
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold(I)-catalyzed cycloisomerization of vinylidenecyclopropane-enes via carbene or non-carbene processes†
De-Yao
Li
a,
Yin
Wei
a,
Ilan
Marek
b,
Xiang-Ying
Tang
*a and
Min
Shi
*a
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: siocxiangying@mail.sioc.ac.cn; mshi@mail.sioc.ac.cn
bSchulich Faculty of Chemistry, Technion–Israel Institute of Technology, Technion City, Haifa 32000, Israel. E-mail: chilanm@tx.technion.ac.il
First published on 24th June 2015
Abstract
Gold catalyzed cycloisomerization of aromatic ring tethered vinylidenecyclopropane-enes provides a divergent synthetic protocol for the construction of O-containing fused heterocycles through controllable carbene or non-carbene related processes. The carbene induced process features a new amphiphilic strategy to generate a gold carbene via a rearrangement of vinylidenecyclopropane. Whereas, the electronic effect of the ortho-substituents switches the reaction mode onto the non-carbene related process, from which five- or six-membered rings are selectively produced through allyl-migration.
Recently, the field of gold catalysis has witnessed significant developments.1 The exploration of new reaction modes in this arena has emerged as one of those at the forefront of current research. Due to the relativistic effect observed with gold and gold carbene complexes,2 these species present unique properties and reactivities. Impressive work has appeared recently,3 providing novel reaction pathways that could be summarized in Scheme 1: (1) Nuc–E strategies have been widely used to form α-functionalized gold carbenes;4 (2) retro-Buchner reaction can be easily used in situ to generate gold carbenes as a potentially common method;5 (3) the Au-σ-activated alkynes could also produce gold vinylidene species, which can act as gold carbenes through versatile reaction pathways.6 However, to the best of our knowledge, there are barely any reports on one precursor being able to rapidly generate molecular complexity7via either carbene or non-carbene pathways in gold catalysis.8 On the basis of our ongoing investigation on metal-catalyzed transformations of vinylidenecyclopropanes (VDCPs),9 we envisaged that VDCPs could be excellent candidates for the exploration of new reaction modes in gold catalysis because of their multiple reaction sites. Previously, Toste and co-workers reported novel intramolecular cyclopropanation and allyl-transfer, both through gold carbene intermediates (Scheme 2).4q–s Interestingly, during our study on the gold catalyzed cycloisomerization of aromatic ring tethered VDCP-enes, we found that cyclopropanation and allyl transfer through controllable carbene/non-carbene related processes could be achieved, respectively, featuring a new gold carbene generation process. The VDCP acts as a nucleophile under activation by the gold(I) complex and then undergoes ring expansion as an electrophile due to its amphiphilic electronic nature to generate gold carbene species. Herein, we wish to report these intriguing new gold-catalyzed transformations, which afford an easy and efficient access to fused five-, six- and eight-membered ring systems (Scheme 2).
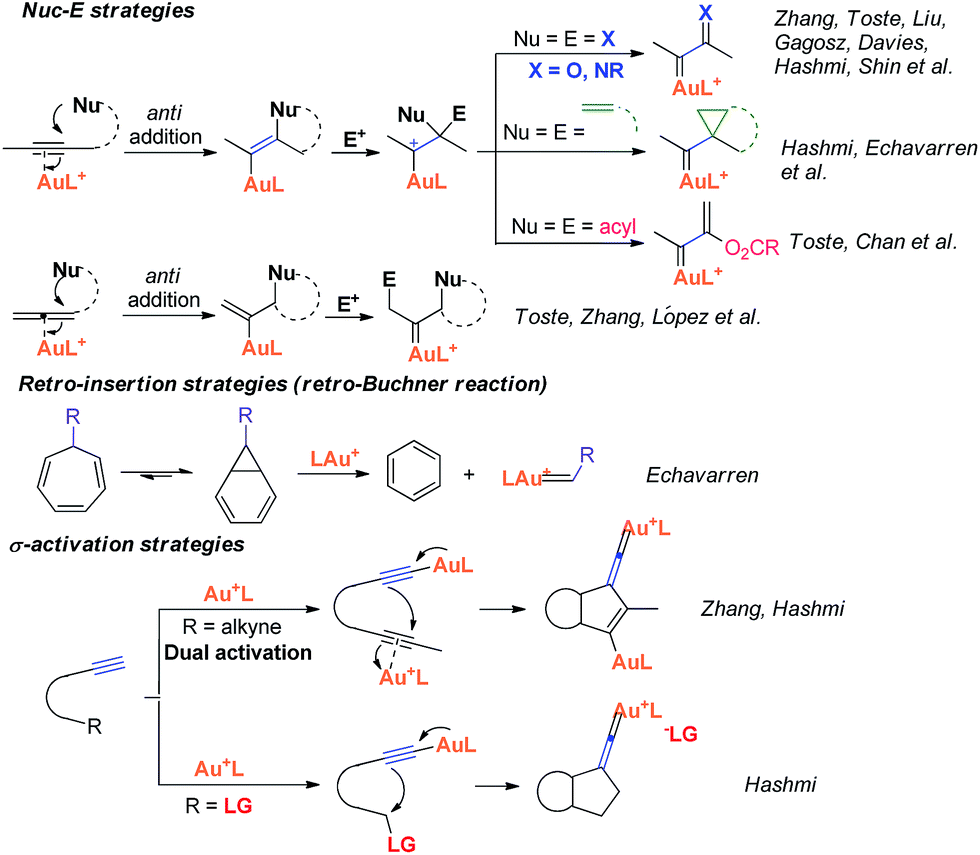 | ||
| Scheme 1 Previous strategies for the generation of gold carbenes. | ||
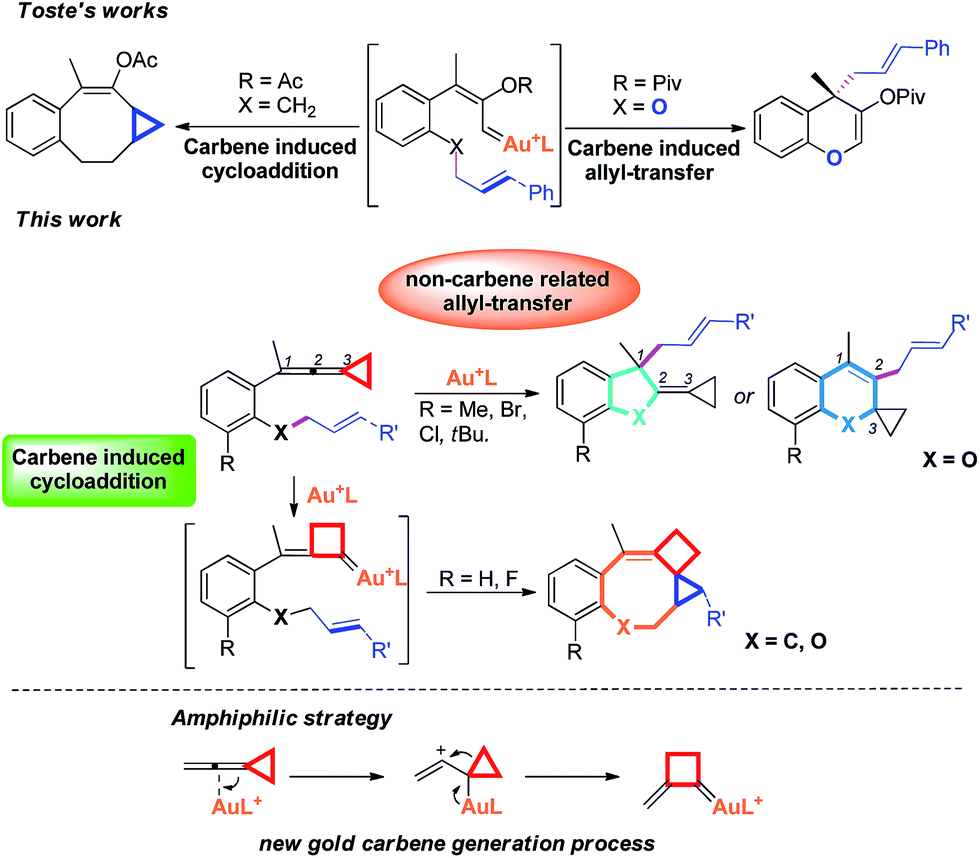 | ||
| Scheme 2 Modulable reaction processes of VDCP-enes catalyzed by gold(I) complexes along with a new gold carbene generation process. | ||
Upon reaction condition screening (see ESI† for the details), we identified JohnPhosAuCl (10 mol%) as the best gold catalyst together with AgNTf2 (10 mol%) to carry out the reaction of 1a. Product 2a10 was formed in 93% yield at ambient temperature in DCE within 5 minutes (Scheme 3).
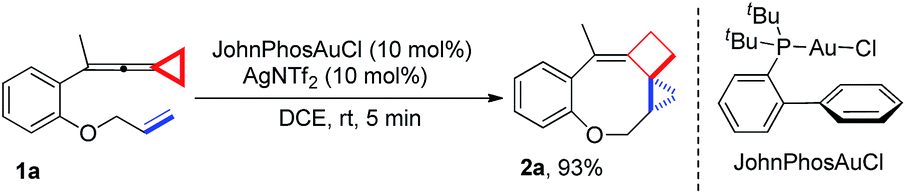 | ||
| Scheme 3 Optimal conditions for the synthesis of 2a. | ||
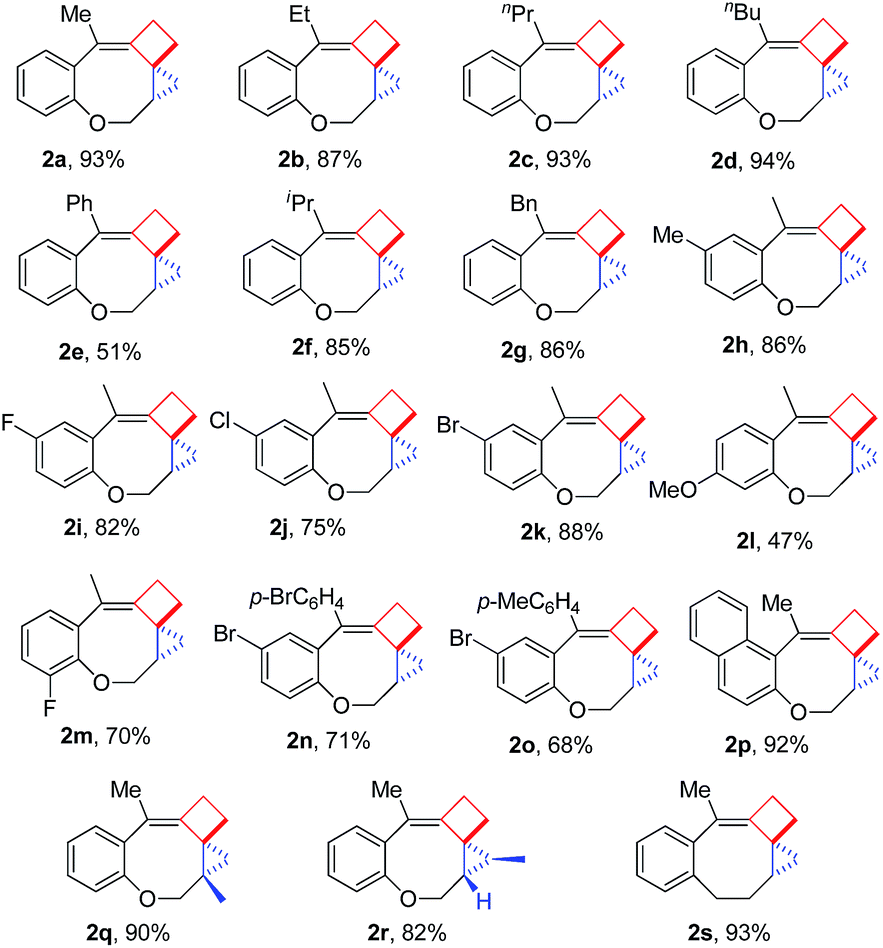
Having the optimal reaction conditions in hand, we next investigated the scope of the reaction with respect to various VDCP-enes. As shown in Table 1, these cyclization reactions proceeded smoothly, affording the desired products in moderate to excellent yields. Although this transformation proceeds in high yields for alkyl-substituted VDCPs (R2 = alkyl), yield is slightly lower for phenyl-substituted VDCP (R2 = Ph, 2e). As for the substituents at the benzene ring, no obvious decrease of yields was observed when different halogen atoms such as F, Cl or Br were introduced. Substrate 1l with electron-donating substituent MeO at the benzene ring afforded product 2l in 47% yield.11 The naphthalene tethered substrate gave the expected product 2p in good yield. Substrates 1q and 1r bearing a methyl substituent at the allyl group also afforded the corresponding products 2q and 2r in 90% and 82% yields, respectively. When a homoallylic group was introduced instead of an allyloxy group, the desired product 2s could be given in 93% yield. It should be noted that only product 2l was obtained in lower yield but this may come from the instability of the starting material. The DFT calculations indicated that the formation of 2 is an exothermic process (see ESI†).
|
|
|---|
| a The reaction free energy ΔGrxn,298 is −17.9 kcal mol−1 (obtained by DFT calculations, for details, see ESI). This result suggests that the formation of product 2 is exothermic. |
|
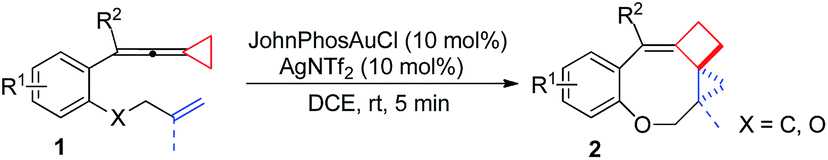
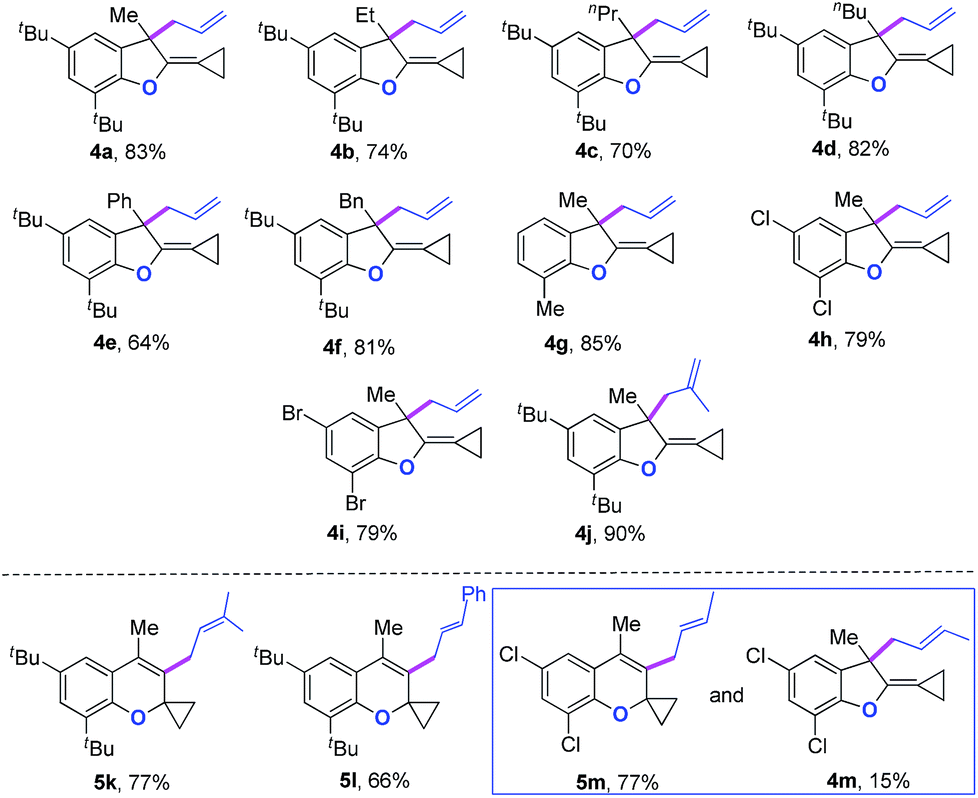
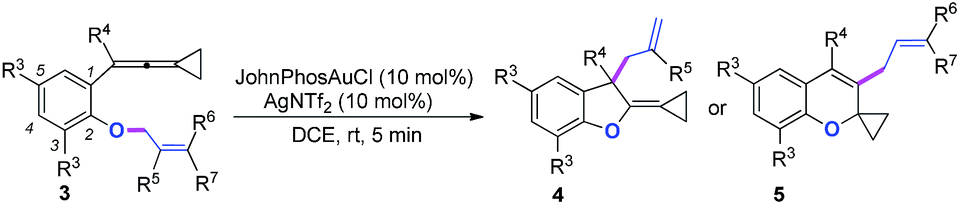
We next synthesized substrate 3a from 3,5-di-tbutyl salicylaldehyde to examine the reaction outcome. However, we found that an allyl-transferred product 4a derived from a non-carbene process was formed in 83% yield rather than the cyclopropanation product (Table 2).12 To have better insight on the substituent effects, we synthesized a library of substrates to clarify the scope. Consistent with the above results, all of these VDCP-enes having a substituent such as tBu, Me or Cl at 3-position gave the allyl-transferred products in good yields. Only when a fluorine atom was introduced at the 3-position, did the reaction proceed via a carbene process, giving 2m in 70% yield as shown in Table 1. As for substrates 3k and 3l having two methyl groups or one phenyl group at the terminal position of the alkene, another type of allyl-transfer took place, affording 5k13 and 5l in 77% and 66% yields, respectively (Table 2). In the case of 3m, two types of allyl-transfer took place at the same time to give 5m and 4m in good total yield as a product mixture (Table 2). 3k's regioisomer, O-(1,1-dimethylprop-2-enyl) derivative, is unavailable via the present synthetic method (see Scheme at page S8 in ESI† for the details).
|
|
|---|
|
Two mechanistic experiments were conducted to clarify the allyl-transfer process of the allyloxyl group (Scheme 4). Treatment of [D]-3a under the standard conditions produced [D]-4a in 79% yield along with >99% D content. The deuterium labeling experiments suggesting that the allyl-transfer might proceed through a 1,3-shift of the allylic oxonium intermediate rather than a 3,3-shift1g,4s because no allylic inversion was observed. Furthermore, the double cross-over experiment using 3k and 3n as substrates under the standard conditions only produced the corresponding products 5k and 5n, respectively and no cross-over products were observed, indicating that the allyl-transfer proceeded via an intimate ion-pair.
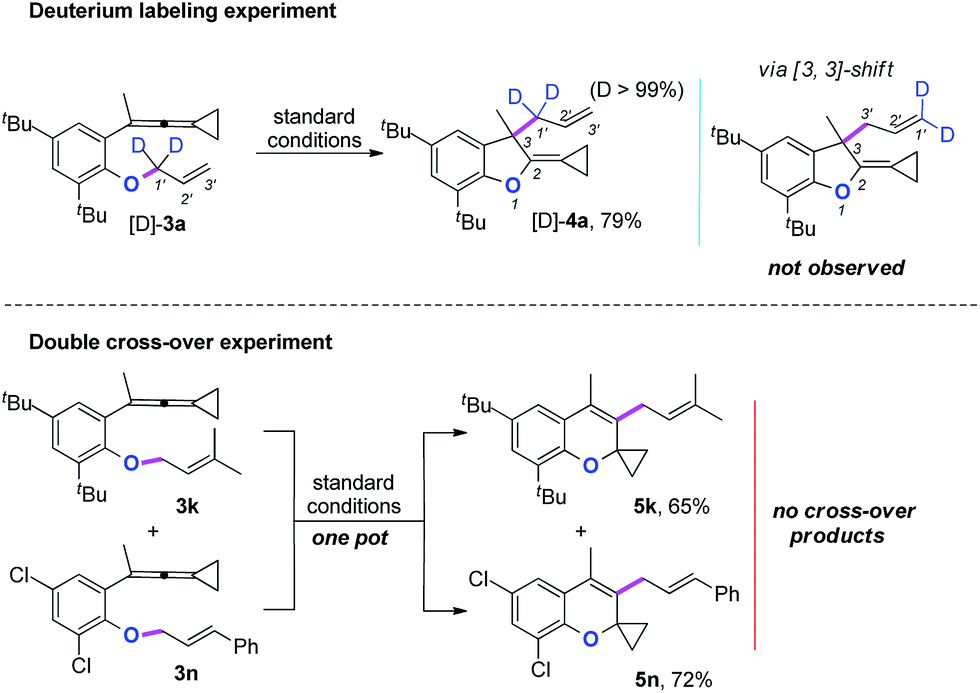 | ||
| Scheme 4 Mechanistic experiments. | ||
Based on Toste's previous work and other groups' results, plausible mechanisms4s,14 for the above gold-catalyzed carbene and non-carbene related processes are outlined in Scheme 5. As for substrate 1a, upon coordination of gold catalyst with VDCP intermediate A was formed, which initiates a ring expansion to give gold stabilized cation B and then carbene species C. Subsequent cyclopropanation produces polycyclic adduct 2a. When the ortho-position is substituted by a Me, Cl, Br or tBu group, due to the increased nucleophilicity of the oxygen atom, 3g attacks the middle carbon of the allenyl moiety in intermediate D to give the corresponding oxonium intermediate E, which undergoes S′E allyl-transfer15 to afford product 4gvia an intimate ion-pair F. In the case of 3k, the oxygen atom exclusively attacks the terminal carbon at the allene moiety of VDCP in intermediate G to give the corresponding oxonium intermediate H presumably due to the steric bulkiness at the alkene site, which similarly undergoes the allyl-transfer to give 5kvia an intimate ion-pair I. Overall, the gold species is finally quenched by the allylic cation to give the allyl-transferred product. The allyl-migration mode is different from previous work,4s,15b,c probably due to the steric hindrance of cyclopropane in the VDCP-type substrates.
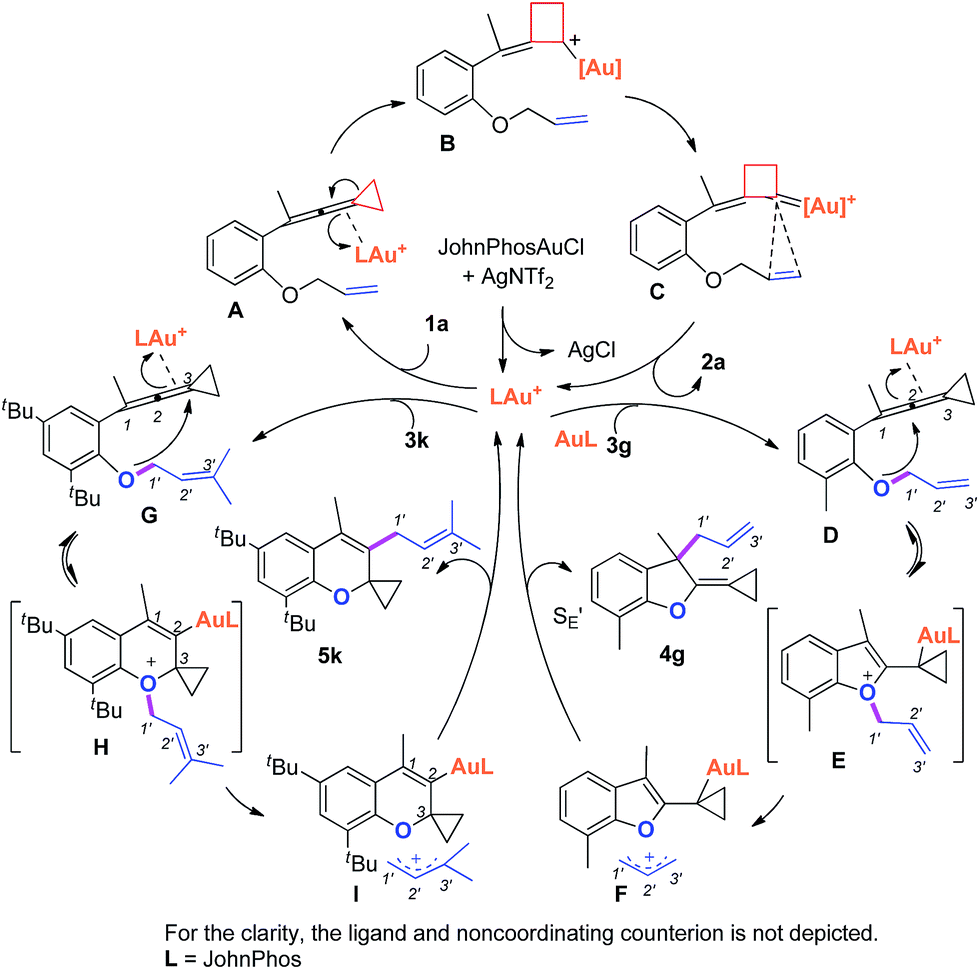 | ||
| Scheme 5 Proposed mechanisms. | ||
The X-ray crystal structures of substrates 1n and 3e indicated that the distances between the oxygen and the central carbon of VDCP are very close (for details, see ESI†), suggesting that it is not the ortho-substituent that induces a proximity effect promoting the nucleophilic attack. We believe that the electronic effects of the ortho-substituents in the substrates influence the nucleophilicity of the oxygen atom, which is the origin for the different reactivities of substrates 2 and 3. Calculation of Mulliken charge on the O atom influenced by X (a substituent or a substituted group at the ortho-position) was carried out on the basis of the B3LYP/6-31+G(d) level, inferring that different substituents led to different electron densities on the oxygen atom in the following order of tBu > Br > Cl > H > F (Table 3). In a similar manner, Mulliken charge on the O atom influenced by Y (a substituent at the para-position) was also calculated at the B3LYP/6-31+G(d) level, and the results show that the substituent at the para-position does not influence the electron density on the oxygen atom significantly (Table 3). These results suggest that the nucleophilicity of the oxygen is tunable by a judicious choice of substituents at the 3-position of the benzene ring, controlling therefore the carbene or non-carbene related processes at the very beginning. The thermodynamic stability of the subsequent oxonium intermediate E with different ortho-substituents was also investigated by DFT calculations (see ESI† for the details). The reactions were probably controlled by kinetic factors judging by the reaction conditions (5 minutes at room temperature). In order to understand why the substituents on the terminal alkene can affect the reaction outcome, we performed DFT calculations on key steps in the reaction of substrate 3k. All calculations have been performed at the B3LYP/6-31+G(d)/SDD level with the Gaussian 09 program (see ESI† for the details). We investigated two reaction pathways starting from gold complex 3k-D in Scheme 6. In path a, the oxygen atom in gold complex 3k-D attacks the terminal carbon at the allene moiety to give an intermediate 3k-Ivia transition state 3k-TS1 with an energy barrier of 7.7 kcal mol−1. Subsequently, the intermediate 3k-I passes through transition state 3k-TS2 with an energy barrier of 2.3 kcal mol−1, giving product complex 5k-P. On the other hand, in path b, the oxygen atom in gold complex 3k-D attacks the middle carbon at the allene moiety to give an intermediate 3k-Fvia transition state 3k-TS1′ with an energy barrier of 11.2 kcal mol−1. The energy of 3k-TS1′ is higher than that of 3k-TS1 by 3.5 kcal mol−1, presumably due to the steric repulsions among the substituents on the terminal alkene, the t-Bu substituent, and the ligand (for optimized structures of 3k-TS1′ and 3k-TS1, see Fig. 1). Subsequently, the intermediate 3k-F passes through transition state 3k-TS2′ with an energy barrier of 9.0 kcal mol−1, giving product complex 4k-P. The calculation results show that all intermediates along path a are thermodynamically more stable than those along path b, meanwhile path a is also kinetically favourable, indicating that the product 5k is the major product. The calculation results are in line with experimental results which obtained the product 5k using 3k as starting material. For comparison, we also investigated two pathways for the reaction of 3a. The relative energies of all intermediates and transitional states along the reaction pathway for the reaction of 3a are shown in Scheme 7. Similarly, the oxygen atom in gold complex 3a-D attacks the terminal carbon at the allene moiety to give an intermediate 3a-Ivia transition state 3a-TS1 with an energy barrier of 9.3 kcal mol−1 in path a′. Subsequently, the intermediate 3a-I passes through transition state 3a-TS2 with an energy barrier of 8.4 kcal mol−1, giving product complex 5a-P. On the other hand, in path b′, the oxygen atom in gold complex 3a-D attacks the middle carbon at the allene moiety to give an intermediate 3a-Fvia transition state 3a-TS1′ with an energy barrier of 8.9 kcal mol−1. The energy of 3a-TS1′ is slightly lower than that of 3a-TS1 by 0.9 kcal mol−1, probably due to the lack of steric repulsions among the substituents on terminal alkene, the t-Bu substituent, and the ligand. Subsequently, the intermediate 3a-F passes through transition state 3a-TS2′ with an energy barrier of 6.3 kcal mol−1, giving product complex 4a-P, which is lower than that of product complex 5a-P by 1.8 kcal mol−1. The calculation results show that the path b′ is also kinetically favourable, indicating that the product 4a is the major product. The calculation results are in line with experimental results which obtained the product 4a using 3a as starting material.
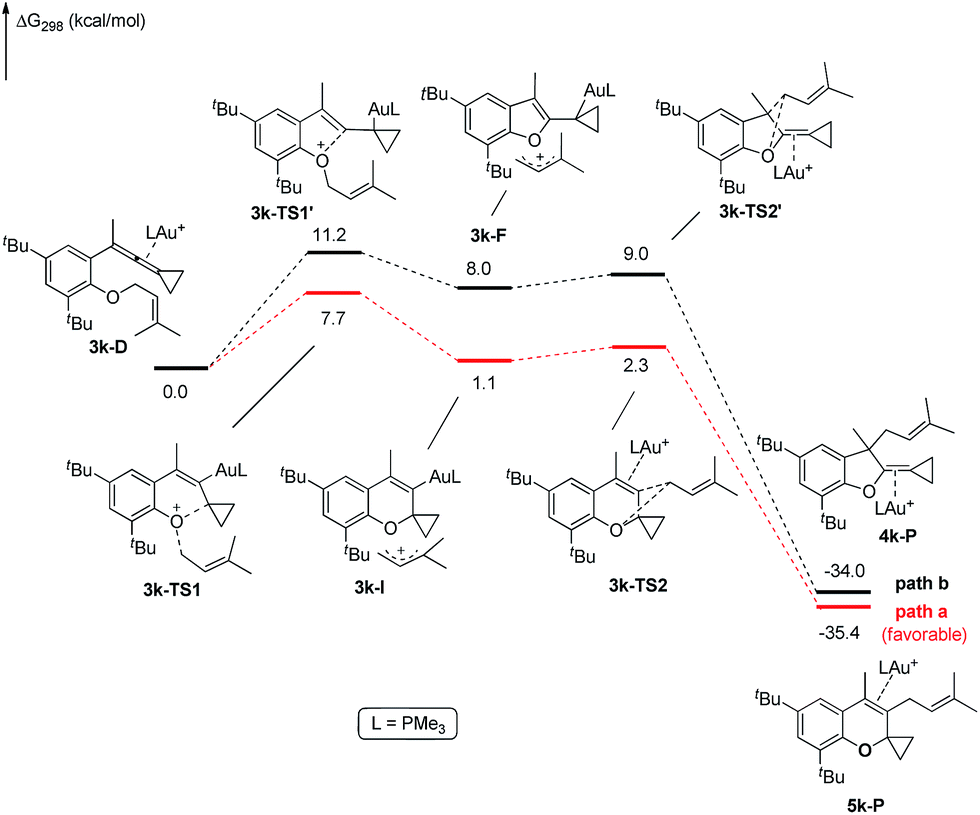 | ||
| Scheme 6 DFT studies on key reaction steps of the reaction of 3k. | ||
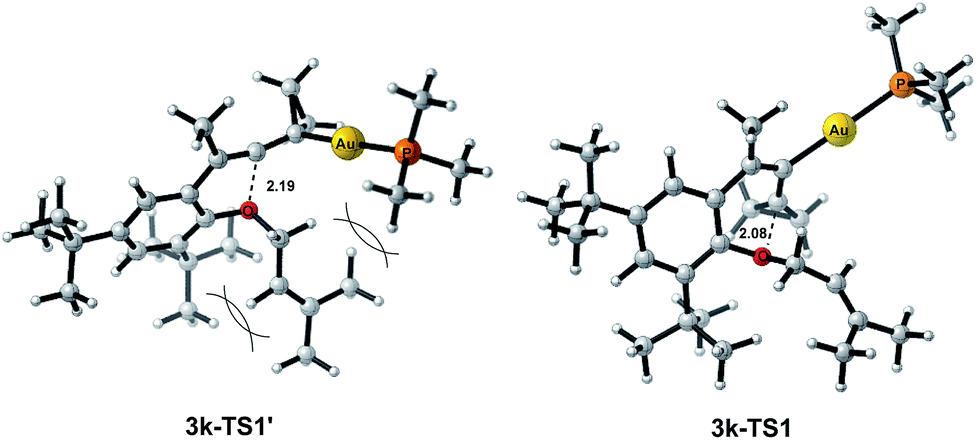 | ||
| Fig. 1 Optimized structures of 3k-TS1′ and 3k-TS1. | ||
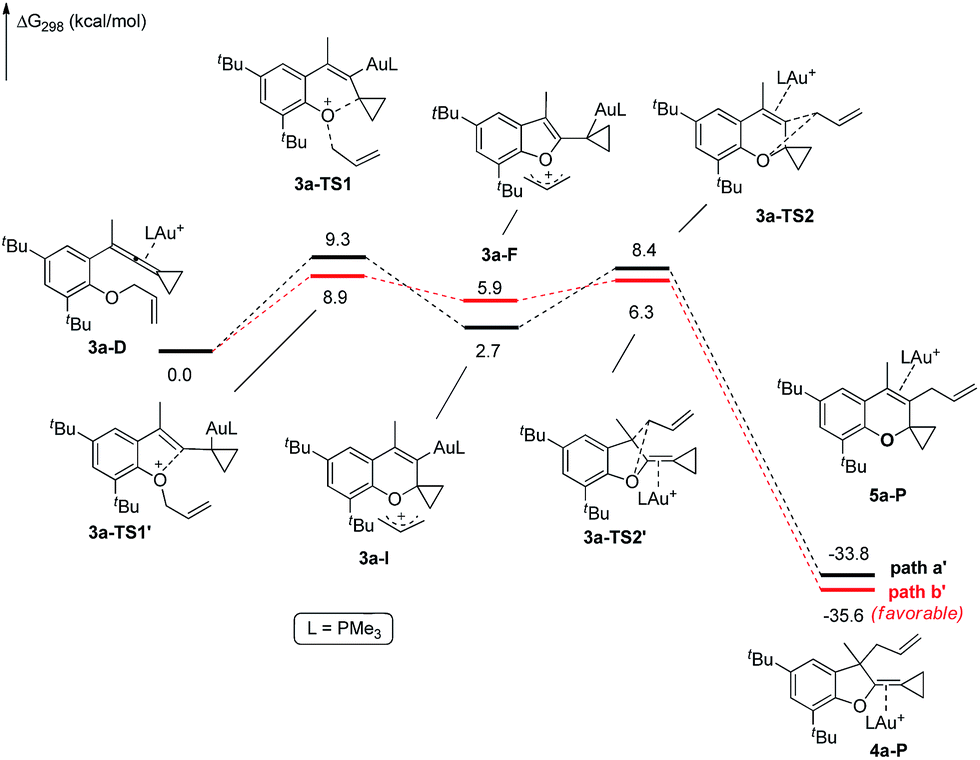 | ||
| Scheme 7 DFT studies on key reaction steps of the reaction of 3a. | ||
On the basis of the above results, we also attempted to develop an asymmetric variant for the two intramolecular cyclizations. The optimization of these asymmetric gold catalyses revealed that xyl-BINAP ligand coordinated gold complex gave the highest ee values in the carbene induced process, while DM-SegPhos ligand coordinated gold complex was the best one for the non-carbene induced process (see ESI† for the details). As shown in Scheme 8, the corresponding product 2 could be obtained in good yields along with 80–87% ee values whereas the allyl-transferred product 4a was obtained in moderate yield along with 67% ee value. Using 20 mol% of AgSbF6 did not improve the ee value of 4a (see ESI†).
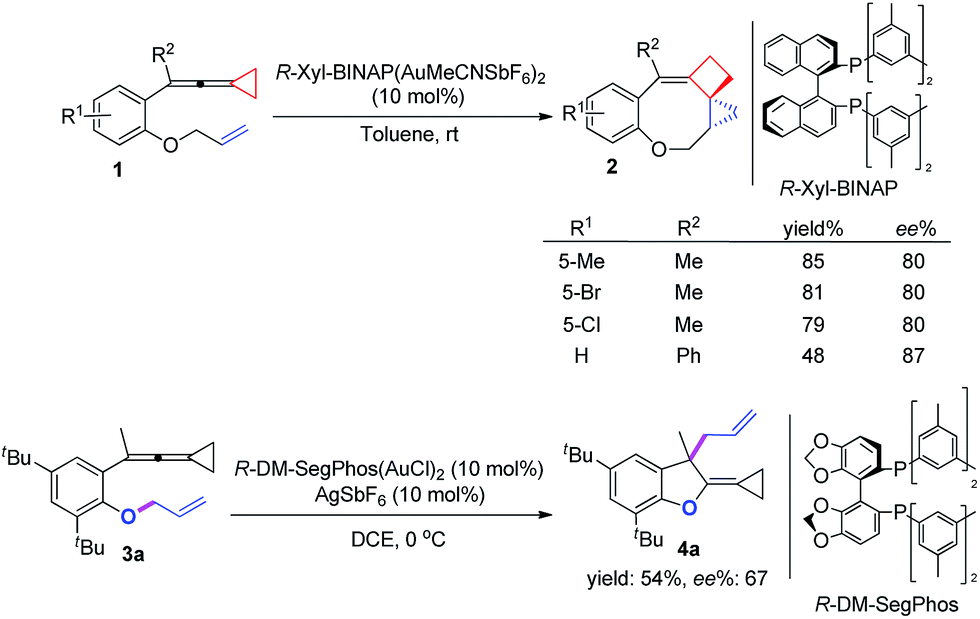 | ||
| Scheme 8 Asymmetric versions of the two processes. | ||
The product 4a could be easily transformed into benzofuran derivative 6 in 82% yield in the presence of a Brønsted acid such as HBr via a [3,3]-sigmatropic rearrangement (Scheme 9) (see ESI† for the details on screening of the reaction conditions).16 The compound 6 could be easily transformed to the masked alcoholic products via normal processes (see ESI† for the details).17
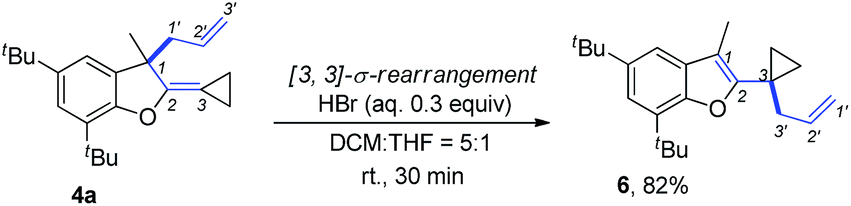 | ||
| Scheme 9 Further transformation of 4a. | ||
In summary, we have explored a novel gold(I)-catalyzed cycloisomerization of VDCP-ene derivatives via carbene or non-carbene processes, in which a substituent adjacent to the oxygen atom could switch the reaction modes. The reaction features a new amphiphilic strategy for gold carbene generation. For non-carbene cyclization, the regioselectivity is dependent upon the steric effect at the alkene moiety. These carbene or non-carbene processes can provide a new synthetic protocol for the divergent synthesis of O-containing heterocyclic scaffolds including a fused tricyclic system and fused five-, and six-membered ring systems. Further investigations to examine the mechanistic details more extensively and exploration of new methodology based on gold catalyzed transformations of VDCPs as well as their asymmetric variants are currently underway in our laboratory.
Acknowledgements
We are grateful for the financial support from the National Basic Research Program of China (973)-2015CB856603, and the National Natural Science Foundation of China (20472096, 21372241, 21361140350, 20672127, 21421091, 21372250, 21121062, 21302203 and 20732008).Notes and references
- For selected reviews, see: (a) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed; (b) A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766 RSC; (c) E. Jiménez-Núňez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; (d) M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC; (e) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed; (f) S. P. Nolan, Acc. Chem. Res., 2011, 44, 91 CrossRef CAS PubMed; (g) Y.-M. Wang, A. D. Lackner and F. D. Toste, Acc. Chem. Res., 2014, 47, 889–901 CrossRef CAS PubMed; (h) C. Obradors and A. M. Echavarren, Acc. Chem. Res., 2014, 47, 902 CrossRef CAS PubMed; (i) L. Zhang, Acc. Chem. Res., 2014, 47, 877 CrossRef CAS PubMed; (j) C. M. Friend and A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 729 CrossRef CAS PubMed; (k) A. S. K. Hashmi, Acc. Chem. Res., 2014, 47, 864 CrossRef CAS PubMed; (l) H.-S. Yeom and S. Shin, Acc. Chem. Res., 2014, 47, 966 CrossRef CAS PubMed; (m) A. Fürstner, Acc. Chem. Res., 2014, 47, 925 CrossRef PubMed.
- (a) P. Pyykko and J. P. Desclaux, Acc. Chem. Res., 1979, 12, 276 CrossRef CAS; (b) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS PubMed; (c) H. G. Raubenheimer and H. Schmidbaur, Organometallics, 2012, 31, 2507 CrossRef CAS.
- For evidence of gold carbenoids, see: (a) A. M. Echavarren, Nat. Chem., 2009, 1, 431 CrossRef CAS PubMed; (b) G. Seidel and A. Fürstner, Angew. Chem., Int. Ed., 2014, 53, 4807 CrossRef CAS PubMed; (c) P. Nösel, L. Nunes dos Santos Comprido, T. Lauterbach, M. Rudolph, F. Rominger and A. S. K. Hashmi, J. Am. Chem. Soc., 2013, 135, 15662 CrossRef PubMed; (d) T. Wang, S. Shi, M. M. Hansmann, E. Rettenmeier, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2014, 53, 3715 CrossRef CAS PubMed.
- For O as Nuc and E, see: (a) B. Lu, Y. Li, Y. Wang, D. H. Aue, Y. Luo and L. Zhang, J. Am. Chem. Soc., 2013, 135, 8512 CrossRef CAS PubMed; (b) Z. Wang, Y. Wang and L. Zhang, J. Am. Chem. Soc., 2014, 136, 8887 CrossRef CAS PubMed; (c) N. D. Shapiro and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 4160 CrossRef CAS PubMed; (d) S. Ghorpade, M.-D. Su and R.-S. Liu, Angew. Chem., Int. Ed., 2013, 52, 4229 CrossRef CAS PubMed; (e) D. Vasu, H.-H. Hung, S. Bhunia, S. A. Gawade, A. Das and R.-S. Liu, Angew. Chem., Int. Ed., 2011, 50, 6911 CrossRef CAS PubMed; (f) S. Bhunia, S. Ghorpade, D. B. Huple and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 51, 2939 CrossRef CAS PubMed; (g) H. S. Yeom, Y. Lee, J. Jeong, E. So, S. Hwang, J. E. Lee, S. S. Lee and S. Shin, Angew. Chem., Int. Ed., 2010, 49, 1611 CrossRef CAS PubMed; (h) P. W. Davies, A. Cremonesi and N. Martin, Chem. Commun., 2011, 47, 379 RSC; (i) G. Henrion, T. E. J. Chavas, X. Le Goff and F. Gagosz, Angew. Chem., Int. Ed., 2013, 52, 6277 CrossRef CAS PubMed; (j) A. S. K. Hashmi, T. Wang, S. Shi and M. Rudolph, J. Org. Chem., 2012, 77, 7761 CrossRef CAS PubMed; (k) S. Shi, T. Wang, W. Yang, M. Rudolph and A. S. K. Hashmi, Chem.–Eur. J., 2013, 19, 6576 CrossRef CAS PubMed; For alkene/aryl as Nuc and E, see: (l) A. S. K. Hashmi, T. M. Frost and J. W. Batsin, J. Am. Chem. Soc., 2000, 122, 11553 CrossRef CAS; (m) A. S. K. Hashmi, M. Rudolph, H. Siehl, M. Tanaka, J. W. Bats and W. Frey, Chem.–Eur. J., 2008, 14, 3703 CrossRef CAS PubMed; (n) V. Lopez-Carrillo and A. M. Echavarren, J. Am. Chem. Soc., 2010, 132, 9292 CrossRef CAS PubMed; (o) P. Perez-Galan, E. Herrero-Gomez, D. T. Hog, N. J. A. Martin, F. Maseras and A. M. Echavarren, Chem. Sci., 2011, 2, 141 RSC; (p) Y. Wang, M. E. Muratore, Z. Rong and A. M. Echavarren, Angew. Chem., Int. Ed., 2014, 53, 14022 CrossRef CAS PubMed; For acyl group as a strategy for gold carbene generation, see: (q) M. J. Johansson, D. J. Gorin, S. T. Staben and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 17616 CrossRef PubMed; (r) I. D. G. Watson, S. Ritter and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 2056 CrossRef CAS PubMed; (s) M. Uemura, I. D. G. Watson, M. Katsukawa and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 3464 CrossRef CAS PubMed; (t) Y. Shi, K. E. Roth, S. D. Ramgren and S. A. Blum, J. Am. Chem. Soc., 2009, 131, 18022 CrossRef CAS PubMed; (u) W. Rao, M. J. Koh, D. Li, H. Hirao and P. W. H. Chan, J. Am. Chem. Soc., 2013, 135, 7926 CrossRef CAS PubMed; For allene as carbene precusor, see: (v) M. R. Luzung, P. Mauleón and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 12402 CrossRef CAS PubMed; (w) G. Zhang, V. J. Catalano and L. Zhang, J. Am. Chem. Soc., 2007, 129, 11358 CrossRef CAS PubMed; (x) I. Alonso, B. Trillo, F. López, S. Montserrat, G. Ujaque, L. Castedo, A. Lledós and J. L. Mascareñas, J. Am. Chem. Soc., 2009, 131, 13020 CrossRef CAS PubMed; (y) F. López and J. L. Mascareñas, Chem.–Eur. J., 2011, 17, 418 CrossRef PubMed.
- (a) C. R. Solorio-Alvarado and A. M. Echavarren, J. Am. Chem. Soc., 2010, 132, 11881 CrossRef CAS PubMed; (b) C. R. Solorio-Alvarado, Y.-H. Wang and A. M. Echavarren, J. Am. Chem. Soc., 2011, 133, 11952 CrossRef CAS PubMed; (c) Y. Wang, P. R. McGonigal, B. Herle, M. Besora and A. M. Echavarren, J. Am. Chem. Soc., 2014, 136, 801 CrossRef CAS PubMed.
- (a) L. Ye, Y. Wang, D. H. Aue and L. Zhang, J. Am. Chem. Soc., 2012, 134, 31 CrossRef CAS PubMed; (b) J. Bucher, T. Wurm, K. S. Nalivela, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2014, 53, 3854 CrossRef CAS PubMed; (c) M. M. Hansmann, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 2593 CrossRef CAS PubMed; (d) A. S. K. Hashmi, I. Braun, P. Noesel, J. Schaedlich, M. Wieteck, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 4456 CrossRef CAS PubMed; (e) A. S. K. Hashmi, T. Haeffner, M. Rudolph and F. Rominger, Chem.–Eur. J., 2011, 17, 8195 CrossRef CAS PubMed; (f) A. S. K. Hashmi, M. Wieteck, I. Braun, P. Noesel, L. Jongbloed, M. Rudolph and F. Rominger, Adv. Synth. Catal., 2012, 354, 555 CrossRef CAS PubMed; (g) A. S. K. Hashmi, M. Wieteck, I. Braun, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 10633 CrossRef CAS PubMed; (h) M. H. Vilhelmsen and A. S. K. Hashmi, Chem.–Eur. J., 2014, 20, 1901 CrossRef CAS PubMed; (i) Y. Yu, W. Yang, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 7586 CrossRef CAS PubMed; (j) A. S. K. Hashmi, I. Braun, M. Rudolph and F. Rominger, Organometallics, 2012, 31, 644 CrossRef CAS; (k) M. M. Hansmann, S. Tšupova, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem.–Eur. J., 2014, 20, 2215 CrossRef CAS PubMed; (l) M. Wieteck, Y. Tokimizu, M. Rudolph, F. Rominger, H. Ohno, N. Fujii and A. S. K. Hashmi, Chem.–Eur. J., 2014, 20, 16331 CrossRef CAS PubMed; (m) P. Nösel, V. Müller, S. Mader, S. Moghimi, M. Rudolph, I. Braun, F. Rominger and A. S. K. Hashmi, Adv. Synth. Catal., 2015, 357, 500 CrossRef PubMed; the possibility of gold allenylidene complexes, the higher homologues, has also been investigated: (n) M. M. Hansmann, F. Rominger and A. S. K. Hashmi, Chem. Sci., 2013, 4, 1552 RSC.
- (a) K. C. Nicolaou, C. R. H. Hale, C. Nilewski and H. A. Ioannidou, Chem. Soc. Rev., 2012, 41, 5185 RSC; (b) M. M. Hann, A. R. Leach and G. Harper, J. Chem. Inf. Comput. Sci., 2001, 41, 856 CrossRef CAS PubMed.
- For carbene or non-carbene processes involved in propargylic ester substrates, see: (a) L. Zhang, J. Am. Chem. Soc., 2005, 127, 16804 CrossRef CAS PubMed; (b) J. Zhao, C. O. Hughes and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 7436 CrossRef CAS PubMed; (c) A. Buzas and F. Gagosz, J. Am. Chem. Soc., 2006, 128, 12614 CrossRef CAS PubMed; (d) A. Correa, N. Marion, L. Fensterbank, M. Malacria, S. P. Nolan and L. Cavallo, Angew. Chem., Int. Ed., 2008, 47, 718 CrossRef CAS PubMed.
- For selected reviews, and articles, see: (a) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317 CrossRef CAS PubMed; (b) G. Audran and H. Pellissier, Adv. Synth. Catal., 2010, 352, 575 CrossRef CAS PubMed; (c) M. Shi, L.-X. Shao, J.-M. Lu, Y. Wei, K. Mizuno and H. Maeda, Chem. Rev., 2010, 110, 5883 CrossRef CAS PubMed.
- The unambiguous structure of 2 has been assigned by X-ray diffraction of 2n.
- The low yield for product 2l is probably due to the instability of starting material 1l under the standard conditions since we found that it partially decomposed under acidic conditions.
- The structure of 4a has been confirmed by NMR spectroscopic analysis (NOESY, DEPT, gCOSY, HSQC and HMBC), see ESI† for the details.
- The structure of 5k has been unambiguously determined by X-ray crystal structure analysis.
- (a) J. Fu, H. Shang, Z. Wang, L. Chang, W. Shao, Z. Yang and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 4198 CrossRef CAS PubMed; (b) L. P. Liu, B. Xu, M. S. Mashuta and G. B. Hammond, J. Am. Chem. Soc., 2008, 130, 17642 CrossRef CAS PubMed.
- (a) A. S. K. Hashmi, A. M. Schuster, S. Litters, F. Rominger and M. Pernpointner, Chem.–Eur. J., 2011, 17, 5661 CrossRef CAS PubMed; (b) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Ackermann, A. D. B. Becker, M. Rudolph, C. Scholz and F. Rominger, Adv. Synth. Catal., 2012, 354, 133 CrossRef CAS PubMed; (c) M. Ackermann, J. Bucher, M. Rappold, K. Graf, F. Rominger and A. S. K. Hashmi, Chem.–Asian J., 2013, 8, 1786 CrossRef CAS PubMed; (d) A. S. K. Hashmi, K. Graf, M. Ackermann and F. Rominger, ChemCatChem, 2013, 5, 1200 CrossRef CAS PubMed.
- (a) H. Zheng, L. L. Adduci, R. J. Felix and M. R. Gagné, Angew. Chem., Int. Ed., 2014, 53, 7904 CrossRef CAS PubMed; (b) R. J. Felix, D. Weber, O. Gutierrez, D. J. Tantillo and M. R. Gagné, Nat. Chem., 2012, 4, 405 CrossRef CAS PubMed; (c) R. J. Felix, O. Gutierrez, D. J. Tantillo and M. R. Gagné, J. Org. Chem., 2013, 78, 5685 CrossRef CAS PubMed.
- The structure of 6 has been confirmed by NMR spectroscopic analysis (NOESY, gCOSY, HSQC and HMBC), see ESI† for the details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds, and CCDC 996502, 980558, 997410 and 996499. See DOI: 10.1039/c5sc01806d |
| This journal is © The Royal Society of Chemistry 2015 |