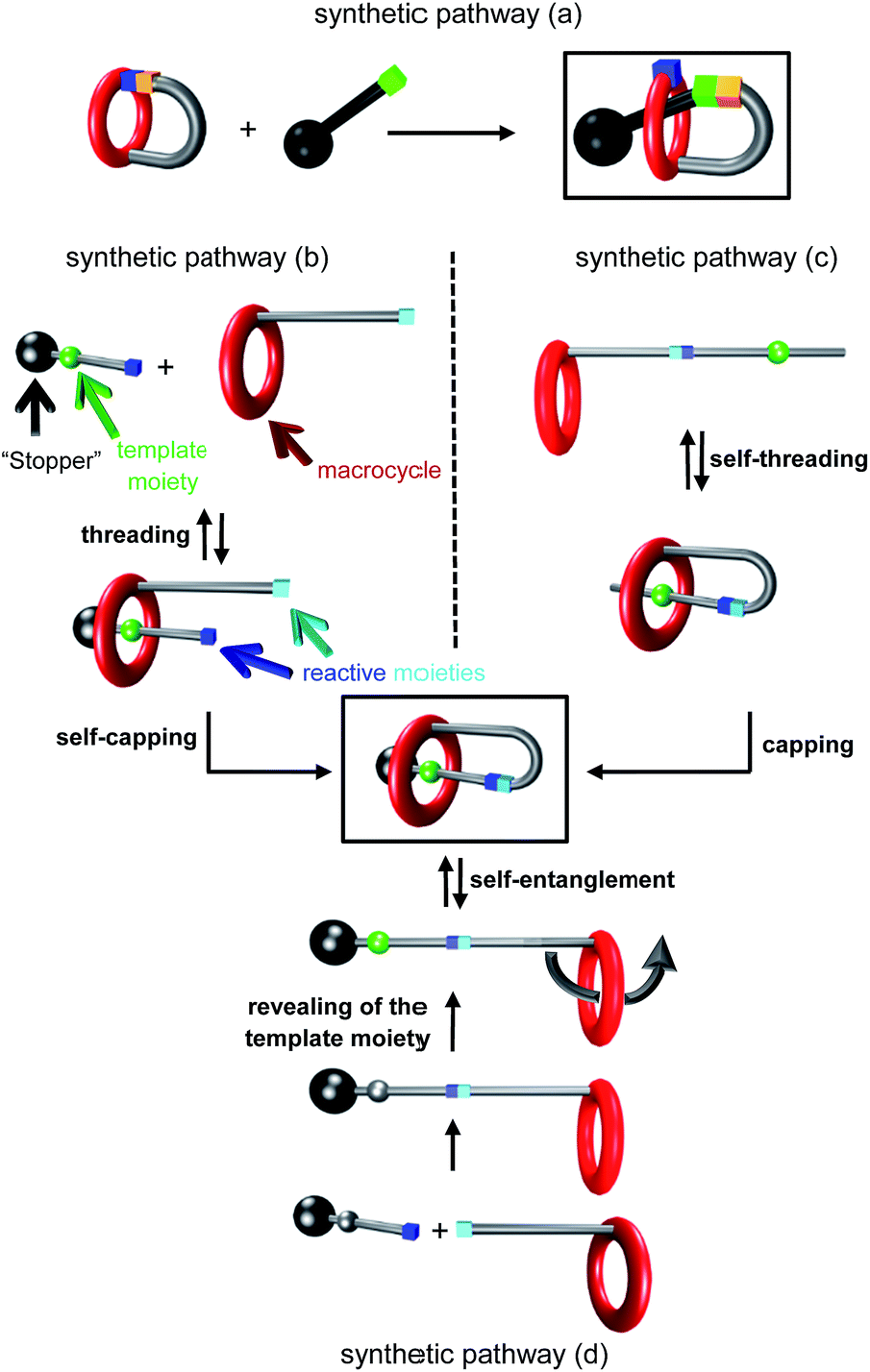
Since the properties of molecules are closely related to their tridimensional structure, mechanically interlocked molecules,1 which adopt very unusual and interesting architectures, seem to be very appealing potential targets in a wide range of research fields. It follows that improving their chemical access is of the highest interest, even though it might be a challenging task in some cases. Among the numerous interlocked molecular architectures, the [1]rotaxane2 molecular architecture holds a unique position, being a single molecule. It consists of a macrocycle that surrounds a molecular thread, in which both components are connected together through a covalent bond. Several different chemical routes have been used to synthesize [1]rotaxanes up to now (Fig. 1). The least used strategy relies on a covalent bond formation and was reported by Hiratani et al. in 2004 (synthetic pathway (a)):3 in that case a key bicyclic intermediate is statistically attacked either by the inner region of the macrocycle to afford the [1]rotaxane, or by the outside of the macrocycle to provide the non-interlocked analogue. More frequently encountered are the other strategies relying on the use of weak interactions as the driving force to assemble the molecular elements into the self-interlocked architecture. They can be divided into three main synthetic pathways. The first one relies on the preliminary template-directed synthesis of a semi [2]rotaxane followed by the chemical connection between the surrounded axle and the macrocycle (synthetic pathway (b)).4 The second one uses a “hermaphrodite” molecule, that is to say a macrocycle that is covalently bonded to a molecular axle which contains a site of interaction for the macrocycle (synthetic pathway (c)). In that case, the non-interlocked molecule remains in equilibrium with the pseudo[1]rotaxane, until the latter is capped either by a hindering5 or a rigid stopper6 at the extremity of the axle. The third templated pathway consists in a self-entanglement strategy of a “hermaphrodite” molecule that already contains a stopper at the extremity of the axle (synthetic pathway (d)). In that particular case, no threading of the bulky extremity is allowed, so that the only way to interlace the molecule is to perform a pirouette of one part of the macrocycle (i.e. the section connected to the axle) with respect to the other.7 This last templated strategy is probably a more demanding chemical pathway to set up since it requires a macrocycle of a bigger cavity to allow the self-entanglement, although enough interactions must be maintained at the same time between the template and the macrocycle to obtain the [1]rotaxane structure. Herein, we report on the synthesis of triazolium-containing mono- and tris-branched [1]rotaxanes which are devoid of any efficient template for the macrocycle (Fig. 2). They only contain a triazolium moiety which serves as a molecular station of poor affinity for the dibenzo-24-crown-8 (DB24C8) derivative in a locked rotaxane.
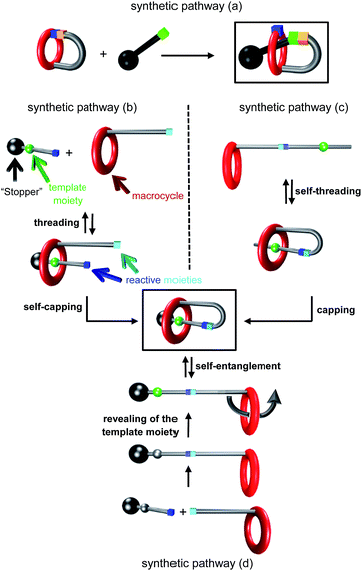 |
| | Fig. 1 Chemical templated routes to [1]rotaxanes. | |
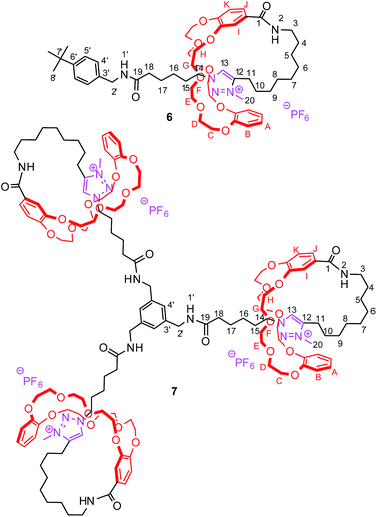 |
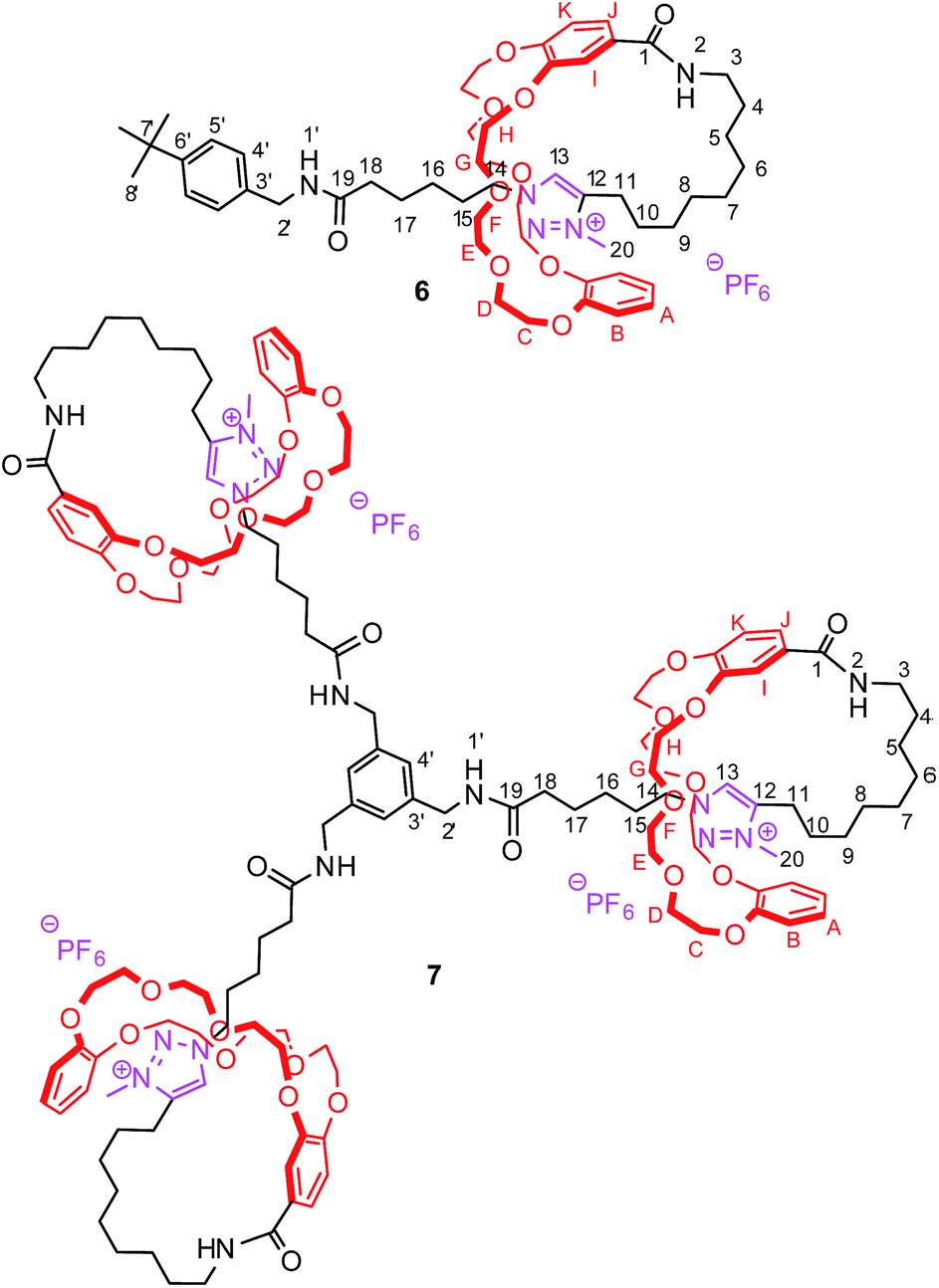
| | Fig. 2 Synthesized [1]rotaxanes 6 and 7 with atom assignments. | |
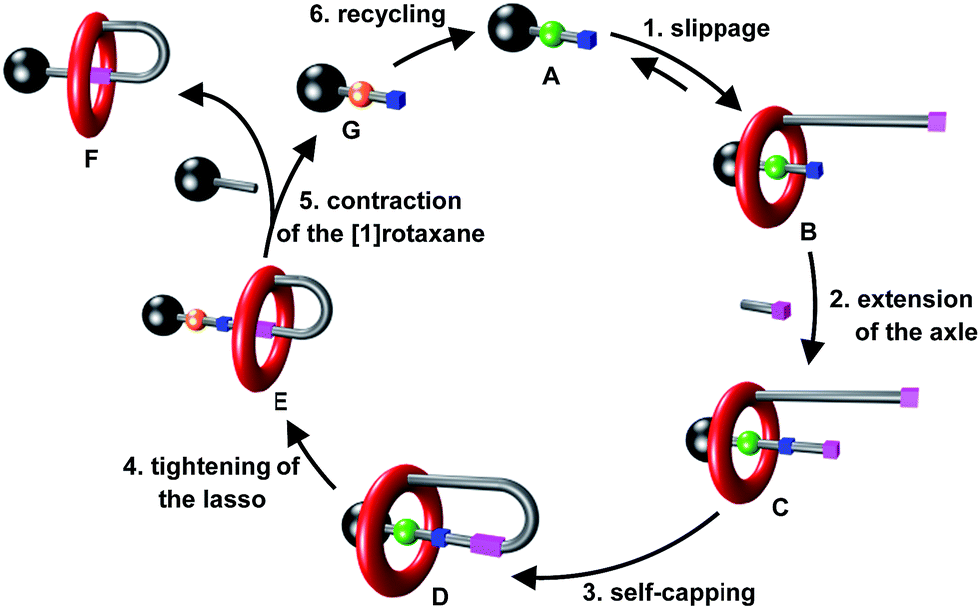
The newly proposed synthetic strategy relies on the use of a recyclable transporter of macrocycles, A, whose utility is to catch a macrocycle and to deliver it to another axle (Fig. 3).8 As it has been designed to contain an efficient template moiety for the macrocycle, A initially efficiently captures the macrocycle so forming the semi[2]rotaxane B in a similar fashion as in the illustrated strategy (b) of Fig. 1. Extension of the encircled axle, then self-capping through the linking between the thread end and the substituted macrocycle provides the locked [1]rotaxane structure D. At this step, a second site of interaction of much poorer affinity for the macrocycle is also created in the encircled axle. Molecular machinery is subsequently used to trigger the tightening of the [1]rotaxane lasso D, through the shuttling of the macrocycle around the final targeted axle. The final step of the strategy consists in the cleavage of the axle of the tightened [1]rotaxane E using a functionalized stopper, thus providing simultaneously the contracted [1]rotaxane F and the initial modified axle G. Eventually, the initial transporter A can be regenerated through the recycling of G. What is here very important to point out is the effectiveness of the strategy. Indeed, the final triazolium-containing[1]rotaxane F (i.e.6 and 7) cannot be efficiently generated using the other already reported strategies. Although the second created site of interaction for the macrocycle (i.e. triazolium) acts as an effective molecular station for the shuttling of the macrocycle in the locked [1]rotaxane structure, it would not permit to drive the rotaxane formation through intermolecular interactions, this being mainly due to entropic factors.9
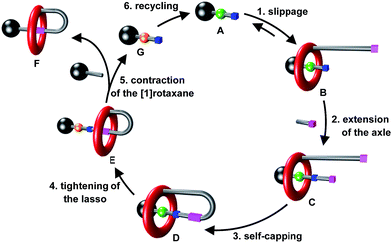 |
| | Fig. 3 A novel route to [1]rotaxane F based on a recyclable macrocycle transporter A. | |
Results and discussion
Preparation of the loosened extended [1]rotaxane D
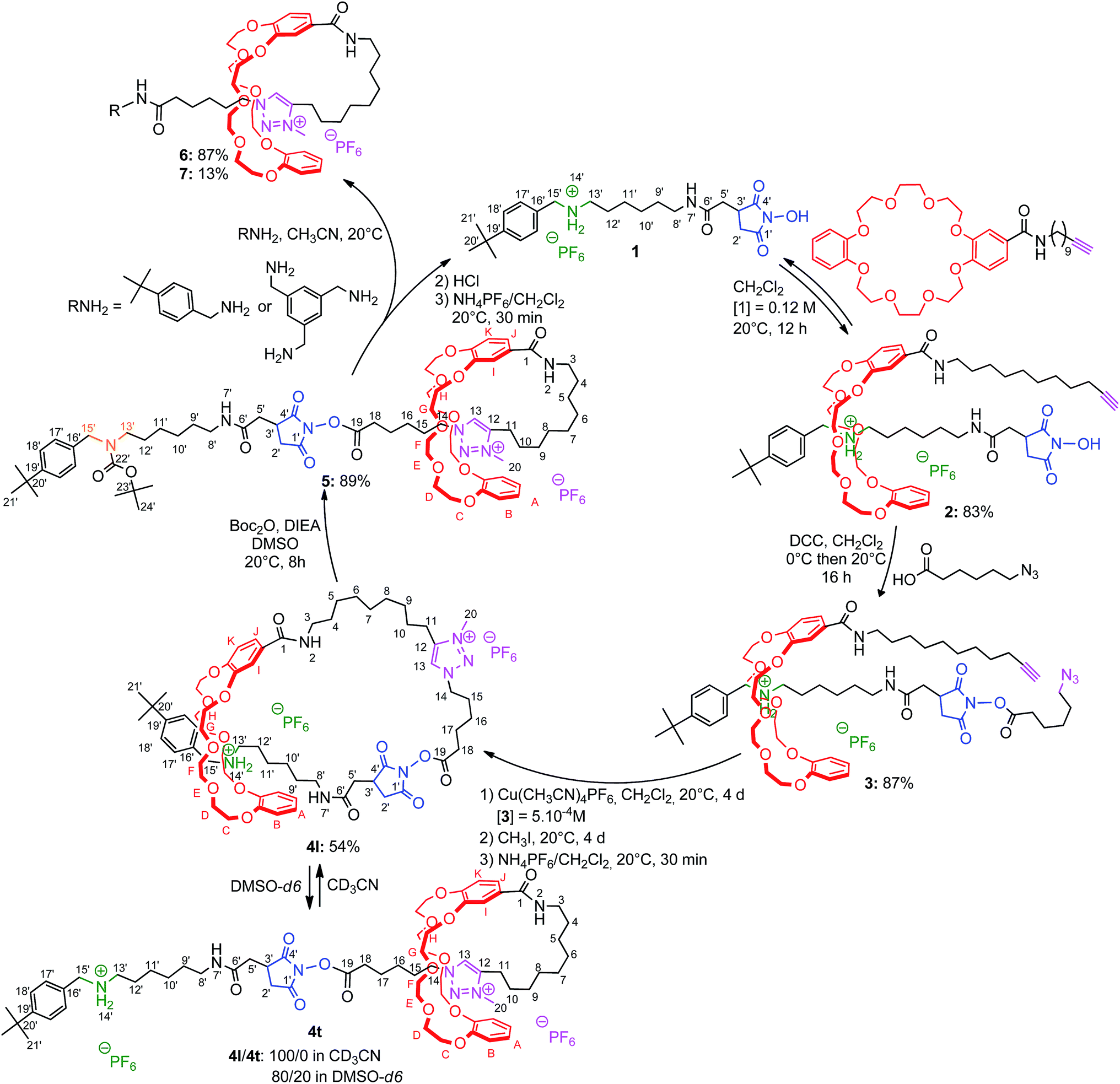
The synthetic sequence begins by the slippage10 of a transporter of macrocycle 1 (Scheme 1). It contains an ammonium moiety (in green) which initially serves as an efficient template for the DB24C8.11 It also contains a tert-butylbenzyl stopper (in black) at one extremity and a N-hydroxysuccinimide (NHS) moiety (in blue) at the other end. The semirotaxane architecture 2 could be obtained in 83% yield after stirring during 12 h in dichloromethane at room temperature, 3 equivalents of the DB24C8 derivative and 1 equivalent of 1 at a concentration of 0.12 M. The bulkiness of the NHS is responsible for the long slippage process and for the stability of the product. Indeed, it is noteworthy that the rotaxane 2 is stable as a powder and can be kept for many days without observing any disassembly. Furthermore, the unthreading is also very slow in a hydrogen-bond promoting solvent such as dichloromethane, which is beneficial here since it allows the purification of 2 by Sephadex chromatography. The extension of the encircled axle was then carried out by esterification of the free hydroxyl group of the NHS-containing rotaxane 2 with the azido hexanoic acid, using dicyclohexylcarbodiimide as coupling reagent. On the one hand, it provides a N-hydroxysuccinimide ester moiety, which will be of utility for the final cleavage step of the strategy. On the other hand, the incorporated azido moiety (in purple), gives the opportunity to self-cap the [1]rotaxane architecture through cyclization. The azido moiety of the extended [2]rotaxane 3 was therefore connected to the alkyne-containing macrocycle, through copper(I)-catalyzed Huisgen12 alkyne-azide 1,3-dipolar cycloaddition.13 High dilution (5 × 10−4 M) conditions were employed to provide the [1]rotaxane architecture in a satisfactory yield of 60%. The so-created triazole moiety was further methylated before being submitted to an anion exchange using ammonium hexafluorophosphate, affording 4l with an overall yield of 54% from 3. This alkylation reveals the N-methyltriazolium in 4 as the second site of interaction for the DB24C8 derivative.14 In hydrogen-bond promoting solvents, as long as the ammonium remains protonated, the DB24C8 derivative is mainly localized around the more favorable ammonium station, resulting in a loosened conformation of the lasso. Since the chemical route to the targeted [1]rotaxanes 6 and 7 requires the large-amplitude shuttling of the macrocycle along the thread toward the triazolium station, the displacement of the equilibrium from the loosened lasso 4l to the tightened lasso 4t was envisaged. We first studied the effect of a variation of the polarity of the medium. Through a more efficient subsequent approach, an N-carbamoylation of the ammonium station was then considered.
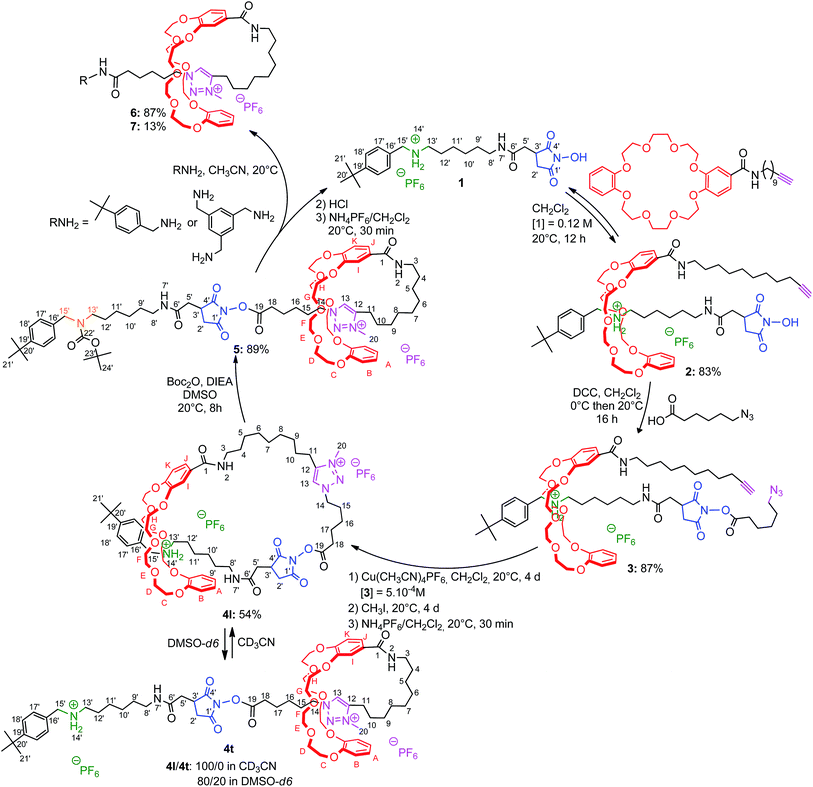 |
| | Scheme 1 Preparation of mono- and tris-branched triazolium-containing [1]rotaxanes using the recyclable macrocycle transporter 1. | |
Tightening of the lasso through solvent-dependent molecular machinery
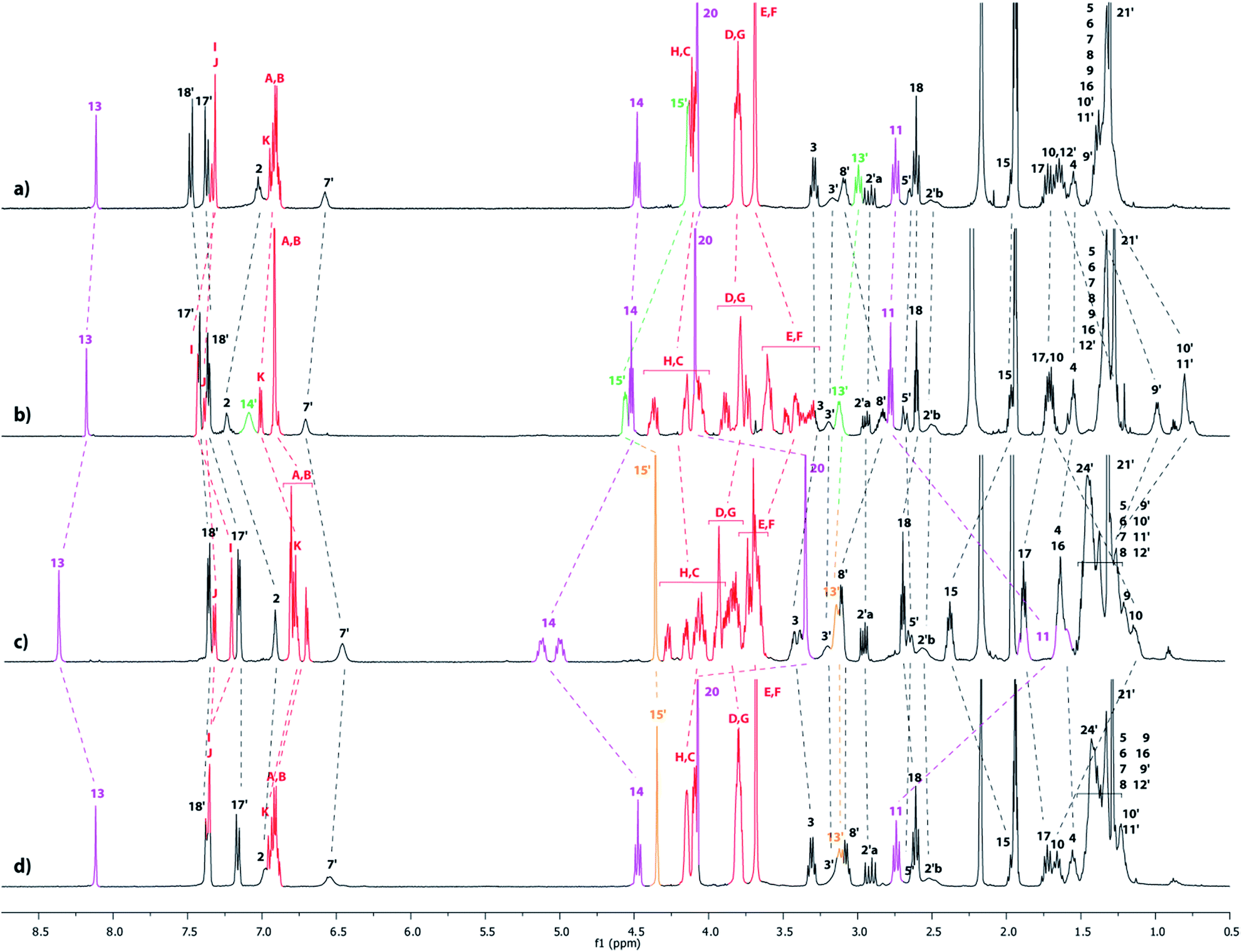
The partial tightening of the loosened lasso 4l was found to be possible through molecular machinery, upon variation in solvent polarity (Scheme 1). In acetonitrile, the DB24C8 derivative resides exclusively around the ammonium station, affording the loosened conformation for the lasso 4l. This was demonstrated by the direct comparison between the 1H NMR spectra of the [1]rotaxane 4l and its non-interlocked analogue 4u (Fig. 4a–b). First of all, only one set of 1H NMR signals can be observed in 4l, which indicates the presence of the DB24C8 only around one of the two stations.15 Moreover, no significant variation of the chemical shifts is observed for the hydrogen atoms (H11,13,14,20) of the triazolium moiety of lasso 4l and the non-interlocked analogue 4u. On the contrary, the hydrogen atoms belonging to the ammonium station, i.e. H15′ and in a lesser extent H13′, are both shifted downfield in 4l (Δδ = 0.42 and 0.13 ppm, respectively), due to their hydrogen-bonding interactions with the oxygen atoms of the DB24C8. At the same time, hydrogens H8′–12′ are all more or less shielded in 4l (from −0.54 to −0.25 ppm) because of their localization in the shielding cavity of the aromatic rings of the DB24C8. Concerning the methylene hydrogens HC–H of the DB24C8 part, they are all split in the interlocked lasso 4l, because they are facing the two non-symmetrical ends of the threaded axle. Besides, we note the upfield shift undergone by hydrogens HE–F in 4l, due to their localization in the shielding cavity of the benzyl ammonium aromatic ring of the encircled axle, therefore corroborating the localization of the DB24C8 around the ammonium station in acetonitrile. In DMSO, the behaviour of the lasso 4 appeared slightly different (Fig. 4c–e).
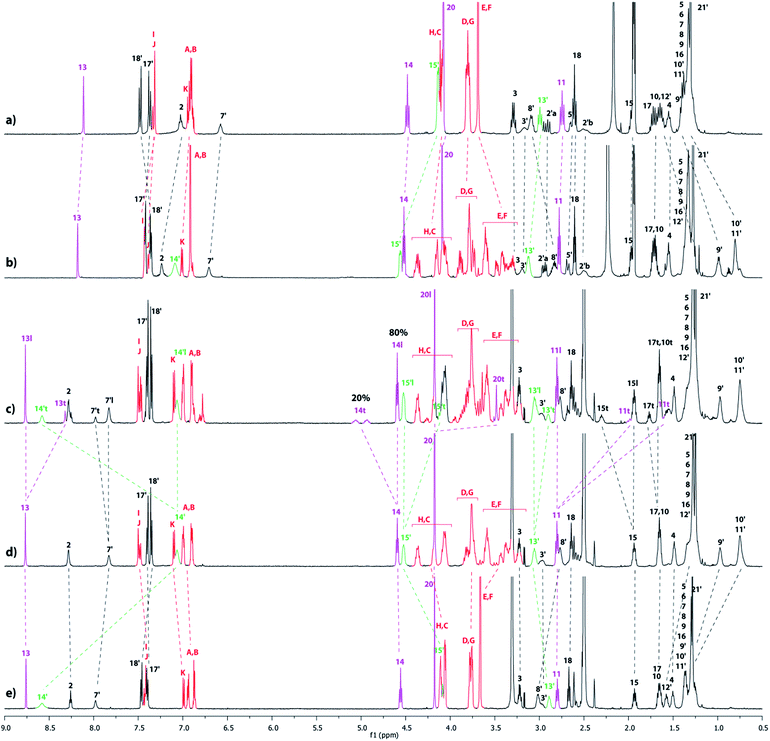 |
| | Fig. 4
1H NMR Spectra (600 MHz, 298 K) of (a) the non-interlocked analogue 4u in CD3CN, (b) the loosened [1]rotaxane 4l in CD3CN, (c) the loosened and tightened [1]rotaxanes 4l and 4t in DMSO-d6 at t = 20 h, (d) the [1]rotaxane 4l in DMSO-d6 at t = 0 h and (e) the non-interlocked analogue 4u in DMSO-d6. The numbering and colorings correspond to the hydrogen assignments indicated in Scheme 1. | |
Indeed, although the exclusive loosened conformation 4l was observed in DMSO at t = 0 (Fig. 4d and e),16 the appearance of a second set of 1H signals was noted with time. This corresponds to the tightened conformation of the lasso (4t). After 22 h, the equilibrium between the two translational conformers was reached and a ratio 4l![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4t of about 80:20 was observed (Fig. 4c). This ratio remained unchanged either upon longer time or by heating the sample. Without any doubt, the new set of signals matches with the tightened conformation 4t, in which the DB24C8 is localized around the triazolium station. This is quite a surprising result, taking into account the much better affinity of the DB24C8 for the ammonium station than for the triazolium. One may suggest that the solvation effect of the DMSO is stronger on the ammonium than on the triazolium, therefore decreasing the difference of affinity between the two respective stations for the DB24C8, hence triggering the slight displacement of the equilibrium toward the tightened lasso 4t. The tightening of the lasso 4 in DMSO was evidenced by the direct comparison between the 1H NMR spectra of the lasso 4t and the non-interlocked analogue 4u (Fig. 4c and e). To summarize, in DMSO-d6 the new set of signals matches with 4t: indeed, the chemical shifts of the hydrogen atoms belonging to the triazolium site (i.e. H11,13,14,15,20) are all affected by the new localization of the DB24C8. In 4t, it is noteworthy to point out both the deshielding and the splitting of H14 (Δδ = 0.37 and 0.5 ppm), the downfield shift of H15 (Δδ = 0.37 ppm), and the shielding of H11, H13 and H20 (Δδ from −0.44 to −1.16 ppm).17 Meanwhile, no variations at all of chemical shifts are noticed between 4t and 4u for the hydrogen atoms H13′,14′,15′ that belong to the ammonium station, which is in accordance with the tightened conformation. Although a partial tightening of the lasso 4 was possible in the more polar solvent DMSO, the obtained ratio (20%) was not found sufficient to efficiently pursue the synthesis of the triazolium-based [1]rotaxanes 6 and 7. We thus decided to use a chemical stimulus to generate the tightened lasso in high yield through the actuation of the molecular machinery.
:4t of about 80:20 was observed (Fig. 4c). This ratio remained unchanged either upon longer time or by heating the sample. Without any doubt, the new set of signals matches with the tightened conformation 4t, in which the DB24C8 is localized around the triazolium station. This is quite a surprising result, taking into account the much better affinity of the DB24C8 for the ammonium station than for the triazolium. One may suggest that the solvation effect of the DMSO is stronger on the ammonium than on the triazolium, therefore decreasing the difference of affinity between the two respective stations for the DB24C8, hence triggering the slight displacement of the equilibrium toward the tightened lasso 4t. The tightening of the lasso 4 in DMSO was evidenced by the direct comparison between the 1H NMR spectra of the lasso 4t and the non-interlocked analogue 4u (Fig. 4c and e). To summarize, in DMSO-d6 the new set of signals matches with 4t: indeed, the chemical shifts of the hydrogen atoms belonging to the triazolium site (i.e. H11,13,14,15,20) are all affected by the new localization of the DB24C8. In 4t, it is noteworthy to point out both the deshielding and the splitting of H14 (Δδ = 0.37 and 0.5 ppm), the downfield shift of H15 (Δδ = 0.37 ppm), and the shielding of H11, H13 and H20 (Δδ from −0.44 to −1.16 ppm).17 Meanwhile, no variations at all of chemical shifts are noticed between 4t and 4u for the hydrogen atoms H13′,14′,15′ that belong to the ammonium station, which is in accordance with the tightened conformation. Although a partial tightening of the lasso 4 was possible in the more polar solvent DMSO, the obtained ratio (20%) was not found sufficient to efficiently pursue the synthesis of the triazolium-based [1]rotaxanes 6 and 7. We thus decided to use a chemical stimulus to generate the tightened lasso in high yield through the actuation of the molecular machinery.
Tightening of the lasso by molecular machinery through the N-carbamoylation of the ammonium
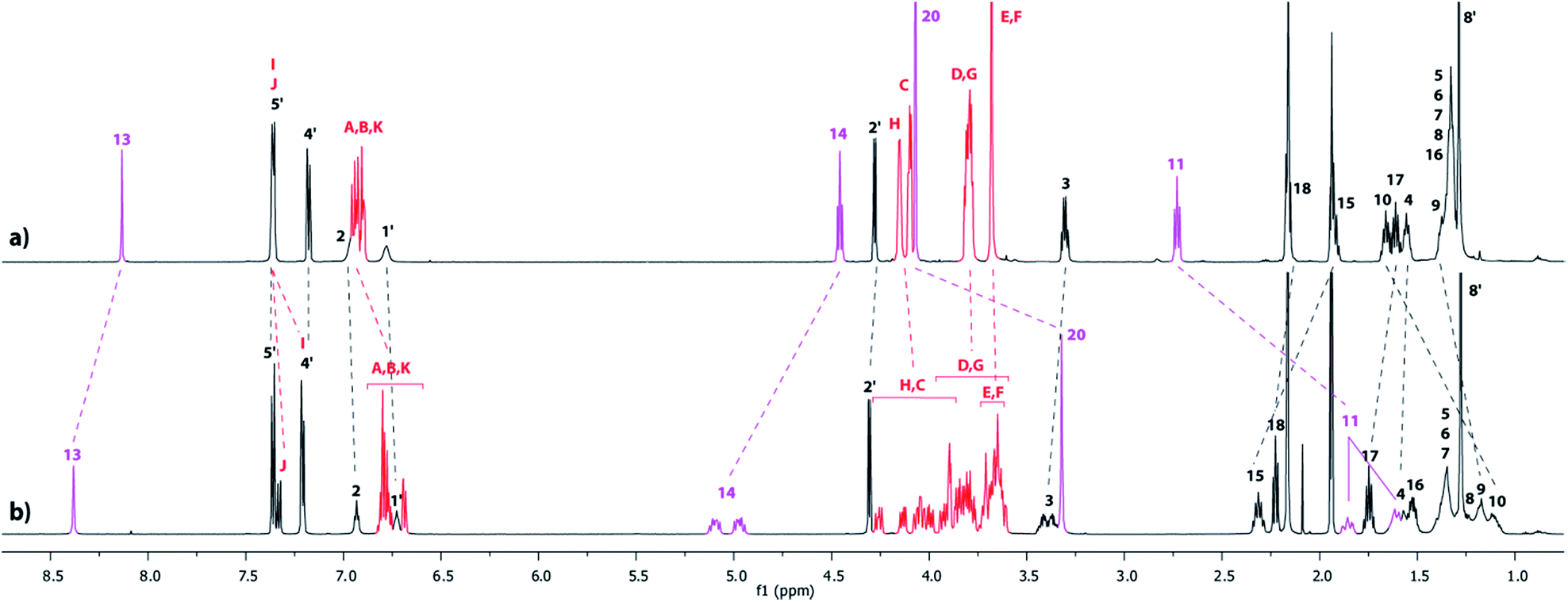
In a second approach, the N-carbamoylation of the ammonium was carried out in order to displace quantitatively the DB24C8 around the triazolium station. Similarly to the N-acetylation,18 the N-carbamoylation19 of a complex ammonium/DB24C8 was reported as a demanding task in acetonitrile because of the strong hydrogen-bonding interactions between the components. This results in the decrease of the acidity of the ammonium, therefore dramatically diminishing the kinetics of the overall reaction. In a previous paper, we improved the N-carbamoylation by using a strong base such as phosphazene, although some side-products resulted from the harsher conditions.8 Here, the N-carbamoylation was successfully achieved without any side-reaction with the help of the weak base diisopropylethylamine and Boc2O, however this is in the more polar solvent DMSO. The DMSO was effectively chosen because its high polarity decreases the binding affinity of the ammonium for the crown ether, hence increasing its acidity and facilitating its deprotonation in very mild conditions. The tightened carbamoylated lasso 5 could be obtained and isolated this way in 89% yield.20 The localization of the crown ether around the triazolium was established by comparing the 1H NMR spectra of the respective loosened and tightened lassos 4l and 5 with their respective non-interlocked analogue 4u and 5u (Fig. 5).
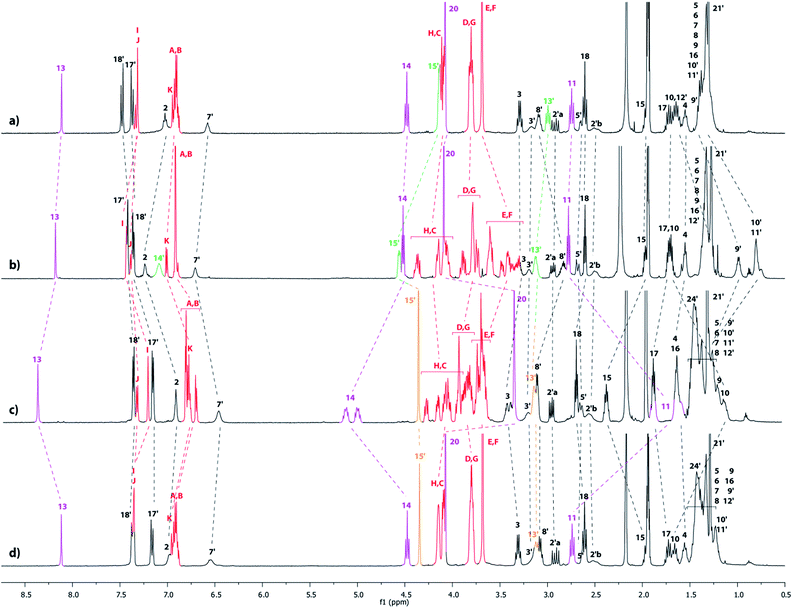 |
| | Fig. 5
1H NMR Spectra (600 MHz, CD3CN, 298 K) of (a) the non-interlocked analogue 4u, (b) the loosened [1]rotaxane 4l, (c) the N-carbamoylated tightened [1]rotaxane 5 and (d) the non-interlocked analogue 5u. The numbering and colorings correspond to the hydrogen assignments indicated in Scheme 1. | |
As already discussed in the preceding section, the comparison between the 1H NMR spectra of 4u and 4l demonstrated the localization of the crown ether around the ammonium station in 4l (Fig. 5a and b). After carbamoylation, the crown ether glides around the triazolium station, resulting in the tightening of the lasso. The conformation of the lasso 5 was evidenced by comparing the NMR spectra of 5 with 4l (Fig. 5b and c). In 5, the chemical shifts of the hydrogen atoms H13′ and H15′, which belong to the ammonium station, are slightly affected by both the displacement of the DB24C8 and the introduction of the carbamoyl moiety. More importantly, significant variations are observed for the hydrogens of the triazolium site. Indeed, in 5, the hydrogens H13 and H14 are both shifted downfield (Δδ = 0.2 and 0.54 ppm, respectively) due to their interaction through hydrogen bonding with the oxygen atoms of the DB24C8. Furthermore, H14 appears split in the tightened lasso 5, probably due to the more constrained asymmetric loop of the lasso. Meanwhile, H11, H20 and in a lesser extent H10 are all shifted upfield in 5 (Δδ = −1.05, −0.75 and −0.53 ppm, respectively), because they experience the shielding effect of the aromatic ring of the DB24C8. This is consistent with the fact that H8′–11′ are all deshielded in 5, because they no longer experience the shielding effect of the aromatic ring of the DB24C8. Besides, and for the same reason that H14, H11 appear importantly split in 5, this is an indicator of the constrained tightened lasso. The localization of the DB24C8 around the triazolium station could also be confirmed by direct comparison between the NMR spectra of 5 and 5u (Fig. 5c and d). Briefly, the only differences of chemical shifts between 5 and 5u concern the hydrogens of the DB24C8 and those of the triazolium station. As already mentioned, the hydrogens HC–H of the crown ether are split in the interlocked architecture. Moreover, H13, H14 and H15 are all shifted downfield in 5 (Δδ = +0.26, +0.59 and +0.41 ppm, respectively) with respect to 5u due to their hydrogen-bonding interactions with the oxygen atoms of the crown ether. Finally, H11, H20 and to a lesser extent H10, are all shifted upfield in 5 (Δδ = −1.01, −0.73 and −0.48 ppm, respectively) because they are localized in the shielding cavity of the aromatic rings of the DB24C8.
Contraction of the tightened lasso 5 and characterization of the mono- and tris-branched triazolium-based [1]rotaxanes 6 and 7
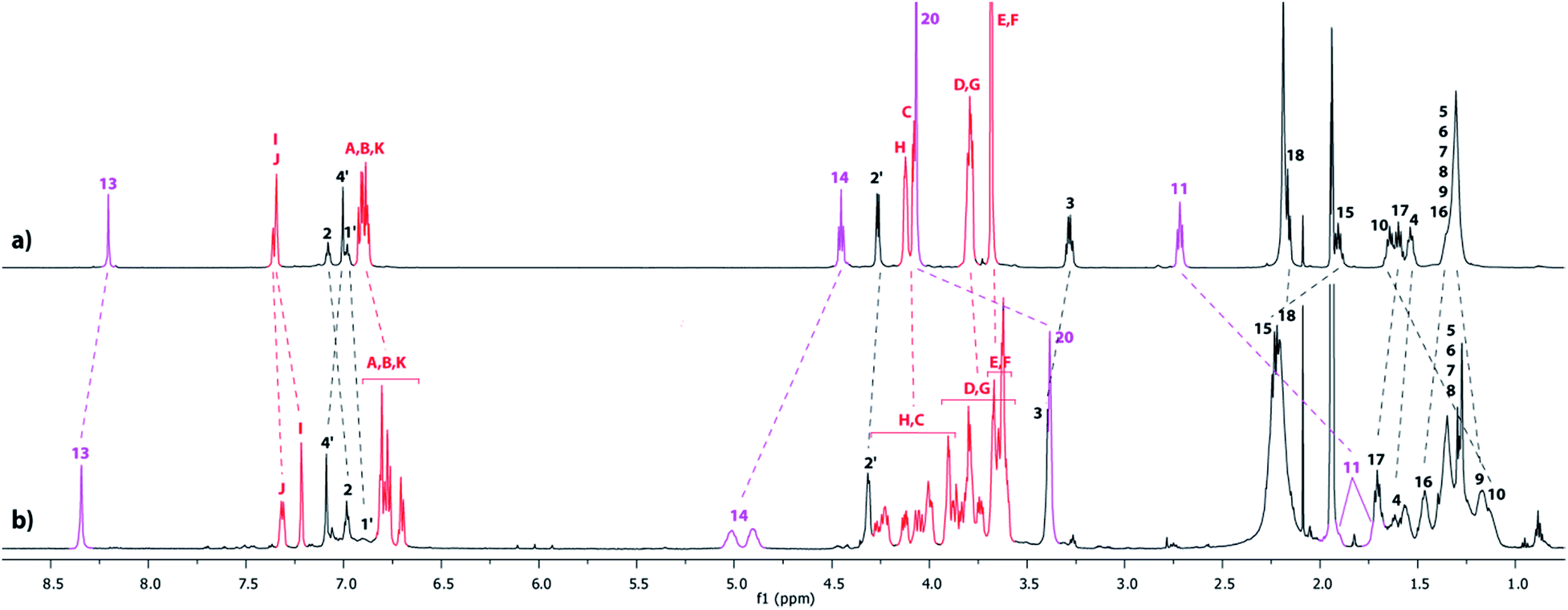
The tightened lasso 5 was designed in such a fashion that the molecular surrounded axle contains a NHS ester that can be cleaved by an amino compound at the end of the multistep sequence. An amine and a triamine were separately added to 5 in order to synthesize the triazolium-based mono and the tris-branched [1]rotaxanes 6 and 7, respectively (Scheme 1 and Fig. 2). Adding 2 equivalents of tert-butylbenzylamine in acetonitrile readily afforded the [1]rotaxane 6 in 87% yield after purification by Sephadex LH 20 chromatography. The synthesis of the tris-branched [1]rotaxane 7 was realized by adding a 0.3 equivalent of the 1,3,5-tris(aminomethyl)benzene to 1 equivalent of 5. The reaction was not as efficient as for the synthesis of 6, since only 13% of pure tris-branched [1]rotaxane 7 was isolated after successive Sephadex and silica gel chromatographies. In this case, the lower yield is not ascribed to a poorer conversion of the contraction reaction but rather to the very tricky purification of the final compound. Characterization of the interlocked molecular architectures 6 and 7 was realized by comparing their 1H NMR spectra with those of their non-interlocked analogues 6u and 7u (Fig. 6 and 7, respectively).
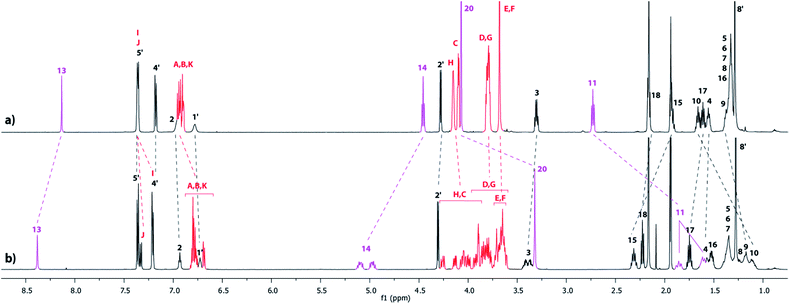 |
| | Fig. 6
1H NMR Spectra (600 MHz, CD3CN, 298 K) of (a) the non-interlocked analogue 6u and (b) triazolium-containing [1]rotaxane 6. The numbering and colorings correspond to the hydrogen assignments indicated in Fig. 2. | |
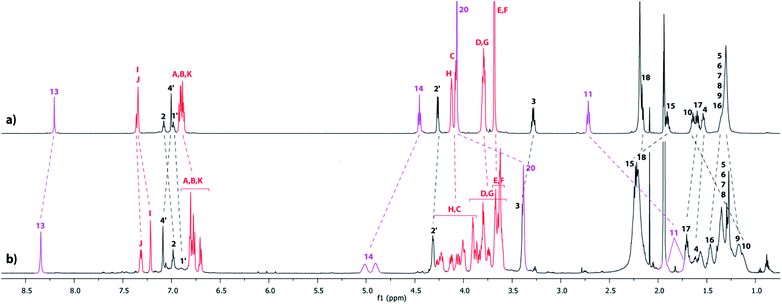 |
| | Fig. 7
1H NMR Spectra (600 MHz, CD3CN, 298 K) of (a) the non-interlocked analogue 7u and (b) triazolium-containing [1]rotaxane 7. The numbering and colorings correspond to the hydrogen assignments indicated in Fig. 2. | |
In the [1]rotaxane 6 (with respect to 6u), and like in all the DB24C8-based interlocked architectures, the methylene hydrogens HC–H are all split because they are facing the two non-symmetrical ends of the threaded axle (Fig. 6). Besides, the hydrogens of the triazolium station are highly affected by the presence of the DB24C8. The hydrogen atoms H13 and H14 and H15 are shifted downfield in 6 (Δδ = 0.24, 0.58 and 0.39 ppm, respectively) because of their interactions through hydrogen bonding with the oxygen atoms of the crown ether. On the contrary, the hydrogens H10, H11 and H20 are significantly shifted upfield (Δδ = −0.45, −0.99 and −0.74 ppm, respectively) because they experience the shielding effect of the aromatic rings of the DB24C8. Of particular interest is the shape of the signals of the hydrogens that are in the loop of the lasso. Indeed, in the [1]rotaxane 6, and as observed in the carbamoylated tightened lasso 5 (Fig. 5c) or in the protonated tightened lasso 4t (Fig. 4c), the hydrogens H14, H11, and to a lesser extent H3, all become split. The chirality of the reported [1]rotaxane architecture (i.e. left- or right-handed helix-type lasso), which is due to the non-symmetrical substitution of the crown ether by the threaded axle, may account for these results. In fact, the hydrogens that are localized in or close to the chiral loop become diastereotopic in the restrained tightened [1]rotaxane, and hence are split in the 1H NMR spectrum. On contrary, no splitting of these hydrogens is observed in the non-interlocked compound 6u, this being obvious due to the absence of the lasso.
Apart from the fact that the 1H NMR spectrum of the tris-branched triazolium-containing [1]rotaxane 7 appears a little bit broader21 than those of 6, exactly the same trend concerning the chemical shift variations and the splitting of signals was observed between the [1]rotaxane 7 and its non-interlocked analogue 7u (Fig. 7).
Conclusions
Even if interlocked components are considered as very attractive molecular targets, the efficient access to a wide range of them is still constrained, especially because they usually need to contain well-chosen moieties that can interact together in order to drive their assembly. In this paper, we have reported the efficient and straightforward preparation of triazolium-containing mono- or tris-branched [1]rotaxanes that could not be obtained through classical methods. Whereas triazolium consists of a quite effective molecular station for DB24C8 derivatives in a locked structure like rotaxane, it cannot be used as a template to drive the formation of a DB24C8-based interlocked component through intermolecular interactions. This is mainly due to the too weak interactions between the moieties. The possibility to hypothetically synthesize a triazolium-based semirotaxane, from a DB24C8 derivative and a triazolium-containing molecular axle, is highly limited by the loss in entropic factor, which becomes here predominant with respect to the weak gain in enthalpy (i.e. the very weak interactions between the two components). Here, the triazolium-containing [1]rotaxane 6 could be obtained using a five-step sequence from the azidohexanoic acid in a 36% overall yield. The possibility to add a polyamine at the last step of the synthesis allows extension of the method to multi-branched22 triazolium-based [1]rotaxanes. As an example, the tris-branched [1]rotaxane 7 was prepared and isolated. The improvement of the chemical access to a wide range of [1]rotaxanes that does not necessarily contain a template is a substantial advance for the synthesis of new structures of interest.
Acknowledgements
P. W. is funded by the LABEX ChemiSyst (ANR-10-LABX-05-01).
Notes and references
-
(a) S. F. M. van Dongen, S. Cantekin, J. A. A. W. Elemans, A. E. Rowan and R. J. M. Nolte, Chem. Soc. Rev., 2014, 43, 99–122 RSC;
(b) M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211–225 RSC;
(c) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed;
(d) J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 11102–11104 CrossRef CAS PubMed;
(e)
J.-P. Sauvage, C. Dietrich-Buchecker and G. Rapenne, in Molecular Catenanes, Rotaxanes and Knots, ed. J.-P. Sauvage and C. Dietrich-Buchecker, Wiley-VCH Weinheim, 1999, ch. 6, pp. 107–139 Search PubMed;
(f) G. A. Breault, C. A. Hunter and P. C. Mayers, Tetrahedron, 1999, 55, 5265–5293 CrossRef CAS;
(g) G. Barin, A. Coskun, M. M. G. Fouda and J. F. Stoddart, ChemPlusChem, 2012, 77, 159–185 CrossRef CAS PubMed;
(h) B. Champin, P. Mobian and J.-P. Sauvage, Chem. Soc. Rev., 2007, 36, 358–366 RSC;
(i) J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC;
(j) J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611–619 CrossRef CAS;
(k) D. H. Qu and H. Tian, Chem. Sci., 2011, 2, 1011–1015 RSC;
(l) D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015 DOI:10.1021/cr5006342.
-
F. Coutrot, in Advances in Atom and Single Molecule Machines, Single Molecular Machines and Motors, ed. C. Joachim and G. Rapenne, Springer International Publishing, Switzerland, 2015, p. 35 Search PubMed.
- K. Hiratani, M. Kaneyama, Y. Nagawa, E. Koyama and M. Kanesato, J. Am. Chem. Soc., 2004, 126, 13568–13569 CrossRef CAS PubMed.
-
(a) P. Franchi, P. Fani, E. Mezzina and M. Lucarini, Org. Lett., 2008, 10, 1901–1904 CrossRef CAS PubMed;
(b) Q. Zhou, P. Wei, Y. Zhang, Y. Yu and X. Yan, Org. Lett., 2013, 15, 5350–5353 CrossRef CAS PubMed.
-
(a) X. Ma, Q. Wang and H. Tian, Tetrahedron Lett., 2007, 48, 7112–7116 CrossRef CAS PubMed;
(b) L. Zhu, H. Yan and Y. Zhao, Int. J. Mol. Sci., 2012, 13, 10132–10142 CrossRef CAS PubMed;
(c) H. Li, J.-N. Zhang, W. Zhou, H. Zhang, Q. Zhang, D.-H. Qu and H. Tian, Org. Lett., 2013, 15, 3070–3073 CrossRef CAS PubMed;
(d) H. Li, X. Li, H. Agren and D.-H. Qu, Org. Lett., 2014, 16, 4940–4943 CrossRef CAS PubMed;
(e) X. Ma, D.-H. Qu, F. Ji, Q. Wang, L. Zhu, Y. Xu and H. Tian, Chem. Commun., 2007, 1409–1411 RSC;
(f) H. Li, H. Zhang, Q. Zhang, Q.-W. Zhang and D. Qu, Org. Lett., 2012, 14, 5900–5903 CrossRef CAS PubMed.
- S. Tsuda, J. Terao and N. Kambe, Chem. Lett., 2009, 38, 76–77 CrossRef CAS.
-
(a) Z. Xue and M. F. Mayer, J. Am. Chem. Soc., 2010, 132, 3274–3276 CrossRef CAS PubMed;
(b) C. Clavel, C. Romuald, E. Brabet and F. Coutrot, Chem.–Eur. J., 2013, 19, 2982–2989 CrossRef CAS PubMed;
(c) C. Clavel, K. Fournel-Marotte and F. Coutrot, Molecules, 2013, 18, 11553–11575 CrossRef CAS PubMed.
- S. Chao, C. Romuald, K. Fournel-Marotte, C. Clavel and F. Coutrot, Angew. Chem., Int. Ed., 2014, 53, 6914–6919 CrossRef CAS PubMed.
- Apart from ref. 8, only one other similar approach has been reported to date, however for the preparation of [2]rotaxanes using a five-component “clipping” rotaxane formation, see: J. S. Hannam, S. M. Lacy, D. A. Leigh, C. G. Saiz, A. M. Z. Slawin and S. G. Stitchell, Angew. Chem., Int. Ed., 2004, 43, 3260–3264 CrossRef CAS PubMed.
-
(a) P. R. Ashton, I. Baxter, M. C. T. Fyfe, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 2297–2307 CrossRef CAS;
(b) Y. Tokunaga, N. Wakamatsu, A. Ohbayashi, K. Akasaka, S. Saeki, T. Hisada, T. Goda and Y. Shimomura, Tetrahedron Lett., 2006, 47, 2679–2682 CrossRef CAS PubMed;
(c) F. M. Raymo, K. N. Houk and J. F. Stoddart, J. Am. Chem. Soc., 1998, 120, 9318–9322 CrossRef CAS;
(d) F. M. Raymo and J. F. Stoddart, Pure Appl. Chem., 1997, 69, 1987–1997 CrossRef CAS;
(e) M. Händel, M. Plevoets, S. Gestermann and F. Vögtle, Angew. Chem., Int. Ed. Engl., 1997, 36, 1199–1201 CrossRef PubMed;
(f) S.-Y. Hsueh, C.-C. Lai, Y.-H. Liu, Y. Wang, S.-M. Peng and C.-C. Hsu, Org. Lett., 2007, 9, 4523–4526 CrossRef CAS PubMed;
(g) J.-J. Lee, A. G. White, J. M. Baumes and B. D. Smith, Chem. Commun., 2010, 46, 1068–1069 RSC;
(h) A. J. McConnell and P. D. Beer, Chem.–Eur. J., 2011, 17, 2724–2733 CrossRef CAS PubMed;
(i) P. R. Ashton, M. Belohradsky, D. Philp and J. F. Stoddart, J. Chem. Soc., Chem. Commun., 1993, 1269–1274 RSC;
(j) M. Asakawa, P. R. Ashton, R. Ballardini, V. Balzani, M. Belohradsky, M. T. Gandolfi, O. Kocian, L. Prodi, F. M. Raymo, J. F. Stoddart and M. Venturi, J. Am. Chem. Soc., 1997, 119, 302–310 CrossRef CAS;
(k) M. A. Bolla, J. Tiburcio and S. J. Loeb, Tetrahedron, 2008, 64, 8423–8427 CrossRef CAS PubMed;
(l) P. Linnartz, S. Bitter and C. A. Schalley, Eur. J. Org. Chem., 2003, 4819–4829 CrossRef CAS PubMed;
(m) H. W. Gibson, N. Yamaguchi, Z. Niu, J. W. Jones, C. Slebodnick, A. L. Rheingold, L. N. Zakharov and J. Polym, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 975–985 CrossRef CAS PubMed.
- A. G. Kolchinski, D. H. Busch and N. W. Alcock, J. Chem. Soc., Chem. Commun., 1995, 1289–1291 RSC.
-
(a) R. Huisgen, Pure Appl. Chem., 1989, 61, 613–628 CrossRef CAS;
(b) R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS PubMed;
(c) R. Huisgen, Angew. Chem., 1963, 75, 604–637 (
Angew. Chem., Int. Ed. Engl.
, 1963
, 2
, 565–598
) CrossRef CAS PubMed;
(d) R. Huisgen, Angew. Chem., 1963, 75, 742–754 (
Angew. Chem., Int. Ed. Engl.
, 1963
, 2
, 633–645
) CrossRef CAS PubMed.
-
(a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS;
(b) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
-
(a) F. Coutrot and E. Busseron, Chem.–Eur. J., 2008, 14, 4784–4787 CrossRef CAS PubMed;
(b) F. Coutrot, C. Romuald and E. Busseron, Org. Lett., 2008, 10(17), 3741–3744 CrossRef CAS PubMed;
(c) F. Coutrot, ChemistryOpen DOI:10.1002/open.201500088.
- The shuttling of the macrocycle between the two stations is slow at the NMR timescale.
- Exactly the same trend of variations of the chemical shifts between hydrogens of 4l and 4u is observed in DMSO-d6 at t = 0 and in CD3CN, corroborating the localization of the DB24C8 around the ammonium station in both solvents.
- For 4t in DMSO, it is interesting to note that H13 is unusually shielded by the aromatic rings of the crown ether, instead of being shifted downfield due to hydrogen-bonding interactions. This is ascribed to two concomitant factors. First, the high polarity of the DMSO solvent decreases the strength of hydrogen bonds. Second, the constrained tightened lasso structure does not allow an optimal tridimensional arrangement between H13 and the oxygen atoms of the crown ether without inducing steric repulsion. Therefore, in this particular case, the oxygen atoms of the crown ether prefer to interact through hydrogen bonds with the sole hydrogen atoms H14 which are located slightly further from the loop. A similar shielding effect underwent by the considered triazolium hydrogen was noticed once in a tightened BMP25C8-based [1]rotaxane. In that case, the larger crown ether (not the solvent) and the constrained lasso both disfavour the hydrogen bond between the triazolium hydrogen and the oxygen atoms of the BMP25C8, resulting in the same shielding effect, even in the less polar dichloromethane, see ref. 7b.
- Y. Tachibana, H. Kawasaki, N. Kihara and T. Takata, J. Org. Chem., 2006, 71, 5093–5104 CrossRef CAS PubMed.
- C. Romuald, E. Busseron and F. Coutrot, J. Org. Chem., 2010, 75, 6516–6531 CrossRef CAS PubMed.
- The [1]rotaxanes 5, 4 and 3, as well as the [2]rotaxane 2, may exist as a mixture of stereoisomers which are not distinguishable by 1H NMR. They arise from both the (R)/(S) stereochemistry of the asymmetric carbon of the NHS moiety and the non-symmetrical substitution of the DB24C8. For recent examples about stereoisomerism in rotaxane structures, see:
(a) C. Reuter, C. Seel, M. Nieger and F. Vögtle, Helv. Chim. Acta, 2000, 83, 630–640 CrossRef CAS;
(b) Q.-F. Luo, L. Zhu, S.-J. Rao, H. Li, Q. Miao and D.-H. Qu, J. Org. Chem., 2015, 80, 4704–4709 CrossRef CAS PubMed;
(c) R. J. Bordoli and S. M. Goldup, J. Am. Chem. Soc., 2014, 136, 4817–4820 CrossRef CAS PubMed.
- The broadness of the 1H NMR spectrum of 7 may result from the existence of several stereoisomers, due to the chiral loop of the lasso (i.e. right- or left-handed helix type lasso).
- For recent multi-branched interlocked structures, see:
(a) J.-N. Zhang, H. Li, W. Zhou, S.-L. Yu, D.-H. Qu and H. Tian, Chem.–Eur. J., 2013, 19, 17192–17200 CrossRef CAS PubMed;
(b) H. Zhang, Q. Liu, J. Li and D.-H. Qu, Org. Lett., 2013, 15, 338–341 CrossRef CAS PubMed;
(c) H. Li, X. Li, Z.-Q. Cao, D.-H. Qu, H. Ågren and H. Tian, ACS Appl. Mater. Interfaces, 2014, 6, 18921–18929 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures and characterization data for all compounds. See DOI: 10.1039/c5sc01722j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence