
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA crystalline sponge based on dispersive forces suitable for X-ray structure determination of included molecular guests†
Elena
Sanna
a,
Eduardo C.
Escudero-Adán
bc,
Antonio
Bauzá
a,
Pablo
Ballester
 bc,
Antonio
Frontera
a,
Carmen
Rotger
a and
Antonio
Costa
*a
bc,
Antonio
Frontera
a,
Carmen
Rotger
a and
Antonio
Costa
*a
aDepartament de Química, Universitat de les Illes Balears, Palma, 07122, Spain. E-mail: antoni.costa@uib.es; Fax: +34 971 172436; Tel: +34 971 173266
bInstitut of Chemical Research of Catalonia (ICIQ), Avda. Països Catalans 16, 43007 Tarragona, Spain
cCatalan Institution for Research and Advanced Studies (ICREA), Passeig Lluis Companys 23, 08010, Barcelona, Spain
First published on 14th July 2015
Abstract
A crystalline porous material showing one-dimensional (1-D) rectangular micropores (12 × 9 Å2) has been assembled from a semirigid macrocyclic tetraimine and EtOAc as the templating agent. The 1-D nature of the material is intrinsic to the conformationally rigid structure of a macrocyclic sub-unit bearing four cyclohexylidene residues. The multiple dispersive forces established among the aliphatic residues glutted the 1-D channels and provided thermal stability to the material at temperatures below 160 °C. Upon removal of the template, the structure of the empty solid exhibited permanent microporosity (SBET = 342 m2 g−1). Being a true molecular sponge, the channel framework of this material allowed the inclusion of a variety of molecular sample guests without compromising its crystalline nature. Remarkably, this crystalline material enabled the structure determination by X-ray diffraction of the included molecules. Theoretical studies demonstrated the vital role played by the dispersive forces in the overall stabilization of the crystal packing.
Introduction
Porous organic materials1 are currently of growing interest owing to their potential applications in diverse fields such as storage and separation of gases,2 molecular sorting3 and catalysis.4 In this context, the synthesis of porous materials constructed from discrete molecules5 is particularly attractive. This approach takes advantage of the structural versatility provided by organic synthesis and the possibility to obtain porous solid materials simply by crystallization.6 The molecular components of porous organic molecular crystals (POMC) lacking an extended directional covalent or coordination bonding are held together only by weak intermolecular forces. Hence, molecular materials based on non-directional intermolecular forces are metastable and tend to collapse upon removal of the template agents used to stabilize the voids of their porous crystalline structures.7 Related materials exclusively stabilized by weak dispersive interactions are scarce. In this regard, Atwood and Barbour's contributions are particularly significant. These authors showed that interstitial van der Waals interactions could be employed as the main component for the design of porous host–guest assemblies.8 A logical development of this approach that produced more stable porous molecular materials relied on the use of molecules interacting through hydrogen bonding and aromatic interactions, either separately or in combination.2d–f,7 Using this strategy, Shimizu and colleagues reported a microporous crystalline bis-urea macrocyclic host that self-assembled by hydrogen bonding and aromatic stacking interactions. In this example, the macrocyclic ureas aligned into columnar 1-D assemblies and maintained its porous structure even upon guest removal.9In this work, we present a new organic material featuring permanent porosity based on the macrocyclic tetraimine 3 (Scheme 1). A key aspect of our design is the use of multiple weak dispersive interactions as the glue element to organize and maintain together the components of the stable porous molecular material. Our report constitutes a paradigmatic example of the so-called Gulliver's principle10 applied to the design of porous organic materials.
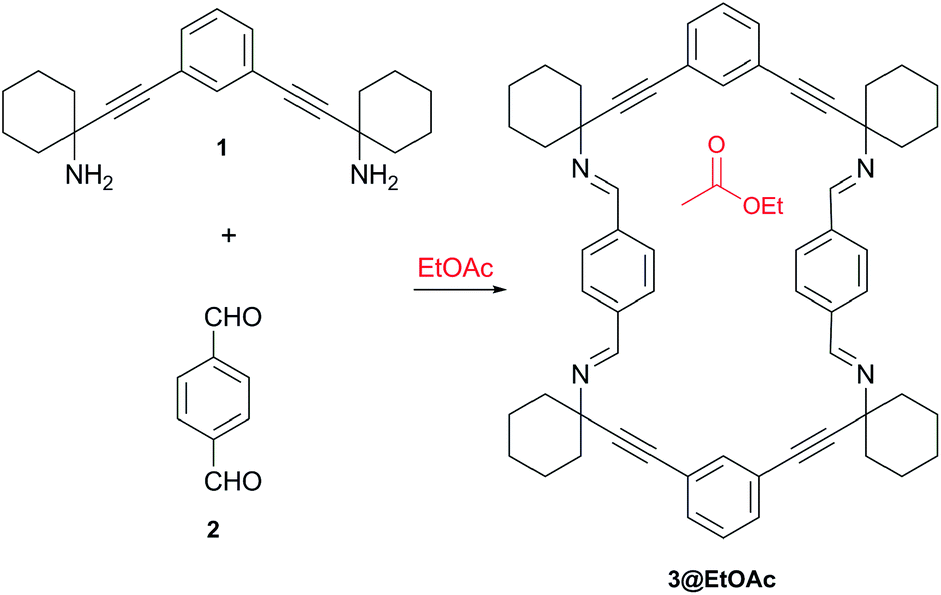 | ||
| Scheme 1 Synthesis of macrocyclic tetraimine 3. | ||
Previously, we have described the synthesis of the 1,3-phenylene-bis-propargylic diamine 1 used in this work as rigid spacer to avoid the collapse of the macrocycle 3.11 The hierarchical self-assembly of the macrocycle 3 into a porous material arises from the combination of multiple van der Waals and aromatic interactions.12 These interactions are established between the phenyl and cyclohexylidene residues, the “sticky groups” of 3. The resulting porous material displays 1-D channels that are large enough for liquid–solid sorption applications.13
More specifically, we were interested in knowing if a crystalline porous material based only on dispersive forces, could be used as the crystalline solid support in X-ray diffraction studies by applying the “crystalline sponge method” developed by Fujita.14 In this method, accurate molecular structures of samples can be obtained by analyzing the diffraction patterns of single crystals without requiring the crystallization of the samples. However, the crystalline sponges reported so far are based on metal–organic frameworks (MOFs) that include electron-rich atoms, such as iodine, bromine, chlorine as well as transition metals in their structures.15 These atoms contribute heavily to the overall diffraction pattern masking the information available for the guest. From this point of view, our system based only in light-atoms, could offer an alternative to MOF-based crystalline sponges.
Results and discussion
Synthesis and structure of the microporous material
The macrocyclic tetraimine 3 was prepared by Schiff base condensation of the diamine 1 with terephthalaldehyde 2 in EtOAc solution. On standing at room temperature, single crystals of 3@EtOAc slowly grew from the solution (yield 50%).‡ The solid material was characterized by single crystal X-ray diffraction,1613C CP MAS NMR and thermogravimetric analysis (TGA).The single crystal X-ray structure showed the macrocycle 3 molecules stacked one on top of another forming a columnar assembly. The solid material displayed one-dimensional channels with distorted rectangular morphology along the b axis (Fig. 1). The total solvent accessible void volume within the channels calculated from PLATON/Squeeze, was 1096 Å3 (274 Å3 per each macrocyclic imine) representing 19.4% of the unit cell volume.17 We observed that the channels were filled with partially disordered EtOAc molecules (Fig. 1a). In the crystal packing, the four aromatic rings of 3 were tilted ca. 60° with regard to the plane of the macrocycle. Furthermore, the two phenyl rings located at the wider sides of 3 oscillated between two positions (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 occupancy) and acted as revolving doors (Fig. 1b).
:25 occupancy) and acted as revolving doors (Fig. 1b).
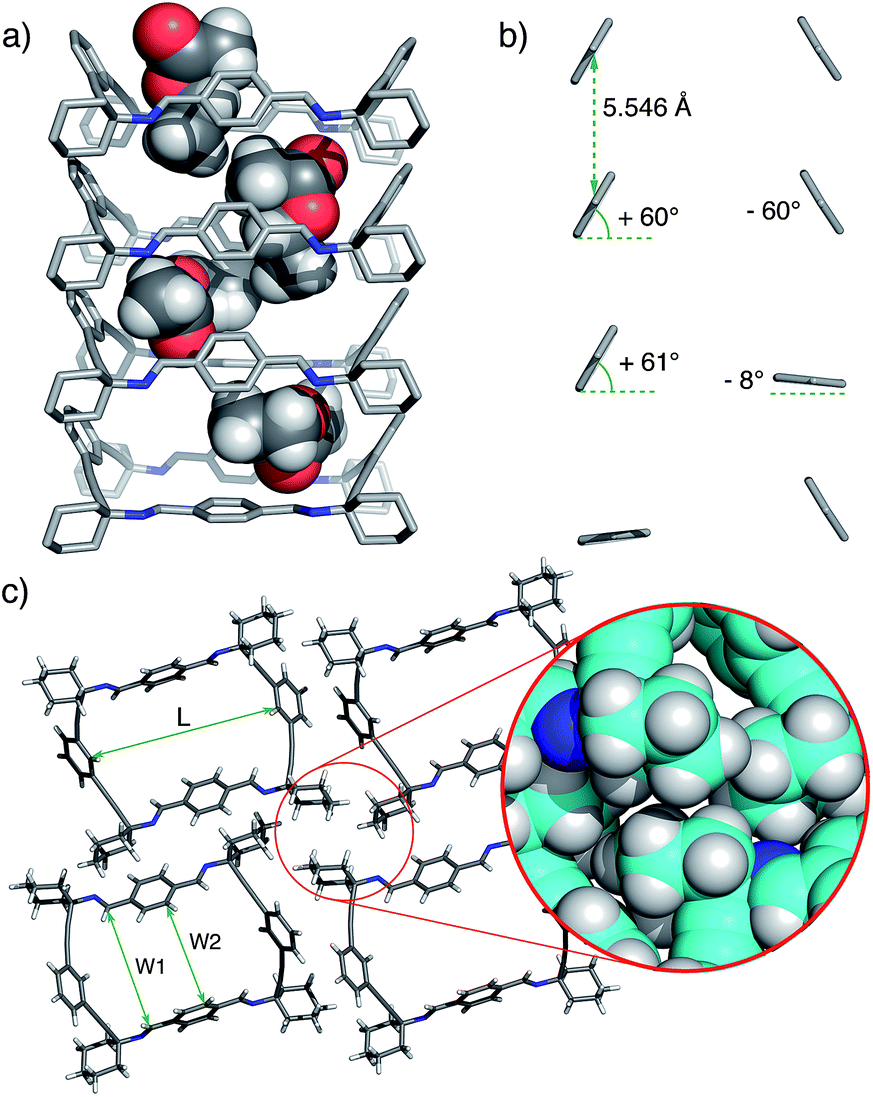 | ||
| Fig. 1 (a) Representative X-ray perspective side-view in b direction of 3@EtOAc crystals displaying disordered EtOAc molecules inside the channel. (b) Partial view showing the relative orientation of the 1,4-phenylene rings (see text). (c) Top view of a molecular cell unit of the monoclinic crystals (Vc = 5642 Å3, Z = 4) of 3@EtOAc (DC = 1.121 g cm−3; L ∼ 11.98 Å, W1 ∼ 9.13 Å; W2 ∼ 6.11–6.90 Å) (CCDC no. 1012389). Disorder and EtOAc solvent molecules are removed for clarity. The inset shows multiple van der Waals contacts between cyclohexylidene residues. | ||
From this structure, it became clear that the combined action of intermolecular displaced π–π aromatic stacking, as well as multiple CH (sp3)–CH (sp3) van der Waals interactions extending throughout the channel, provided the necessary stabilization of the assembly.
The results obtained in 13C CP-MAS NMR experiments confirmed the role of EtOAc as guest template. The carbon signals of the relatively mobile molecules of EtOAc included in the solid, but not those of the host tetraimine 3 that formed the porous framework were observed in the standard mode 13C MAS spectra (Fig. S7 in ESI†).
Stability and dynamic behaviour of the porous material
Guest solvent molecules were completely removed by treating the solvate 3@EtOAc under vacuum for several hours at room temperature (12 h, 2 × 10−2 mm Hg). This treatment afforded a new crystalline material, the apohost 3(I). The X-ray structure of 3(I) (Fig. 2) revealed the absence of electron density in the channels previously filled with EtOAc. The lack of EtOAc in the crystalline structure was also confirmed by registering the 1H NMR spectra of single crystals of 3(I) dissolved in CDCl3.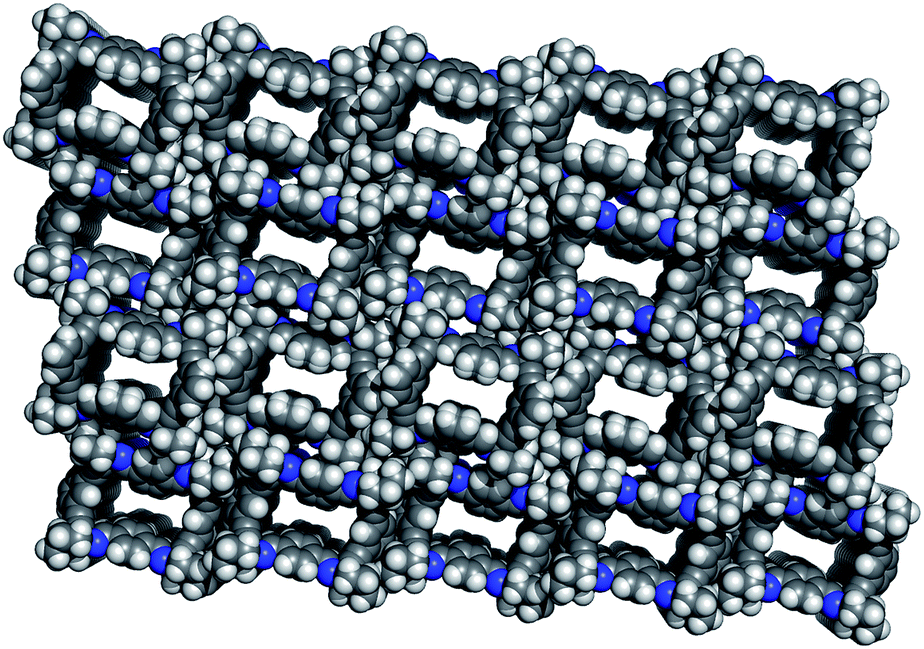 | ||
| Fig. 2 X-ray perspective top-view of the apohost 3(I) (Vc = 5639 Å3, Z = 4) (DC = 0.9859 g cm−3; L ∼ 11.98 Å, W1 ∼ 9.20 Å; W2 ∼ 6.90 Å) (CCDC no. 1012398). | ||
More important, the crystal packing of 3(I) was almost identical to that of the 3@EtOAc solvate (see Table S1 in ESI†), indicating that the porous structure remained intact after vacuum treatment.
The relatively easy removal of the included EtOAc molecules at room temperature implied that they were weakly retained in the channels.
Remarkably, the 3@EtOAc solvate could be regenerated by simply soaking crystals of the empty material 3(I) in EtOAc. These findings were consistent with the absence of strong dipole–dipole or hydrogen bonding interactions between the cycloimine 3 and the included EtOAc molecules.
The dynamic nature of the guest exchange constituted an essential feature of this material. The kinetics of the guest exchange process was monitored by using 13C NMR. Crystals of 3@13C-[1.2]-EtOAc obtained by treating 3(I) with 13C labelled EtOAc were suspended in unlabelled EtOAc.
We observed that the combined integral values of the isotopic 13C doublet grew steadily with time and reached a plateau after 8–10 h (Fig. 3). This experiment demonstrated that the almost complete exchange of the included 13C labeled EtOAc was slow in the human time-scale. Desolvation of EtOAc was also monitored using thermogravimetry. The TGA trace of 3@EtOAc revealed an 11.8% weight loss in a temperature range from room temperature to 175 °C. This weight loss corresponded to the vaporization of 1.3 molecules of EtOAc per macrocycle, therefore indicating the 3:4 stoichiometry found for this complex (see Fig. S9 in ESI†). In complete agreement, the differential scanning calorimetry (DSC) thermogram of 3@EtOAc exhibited an endothermic peak at around 160 °C due to vaporization of the entrapped guest solvent (Fig. 4b).
 | ||
| Fig. 3 Plot representing the fraction of 13C-[1.2]-EtOAc in a solution of EtOAc. This plot was obtained from 50 mg of labelled 3@13C-[1.2]-EtOAc suspended in 0.5 mL of EtOAc at 298 K, by integration of the 13C satellites with respect to the 12C signal and assuming the complete replacement of 13C-[1.2]-EtOAc. The inset shows representative increasing carbonyl doublet resonances (169.7 ppm, JC–C = 59.5 Hz) used to extract the integral data. | ||
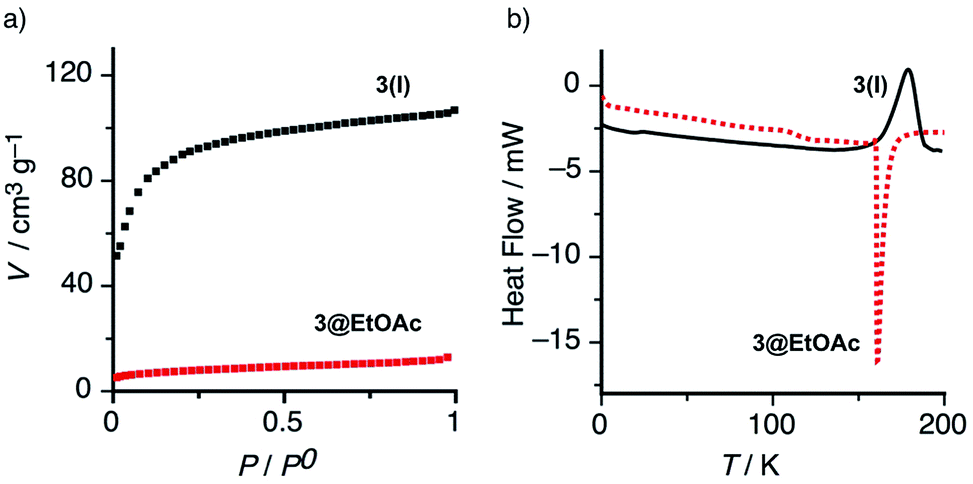 | ||
| Fig. 4 (a) Adsorption isotherm of N2 for the solvate 3@EtOAc compared to the empty solid 3(I) (T = 77 K, P0 = 1 atm). (b) DSC traces of 3@EtOAc displaying an endothermic peak at 160 °C due to solvent vaporization (red dotted line) and of 3(I) showing an exothermic peak at 177 °C assigned to irreversible phase transition to 3(II). | ||
On the other hand, the porosity of 3(I) was also confirmed by gas absorption of nitrogen at 77 K (Fig. 4a). The empty solid 3(I) exhibited a typical type I adsorption isotherm indicative of a microporous (<2 nm) structure with significant specific surface area (SBET = 342 m2 g−1).18 In addition, the DSC trace of the apohost 3(I) displayed an exothermic and irreversible process at around 175 °C (Fig. 4b and S10†), which we assigned to a monotropic solid-phase transition of 3(I). In both cases, the X-ray structure analysis of the material obtained after thermal treatment above 160 °C, revealed the formation of a new non-porous polymorphic form 3(II).
The X-ray structure of 3(II) (see Fig. S11 in ESI†) showed densely packed layers of displaced macrocyclic imine units 3 (DC = 1.127 g cm−3) and the concomitant loss of the columnar assembly characteristic of the 3(I) polymorphic form.
Liquid–solid sorption experiments
The void volumes of the porous frameworks exhibited by the crystalline materials 3@EtOAc and 3(I) are sufficiently large to include a variety of molecular guests. This characteristic, together with the absence of polar groups that can be displayed to the included guests and the high thermal stability of the crystalline materials, suggested us the potential use of 3@EtOAc and 3(I) as suitable supports for X-ray diffraction studies by the crystalline sponge method.14,15To test this functionality, we soaked single crystals of 3@EtOAc and 3(I) in different neat liquid samples with molecular volumes ranging from 55–60 Å3i.e. nitromethane and ethylene glycol, to >200 Å3 of Z-stilbene (see, Table S2 in ESI†). In favourable cases, i.e. less than 5 min for nitromethane, the initially floating crystals of 3@EtOAc gradually sank to the bottom of the vial providing a visual indication of the exchange status. During the process, we did not observe any apparent degradation of the crystal integrity. The exchange of the guest samples was confirmed by 1H NMR of CDCl3 solutions of the solvates. Also, 13C CP-MAS NMR experiments confirmed the inclusion of the liquid guests within the porous framework. The carbon signals of non-volatile guests (i.e. diethyl squarate) appeared slightly upfield shifted (0.5–2 ppm) with respect to those of diethyl squarate in the bulk (Fig. S8 in ESI†).
Owing to the poor solubility of the solvates in CDCl3, the molar ratios were better evaluated by digesting the solvates with DCl/D2O (50 μL) before registering the 1H NMR spectra in a mixture of DMSO-d6:CDCl3 (2.5:1 v/v, 600 μL) (see, ESI†).15c In general, the molar ratios of 3:sample evaluated by 1H NMR after a common period of 72 h, agree well with the crystallographic stoichiometries (Table S2 in ESI†). In one case, the average experimental molar ratio calculated for Z-stilbene of 0.8 was consistently inferior to that expected for a 1:1 3@Z-stilbene solvate. This result is likely due to the incomplete inclusion of Z-stilbene within the channels of the 3(I). Therefore, the molecular volume of Z-stilbene of 216 Å3, approaching the maximum available void volume of 274 Å3 per macrocycle calculated using PLATON/Squeeze,17 suggested us a practical upper limit for the effective inclusion of samples in 3(I).
Fig. 5a–h, depict ORTEP views of representative X-ray structures obtained by guest exchange from single crystals of 3@EtOAc or alternatively, by soaking a single crystal of the empty apohost 3(I) with ca. 1 mL of the liquid sample. Both methods produced identical results in all tested cases (see ESI for X-ray structural data†). Both, the empty material 3(I) and the solvate 3@EtOAc allowed the formation of inclusion solid complexes (solvates) with aromatic, unsaturated and aliphatic neutral substrates bearing a variety of functional groups such as hydroxy, aldehyde, ester and nitro groups, among others.
 | ||
| Fig. 5 (a to h) ORTEP views of different liquid molecules diffused into the channel structure of 3(I) with thermal ellipsoids set at 50% probability. Disorder is removed for clarity. CCDC codes 1012389 to 1012397 and 1063714 contain the structural X-ray data (see ESI for RX structural details†). | ||
Since the included guests could, eventually, establish hydrogen bonds or dipole–dipole interactions with 3 that would lead to degradation of the porous structure of the solids, the obtained results in the guest exchange/inclusion experiments confirmed that the interactions established between 3 and the incoming sample guests must be feeble.
Remarkably, the inclusion of the different guests into the crystal lattice did not disrupt the pore size significantly (ΔL < 2%; ΔW1 < 2.6%). However, to obtain a better fit between the size and shape of the pore and those of the included molecules (induce fit), the two opposite phenyl rings located on the wider sides of the macrocycle 3 oscillated with a mechanism that resembles the functioning of the revolving doors. The adaptation process experienced by the pore was revealed by careful analysis of the X-ray structures of 3@EtOAc, 3@p-anisaldehyde, and 3(I)@S-(−)-nicotine (ΔW2 ∼ 20%).
The induced fit process, in which the width of the channels responded to the size and shape of the included sample guests took place without any loss of crystal integrity (Table S1 in ESI†). In contrast, the X-ray structure of the non-porous polymorphic form 3(II) displayed a narrowed entrance of the pore that can be referred as “closed” doors. Most likely, this structural change was responsible for inhibiting the guest sorption of liquid guests (W1 ∼ 9.91 Å; W2 ∼ 5.54 Å).
Theoretical calculations
 | ||
| Fig. 6 Comparison of the X-ray and DFT-D3 optimized structures of 3(I) and interaction energies at the BP86-D3/def2-TZVP level of theory (value in parenthesis corresponds to the BP86-D3/def2-SVP level). | ||
In addition, the interaction energy computed for the dimer using the crystallographic coordinates (without optimization) gave a very similar value (−32.9 kcal mol−1). Removal of the cyclohexylidene residues led to a significant reduction of the interaction energy (−14.5 kcal mol−1). This finding supported the strong influence exerted by the C–H⋯H–C interactions to the stability of the porous material.19
The energy difference calculated for the two energy-minimized dimers (18.4 kcal mol−1) represented an estimation of the contribution of these interactions to the overall dimerization process. Since four cyclohexylidene residues are present in the macrocycle 3, each intermolecular cyclohexylidene⋯cyclohexylidene interaction contributes in 4.6 kcal mol−1. This value was in good agreement with previously reported high level ab initio results for dodecahedrane.19
To further analyze the non-covalent interactions in the dimer 32 we used the NCI (Non Covalent Interaction) index.20 This visualization index enables the identification and visualization of weak non-covalent interactions efficiently.
The isosurfaces corresponded to both favourable and unfavourable interactions, as differentiated by the sign of the second density Hessian eigenvalue and defined by the isosurface color. NCI analysis allowed an assessment of host–guest complementarity and the extent to which weak interactions stabilize a complex. Fig. 7, shows the representation of the NCI plot computed for the dimer 32. In addition to a panoply of C–H⋯H–C interactions, two weak equivalent and oppositely located parallel-displaced π–π interactions and four C H/π interactions involving the π-system of the triple bond were also observed. The largest isosurface was located around the cyclohexylidene rings that established a multitude of C–H⋯H–C interactions with another cyclohexylidene and the aromatic ring.
 | ||
| Fig. 7 NCI surfaces of a dimer of 3 used as a minimalist model of the 1-D channel. The isosurfaces (pale and dark green) correspond to favourable. | ||
The process was favourable by 4.1 kcal mol−1 (Fig. 8). Subsequently, we assessed the energy required to exchange the expelled EtOAc by a new SQA molecule. This second process was also favourable by 3 kcal mol−1. This sequence was repeated to evaluate the energy of the exchange processes of the second and third molecules of EtOAc by SQA molecules (see Fig. S15–S17 in ESI†).
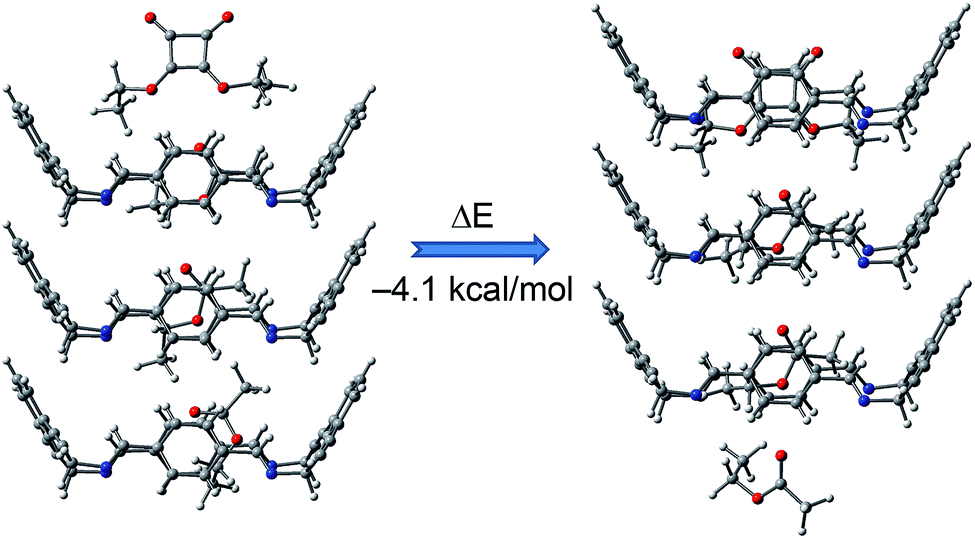 | ||
| Fig. 8 Favourable energy change due to the substitution of one EtOAc molecule by one diethyl squarate molecule in a minimalist representative channel of 3@EtOAc. | ||
These processes were also energetically favourable and showed similar magnitudes for the exchange energies. Hence, the results of this theoretical study agreed well with the aforementioned experimental results and supported the viability of the liquid–solid guest exchange process.
Conclusions
In summary, we have synthesized an unprecedented conformationally semirigid macrocyclic tetraimine 3 that is decorated with four peripheral cyclohexylidene substituents. The tetraimine 3 crystallized from EtOAc solutions affording a porous crystalline material 3@EtOAc composed of one-dimensional channels filled with solvent molecules. The crystal packing of 3@EtOAc was stabilized mainly by dispersive interactions. We have demonstrated that the templating EtOAc solvent molecules can be removed in vacuo to yield an empty but crystalline and porous solid 3(I), which can be considered as a polymorphic form of 3@EtOAc. Both solid materials 3@EtOAc and 3(I), are rare examples of porous crystalline structures stabilized exclusively by dispersive forces. We confirmed the use of these solid crystalline materials for X-ray structure determination of molecular guests included in their pores by the crystalline sponge method. Finally, using high-level theoretical calculations we evaluated the strength of the cohesive interactions responsible for the porous structure, as well as the favourable energy change produced by guest exchange of molecules in the pores. Because porous materials based on dispersive forces have energetic advantages for the reversible storage of polar molecules in comparison to porous materials containing polar groups that can be presented to the included guests, we anticipate an emergence of materials of the former type in the near future.Acknowledgements
This work was supported by Ministry of Economy and Competitiveness grants (CTQ2014-57393-C2-1-P, Severo Ochoa Excellence Accreditation 2014–2018 SEV-2013-0319 and Consolider-Ingenio CSD2010-00065, FEDER funds). E. S. thanks CAIB and FSE for a predoctoral fellowship.Notes and references
- (a) N. B. McKeown, J. Mater. Chem., 2010, 20, 10588–10597 RSC; (b) J. T. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed; (c) G. D. Pantoş, P. Pengo and J. K. Sanders, Angew. Chem., Int. Ed., 2007, 46, 194–197 CrossRef PubMed.
- (a) H. Kim, Y. Kim, M. Yoon, S. Lim, S. M. Park, G. Seo and K. Kim, J. Am. Chem. Soc., 2010, 132, 12200–12202 CrossRef CAS PubMed; (b) Y. Jin, B. A. Voss, R. D. Noble and W. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6348–6351 ( Angew. Chem. , 2010 , 122 , 6492–6495 ) CrossRef CAS PubMed; (c) O. K. Farha, Y.-S. Bae, B. G. Hauser, A. M. Spokoyny, R. Q. Snurr, C. A. Mirkin and J. T. Hupp, Chem. Commun., 2010, 46, 1056–1058 RSC; (d) Y. He, S. Xiang and B. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed; (e) W. Yang, A. Greenaway, X. Lin, R. Matsuda, A. J. Blake, C. Wilson, W. Lewis, P. Hubberstey, S. Kitagawa and N. R. Champness, J. Am. Chem. Soc., 2010, 132, 14457–14469 CrossRef CAS PubMed; (f) P. Li, Y. He, J. Guang, L. Weng, J. C. Zhao, S. Xiang and B. Chen, J. Am. Chem. Soc., 2014, 136, 547–549 CrossRef CAS PubMed.
- T. Mitra, K. E. Jelfs, M. Schmidtmann, A. Ahmed, S. Y. Chong, D. J. Adams and A. I. Cooper, Nat. Chem., 2013, 5, 276–281 CrossRef CAS PubMed.
- (a) Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC; (b) P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- M. Mastarlerz, Chem.–Eur. J., 2012, 18, 10082 CrossRef PubMed.
- J. R. Holst, A. Trewin and A. I. Cooper, Nat. Chem., 2010, 2, 915–920 CrossRef CAS PubMed.
- (a) K. Raatikainen and K. Rissanen, Chem. Sci., 2012, 3, 1235–1239 RSC; (b) L. J. Barbour, Chem. Commun., 2006, 1163–1168 RSC; (c) P. Brunet, M. Simard and J. D. Wuest, J. Am. Chem. Soc., 1997, 119, 2737–2738 CrossRef CAS; (d) M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed.
- (a) S. J. Dalgarno, P. K. Thallapally, L. J. Barbour and J. L. Atwood, Chem. Soc. Rev., 2007, 36, 236–245 RSC; (b) J. L. Atwood, L. J. Barbour and A. Jerga, Science, 2002, 296, 2367–2369 CrossRef CAS PubMed.
- M. B. Dewal, M. W. Lufaso, A. D. Hughes, S. A. Samuel, P. Pellechia and L. S. Shimizu, Chem. Mater., 2006, 18, 4855–4864 CrossRef CAS.
- L. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS.
- B. Soberats, L. Martínez, M. Vega, C. Rotger and A. Costa, Adv. Synth. Catal., 2009, 351, 1727–1731 CrossRef CAS PubMed.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- (a) R. Natarajan, L. Bridgland, A. Sirikulkajorn, J.-H. Lee, M. F. Haddow, G. Magro, B. Ali, S. Narayanan, P. Strickland, J. P. H. Charmant, A. G. Orpen, N. B. McKeown, C. G. Bezzu and A. P. Davis, J. Am. Chem. Soc., 2013, 135, 16912–16925 CrossRef CAS PubMed; (b) T. Jacobs and L. J. Barbour, CrystEngComm, 2013, 15, 1512–1514 RSC.
- (a) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS PubMed; (b) Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780–783 CrossRef CAS PubMed.
- (a) S. Yoshioka, Y. Inokuma, M. Hoshino, T. Sato and M. Fujita, Chem. Sci., 2015, 6, 3765 RSC; (b) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai and M. Fujita, Nat. Protoc., 2014, 9, 246 CrossRef CAS PubMed; (c) R. Kubota, S. Tashiro, M. Shiro and M. Shionoya, Nat. Chem., 2014, 6, 913 CrossRef CAS PubMed; (d) T. R. Ramadhar, S.-L. Zheng, Y.-S. Chen and J. Clardy, Chem. Commun., 2015, 51, 11252–11255 RSC; (e) N. Zigon, M. Hoshino, S. Yoshioka, Y. Inokuma and M. Fujita, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201502302.
- CCDC codes 1012389, 1012398 and 1012399, contain the crystal data for 3@EtOAc, 3(I) and 3(II), respectively.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, A46, 194–201 CAS.
- K. S. V. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and J. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- J. Echeverría, G. Aullón, D. Danovich, S. Shaik and S. Alvarez, Nat. Chem., 2011, 3, 323–330 CrossRef PubMed.
- E. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of macrocycle 3 together with characterization data. TGA and DSC traces. CIF files of all structures. Computational details and additional calculations. CCDC 1012389–1012397 and 1063714. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01838b |
‡ Crystal data for 3@EtOAc (CCDC no. 1012389): C65.36H70.72N4O2.68, M = 955.18, monoclinic, a = 34.794(16) Å, b = 5.836(3) Å, c = 28.131(14) Å, α = 90.00°, β = 97.862(11)°, γ = 90.00°, V = 5659(5) Å3, T = 100(2) K, space group C2/c, Z = 4, 35400 reflections measured, 10696 independent reflections (Rint = 0.0434). The final R1 values were 0.0597 (I > 2σ(I)). The final wR(F2) values were 0.1624 (I > 2σ(I)). The final R1 values were 0.0931 (all data). The final wR(F2) values were 0.1931 (all data). Crystal data for 3(I), (CCDC no. 1012398): C60H60N4, M = 837.12, monoclinic, a = 34.8696(19) Å, b = 5.8099(3) Å, c = 28.1212(16) Å, α = 90°, β = 98.143(2)°, γ = 90°, V = 5639.6(5) Å3, T = 100(2) K, space group C2/c, Z = 4, 26236 reflections measured, 8377 independent reflections (Rint = 0.0577). The final R1 values were 0.0602 (I > 2σ(I)). The final wR(F2) values were 0.1686 (I > 2σ(I)). The final R1 values were 0.0825 (all data). The final wR(F2) values were 0.1883 (all data). Crystal data for 3(II), (CCDC no. 1012399): C30H30N2, M = 418.56, triclinic, a = 6.5695(4) Å, b = 13.4759(8) Å, c = 14.1239(9) Å, α = 91.0310(10)°, β = 99.2650(10)°, γ = 91.6850(10)°, V = 1233.21(13) Å3, T = 100(2) K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, 13275 reflections measured, 13275 independent reflections (Rint = 0.0000). The final R1 values were 0.0529 (I > 2σ(I)). The final wR(F2) values were 0.1334 (I > 2σ(I)). The final R1 values were 0.0860 (all data). The final wR(F2) values were 0.1550 (all data). , Z = 2, 13275 reflections measured, 13275 independent reflections (Rint = 0.0000). The final R1 values were 0.0529 (I > 2σ(I)). The final wR(F2) values were 0.1334 (I > 2σ(I)). The final R1 values were 0.0860 (all data). The final wR(F2) values were 0.1550 (all data). |
| This journal is © The Royal Society of Chemistry 2015 |