
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cooperative benzylic–oxyallylic stabilized cations: regioselective construction of α-quaternary centers in ketone-derived compounds†
Nitin S.
Dange
,
Jacob R.
Stepherson‡
,
Caitlan E.
Ayala‡
,
Frank R.
Fronczek§
and
Rendy
Kartika
*
Department of Chemistry, Louisiana State University, 232 Choppin Hall, Baton Rouge, LA 70810, USA. E-mail: rkartika@lsu.edu
First published on 22nd July 2015
Abstract
We describe a novel reactivity of benzylic–stabilized oxyallyl cations towards regioselective construction of carbon quaternary centers. These synthetically useful intermediates were readily generated upon ionization of aryl substituted α-hydroxy methylenol ethers with catalytic, mild Brønsted acid. The emerging unsymmetrical oxyallyl cations were then directly captured by indoles and other nucleophiles with exquisite control of regioselectivity, predictably at the electrophilic carbon bearing the alkyl substituent to produce highly functionalized, value-added enol ethers.
Introduction
Oxyallyl cation is a reactive intermediate with profound roles in many synthetic applications. It exhibits unique reactivity due to the distribution of its electrophilic character over three carbon atoms. For example, this species is universally regarded as the participating intermediate in the Nazarov cyclization,1 involving electrocylic ring closure of divinyl ketone (Scheme 1), which could be intercepted via facile processes, such as [4+3] cycloaddition1b,2 and [3+2] cycloaddition,3 to expediently generate a variety of complex molecular architectures. Recently, studies on the generation of oxyallyl cations via acid or base-induced departure of a leaving group at the α-carbon of ketone-derived compounds, followed by immediate nucleophilic capture began to emerge in the literature.4 These new bond-forming processes successfully introduced various α-functionalities that otherwise would not have been feasible using classical strategies. While these seminal reports largely employed symmetrical systems, the use of unsymmetrical oxyallyl cations 2 appeared to be rather problematic, as addition of nucleophiles to this intermediate had been shown to occur competitively at both α and α′ positions, producing a mixture of regioisomers. Our group recently contributed a solution to this fundamental problem.5 We discovered that simple protection of oxyallyl cation 2 to the corresponding O-TBS variant 6 enabled a highly regioselective addition of indoles to furnish α,α′-disubstituted silylenol ethers 7.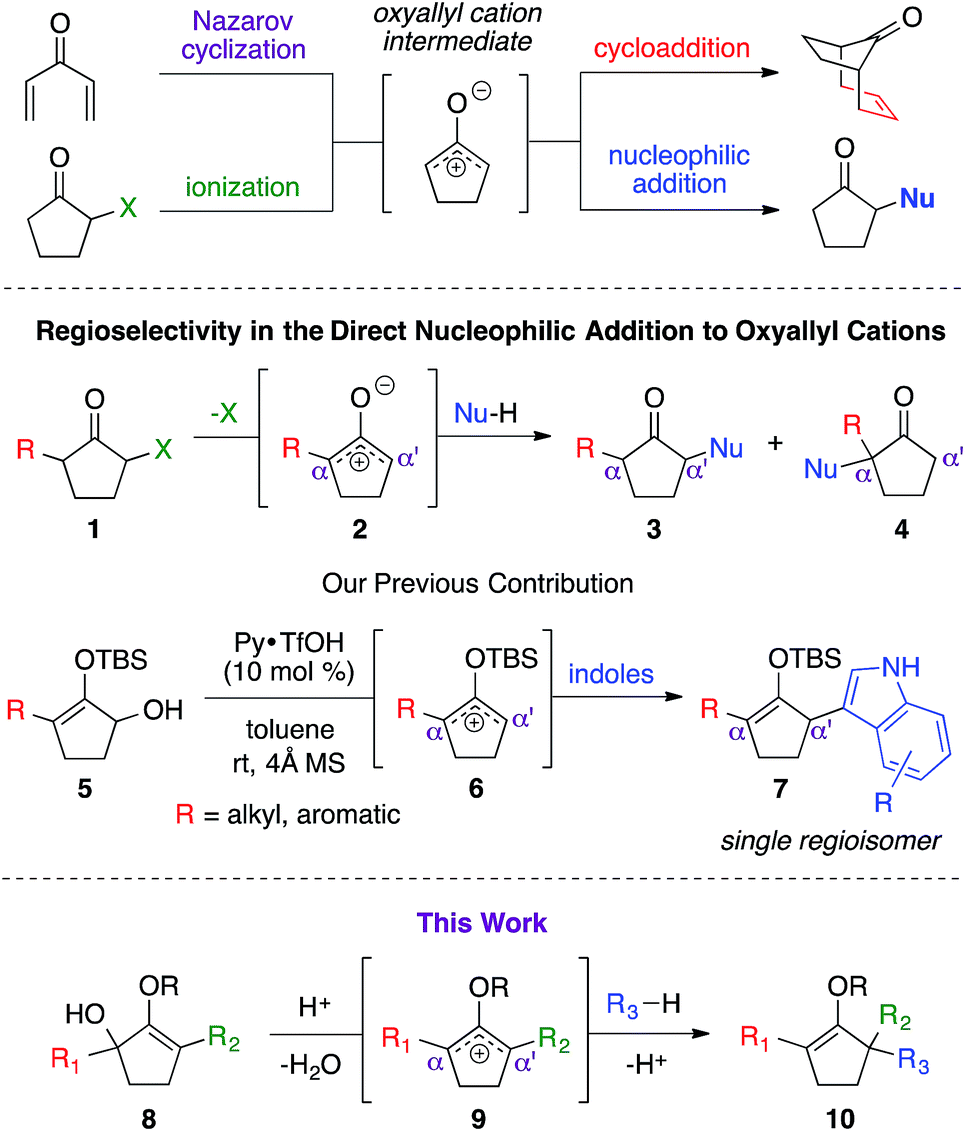 | ||
| Scheme 1 Direct nucleophilic addition to oxyallyl cation. | ||
Driven by our interest in developing de novo synthesis of quaternary carbon centers, our unique tactic in controlling regioselectivity under exceedingly mild conditions encouraged us to investigate a direct nucleophilic addition to much more complex unsymmetrical α,α′-disubstituted oxyallyl cations, viz.9. In fact, the success of this novel chemistry would introduce a powerful paradigm in the regioselective construction of an α-quaternary carbon center in ketone-derived compounds, which remain a significant synthetic challenge.6 While formation of quaternary centers via nucleophilic trapping of α,α′-disubstituted oxyallyl cations generated via Nazarov cyclization are precedented,7 there are only a very few examples on the use of unsymmetrical oxyallyl cations that proceeded in a regioselective fashion. Our hypothesis followed the assumption that ionization of disubstituted α-hydroxy enol ether 8 promoted by Brønsted acid in a non-polar medium should generate oxyallyl cation 9. Assuming that the substituents in the α and α′ positions are electronically dissimilar, we proposed that the ensuing nucleophilic addition to this reactive intermediate should proceed with a distinct regioselective preference.
Results and discussion
We commenced our investigation by rapidly assembling model substrates 12a–12d in a divergent manner from 3-methyl-cyclopentane-1,2-dione (Scheme 2). This commercially available compound was subjected to an initial treatment with TBSCl and imidazole to give silylenol ether 11a, or methyl iodide and potassium carbonate to yield the methoxy variant 11b. The residual ketone functionality in these adducts was then either reduced with DIBAL to generate secondary alcohol 12a and 12b, or reacted with phenylmagnesium chloride to afford tertiary alcohol substrates 12c and 12d.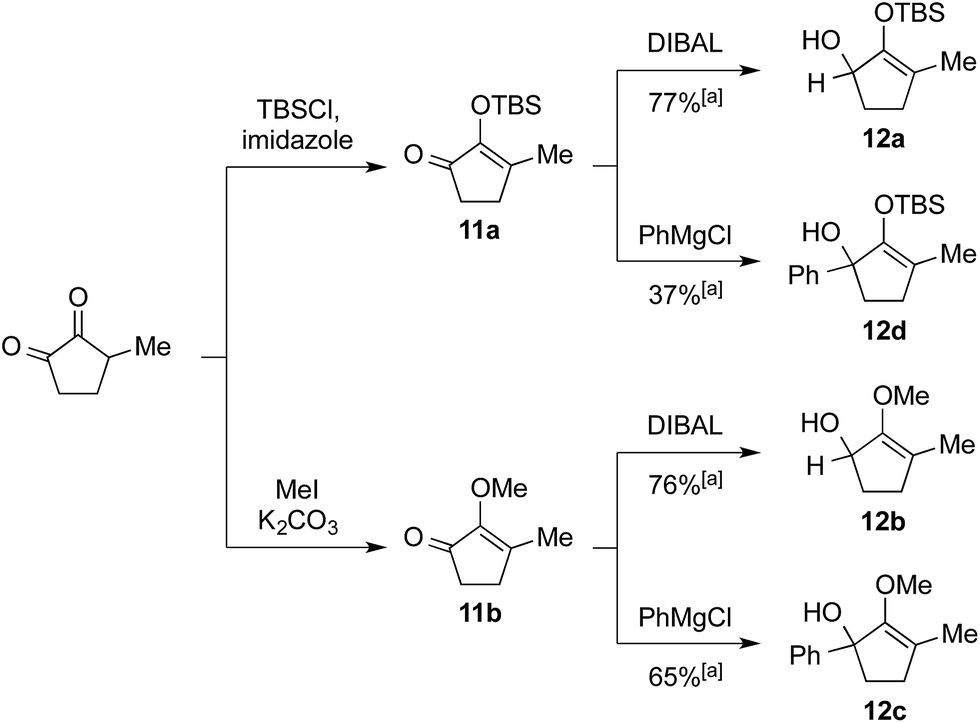 | ||
| Scheme 2 Synthesis of starting materials 12a–12d. [a] Isolated yield over two steps after flash chromatography. | ||
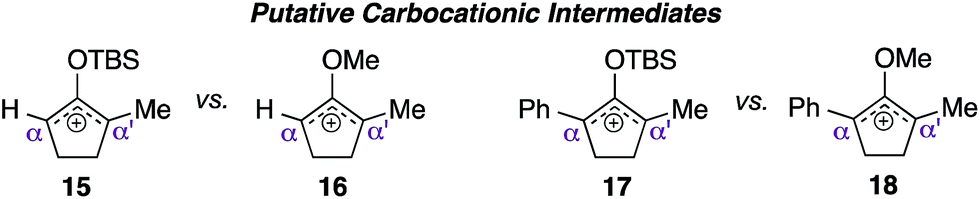
With the availability of these crucial starting materials, we then proceeded towards screening studies. As depicted in Table 1, entries 1 and 2, we began by deliberately increasing the catalyst loading to 50 mol% and the reaction concentration to 0.2 M from the previously established conditions to accelerate the reaction.5 We suspected that the TBS enol ether moiety impeded the rate of reaction, perhaps through destabilization of the emerging silyloxyallyl cation intermediate 15 by its electron-withdrawing character. To test this hypothesis, we subjected methylenol ether analogue 12b to identical conditions, and activation of this electron rich substrate was indeed complete in just one hour. Surprisingly, the corresponding methylenol ether adducts were produced in an essentially 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomers, indicating that addition of indole to the presumed oxyallyl cation 16 was not selective.
:1 mixture of regioisomers, indicating that addition of indole to the presumed oxyallyl cation 16 was not selective.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Catalyst | Loading (mol%) | Conc.a (M) | Time (h) | Yieldb |
13:14c |
| a Reaction concentration was based on starting material 12. b Isolated yield of products 13 or 14 after flash chromatography. c The ratio of regioisomers was determined by 1H NMR of the crude reaction mixture. d Combined yield for both regioisomers as they were not separable by flash chromatography. e The corresponding ketones were isolated upon aqueous workup. f Starting material 12d was never fully consumed. | |||||||||
| 1 | 12a | –TBS | –H | Py·TfOH | 10 | 0.05 | 66 | 91% | 99:1 |
| 2 | 12a | –TBS | –H | Py·TfOH | 50 | 0.2 | 3 | 70% | 99:1 |
| 3 | 12b | –Me | –H | Py·TfOH | 50 | 0.2 | 1 | 71%d | 56:44e |
| 4 | 12c | –Me | –Ph | Py·TfOH | 50 | 0.2 | 1 | 86% | 1:99 |
| 5 | 12c | –Me | –Ph | Py·TfOH | 10 | 0.2 | 2 | 81% | 1:99 |
| 6 | 12c | –Me | –Ph | Py·TsOH | 10 | 0.2 | 2 | 78% | 1:99 |
| 7 | 12c | –Me | –Ph | CSA | 10 | 0.2 | 2 | 40% | 1:99 |
| 8 | 12c | –Me | –Ph | TfOH | 10 | 0.2 | 1 | 14% | 1:99 |
| 9 | 12d | –TBS | –Ph | Py·TfOH | 10 | 0.2 | 110 | 38%f | 1:99 |
|
|||||||||

Despite the complete loss of regioselectivity, we were enthused to observe a possible formation of quaternary center 14. We subsequently incorporated an aromatic ring in the R2 position via starting material 12c, believing that the α-phenyl and α′-methyl substituents would provide sufficient electronic bias to differentiate the two electrophilic centers in unsymmetrical oxyallyl cation 18 towards nucleophilic attack. Indeed, exposure of substrate 12c to the activation conditions afforded quaternary center 14 in 86% yield, remarkably as a single regioisomer, where indole addition occurred exclusively at the α′-position (entry 4). Gratifyingly, reducing the catalyst loading back to 10 mol% essentially furnished the same product with an identical isolation yield and regioselectivity, albeit with a slightly longer but manageable reaction time (entry 5).
As indicated in entry 6, activation of starting material 12c with 10 mol% pyridium tosylate produced the corresponding indole adduct 14 in an essentially identical yield from that obtained using catalytic pyridinium triflate. This result suggested that the counter anion perhaps merely served as a spectator in this methodology. Nevertheless, the quality of our reaction appeared to be directly dependent on the acidity of the catalysts. For instance, the use of stronger Brønsted acids,8 such as CSA (entry 7) and triflic acid (entry 8) led to significant decomposition of materials, thus leading to much lower isolation yields of the target product. These studies also demonstrated that free pyridine, liberated from pyridinium ion catalyst upon ionization, did not play a role in controlling regioselectivity,5 as addition of indole to starting material 12c, catalyzed by either CSA or triflic acid, also produced the corresponding quaternary center 14 as a single regioisomer. To further signify the role of O-methyl group in accelerating the rate of reaction, we then exposed TBS enol ether variant 12d to the optimal reaction conditions. This compound, which presumably proceeded to generate electron deficient silyloxyallyl cation 17, severely lacked reactivity and in fact failed to reach completion even after 110 hours of reaction time (entry 9).
With these preliminary results in hand, our investigation continued with a scope of indoles towards α′-quaternarization of methylenol ether 19 (Table 2).9 Electron-donating substituents, such as methyl and methoxy groups, produced quaternary stereocenters 21b and 21c in 90% and 74% yields, respectively. N-Methyl indole was also a suitable nucleophile, which afforded product 21d in 74% yield. Halogen substituents, such as 6-chloro and 5-bromo indoles, were found robust under the optimized reaction conditions, leading to formation of the corresponding methylenol ethers 21e and 21f in 75% and 85% yields, respectively. Electron-deficient methyl-5-carboxylate indole furnished quaternary center 21g in 68% yield despite a longer reaction time. Structurally elaborate 5-methoxy-1H-benzo[g]indole was also found to produce the corresponding quaternary center 21h in near quantitative yield.
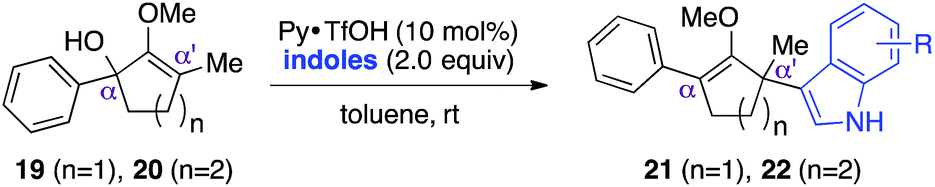

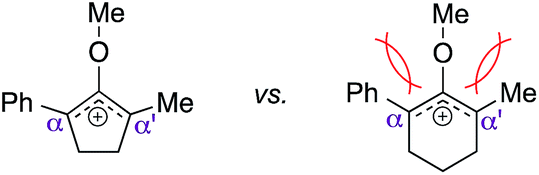
This synthetic method was also applicable to analogous 6-membered α-hydroxy methylenol ether 20, which yielded the corresponding α′-quaternary center in methylenol ether 22 in high yields as a single regioisomer despite the longer reaction time.10 Functionalization of starting material 20 with indole cleanly produced α′-indoyl product 22a in 80% yield.9 The use of electron donating 5-methoxyindole and halogenated 5-bromoindole furnished the corresponding methylenol ethers 22b and 22c in 69% and 85%. The structure of compound 21c and 22b was conclusively confirmed by X-ray analysis.11 Interestingly, electron deficient methylindole-5-carboxylate led to an isolation of the resulting product 22d in modest 35% yield. We also explored the applicability of sterically congested 2-phenylindole and N-methyl indole, which led to an installation of α′-quaternary center in 22e and 22f in good yields.
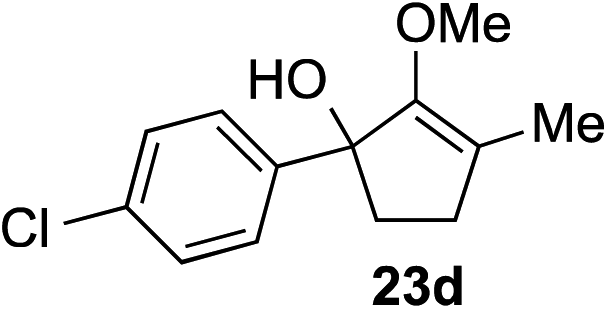
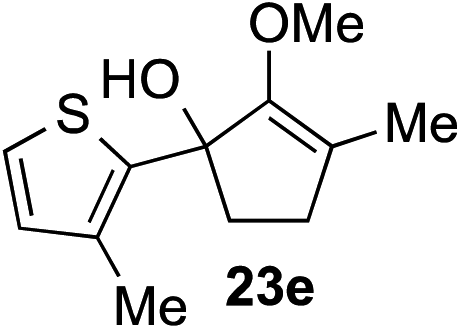
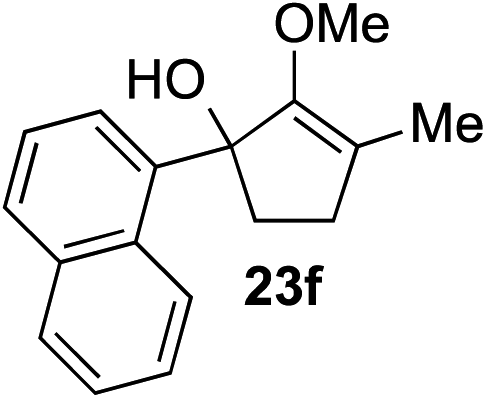
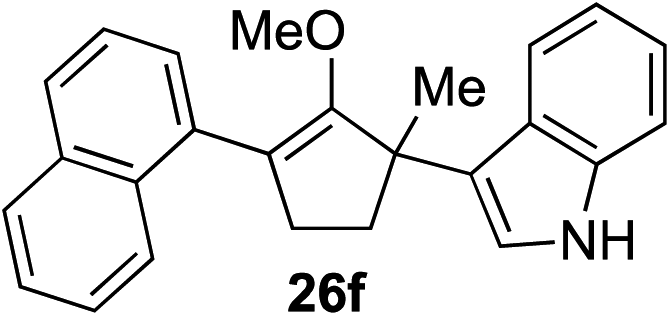
The role of aromatic and alkyl substituents at the α and α′-positions, respectively, was then examined. As shown in Table 3, we proposed that ionization of either α-hydroxy methylenol ether 23 or α′-hydroxy methylenol ether 24 with pyridinium triflate should both generate methyloxyallyl cation 25, which would be captured by indole, predictably at the α′-position, to give the corresponding product 26. As illustrated in entries 1–6, various aromatic rings, such as electron-donating p-anisole 23a and p-toluene 23b, cleanly furnished α′-indoyl adducts 26a and 26b in 90% and 84% yields, respectively. Halogen substituents, such as 4-fluorophenyl 23c and 4-chlorophenyl 23d, were also tolerated to produce methylenol ethers 26c and 26d in excellent yields. An aromatic heterocycle, such as 3-methylthiophene 23e, furnished quaternary center 26e in near quantitative yield. A larger aromatic system, such as the 2-napthyl group in 23f, was also found to be suitable in this chemistry.
|
|
|||
|---|---|---|---|
| Entry | Starting material | Product | Yielda |
| a Isolated yield of products 26 after flash chromatography. b The low yield was attributed to poor solubility of the product in most organic solvents. c Combined yield for both regioisomers. d The major regioisomer was not assigned, as these compounds were not separable by chromatography. e The ratio of regioisomers was determined by 1H NMR of the crude reaction mixture. | |||
| 1 |
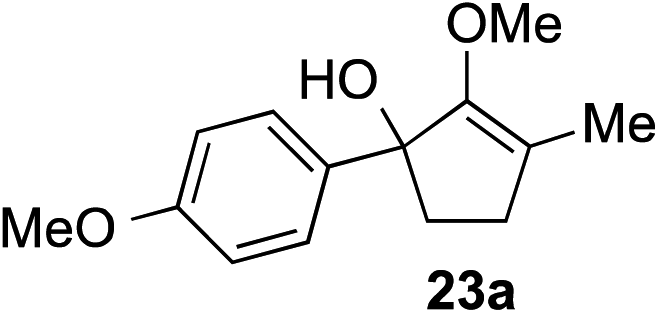
|
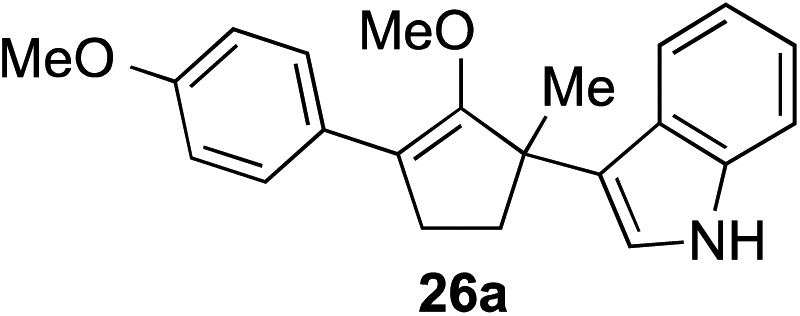
|
90% (1 h) |
| 2 |

|
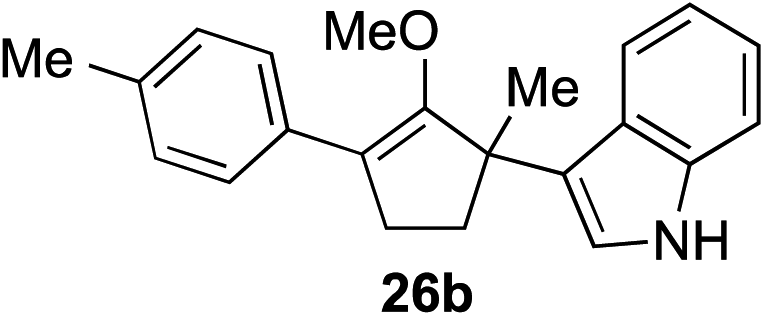
|
84% (1 h) |
| 3 |

|
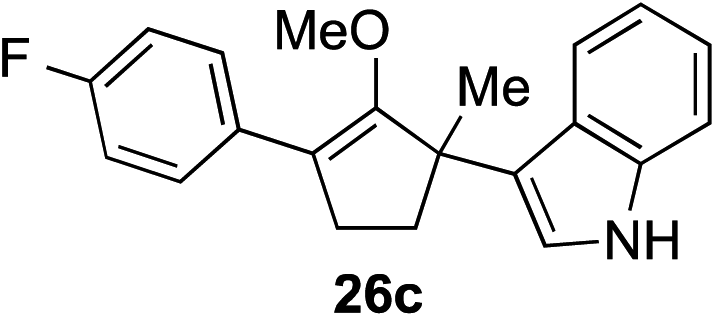
|
88% (1 h) |
| 4 |
|

|
90% (1 h) |
| 5 |
|

|
93% (1 h) |
| 6 |
|
|
31%b (1 h) |
| 7 |
|

|
80% (1 h) |
| 8 |
|

|
80% (18 h) |
| 9 |
|

|
46%c,d 3:2 rre (26 h) |
| 10 |

|
|
65%c 1:1 rre (22 h) |
| 11 |
|
|
88% (1 h) |
| 12 |
|
|
80%c 3:1 rre (1 h) |
| 13 |

|
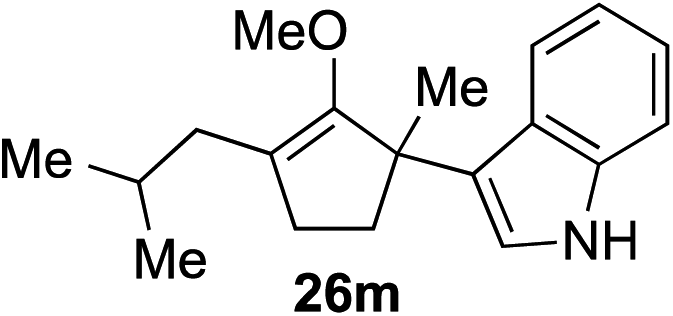
|
78%c 4:1 rre (1 h) |

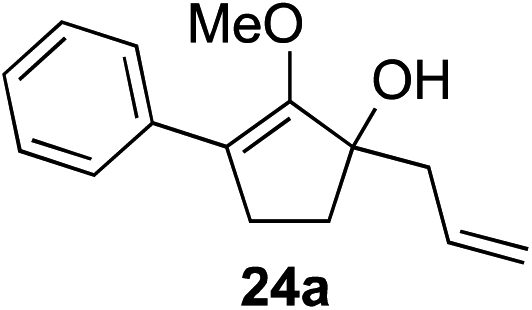
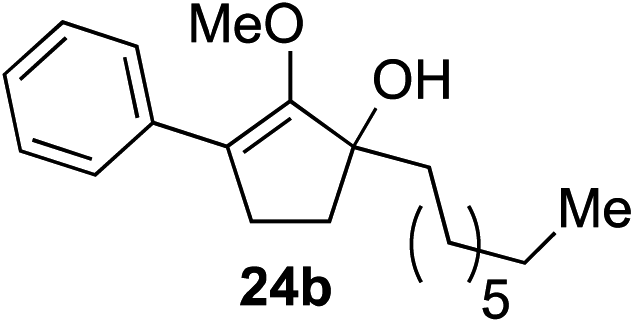
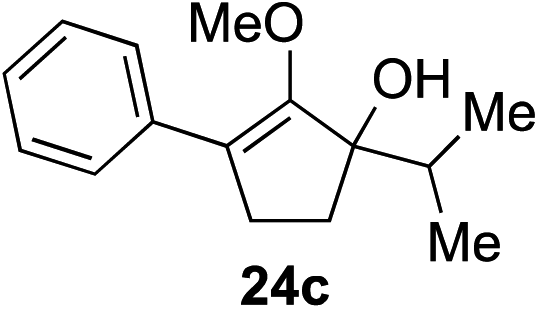
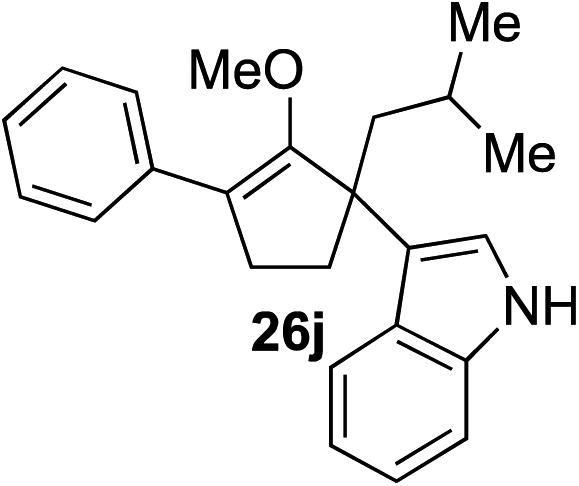
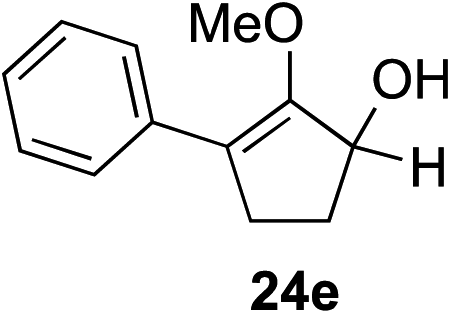
Our investigation continued on with efforts to identify the effect of various α′-alkyl substituents in this enol ether functionalization reaction. As shown in Table 3, entries 7 and 8, allylic and aliphatic side chains 24a and 24b gave the corresponding α′-indoyl enol ethers 26g and 26h, respectively, in excellent yields as a single regioisomer. The regioselectivity induction in this methodology appeared to be sensitive to simple modulation in the steric effect. For instance, a significant erosion in regioselectivity was observed when sterically encumbered isopropyl or isobutyl groups were incorporated to the α′-position in starting materials 24c and 24d. The longer reaction time involving substrates 24b–24d suggested that an increasing size of the alkyl substituents substantially impeded the rate of reaction.
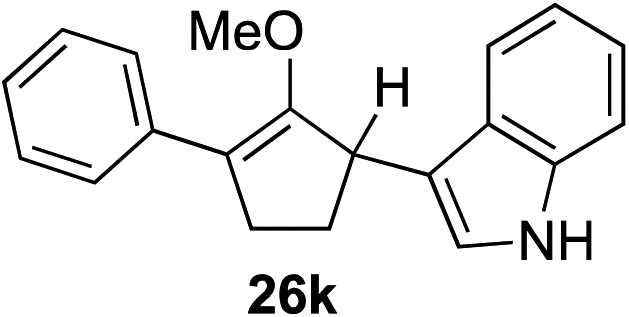
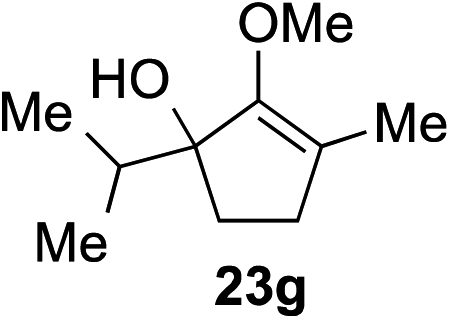
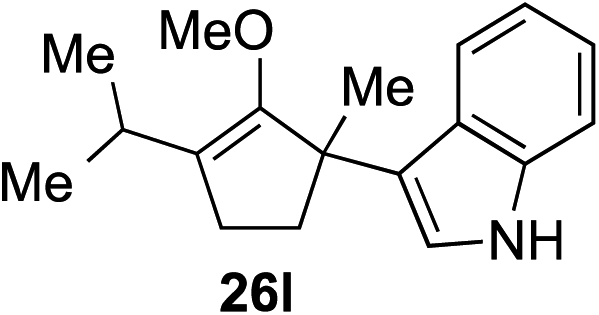
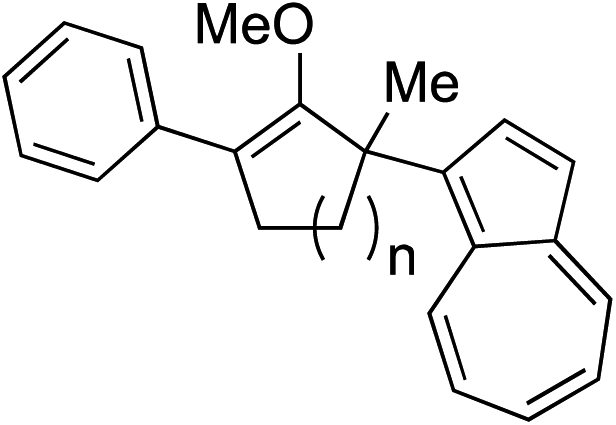
As opposed to the methyl variant 12b (Scheme 1, entry 3), α-phenyl substituted starting material 24e exclusively produced α-phenyl-α′-indole 26k as a single regioisomer in quantitative yield. This astounding result strongly suggests that the unusual benzylic-oxyallylic carbocation stabilization appeared to have cooperatively directed in these direct nucleophilic addition reactions.12 The involvement of the aromatic substituents in facilitating regioselectivity control was further confirmed by the use of starting materials bearing α-isopropyl 23g and α-isobutyl 23h (entries 12 and 13). Exposure of these substrates to the reaction conditions produced the corresponding methylenol ether adducts 26l and 26m with the indole addition occurring at the less sterically congested methylated α′-carbon. The observed regioselectivity in these products, however, was rather modest, suggesting that while steric influence readily offered some degrees of regiochemical induction, these effects alone were not sufficient in furnishing exclusive regioselectivity in the addition of nucleophiles to the putative oxyallyl cation intermediates without reinforcement from the benzylic-type stabilization provided by aryl substituents.
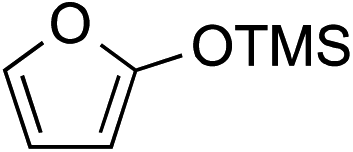
While we and others have extensively utilized substituted indoles in studies concerning a direct nucleophilic addition to oxyallyl cations,4f,4g,5 there are only a few precedents on the utility of other carbon and heteroatom nucleophiles in this type of chemistry.4g,4h As a guideline for us to judiciously select other potential carbon nucleophiles beyond indole, we consulted Mayr's nucleophilicity parameters (N values)13 and extracted the N values of various substituted indoles, which ranged from N = 2.2 for electron poor indoles to N = 7.2 for electron rich indoles,13d as a reference. As illustrated in Table 4, this strategy enabled us to identify that pyrrole (N = 4.6) successfully reacted with starting materials 19 and 20 to afford the corresponding methylenol ethers 27a and 28a in 45% and 89% yields, respectively, although these reactions were rather sluggish.13e In contrast, the use of much more nucleophilic 2,4-dimethylpyrrole (N = 10.7) rapidly converted five-membered methylenol ether 19 to the corresponding quaternary center 27b in 83% yield within 30 minutes.13f Azulene (N = 6.7), a highly nucleophilic neutral aromatic hydrocarbon, was found to react in this method and yielded methylenol ethers 27c and 28c as a single regioisomer.13h We were also able to demonstrate the utility of 2-(trimethylsiloxy)furan (N = 7.2) in this methodology.13c This nucleophile readily functionalized starting material 19 to furnish stereochemically congested adduct 27d in 62% yield, surprisingly as a single diastereomer.11
|
|
|||
|---|---|---|---|
| Entry | Nucleophile | Product | Yielda |
| a Isolated yield of products 27 and 28 after flash chromatography. b 50 mol% of pyridinium triflate was added. c 1.1 equivalent of azulene was employed. d The starting material was not fully consumed. e 4 Å molecular sieves were added. | |||
| 1 |
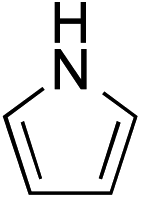
|

|
27a 45% (1.5 h), 28a 89%b (18 h) |
| 2 |
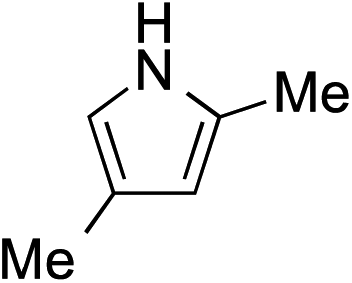
|
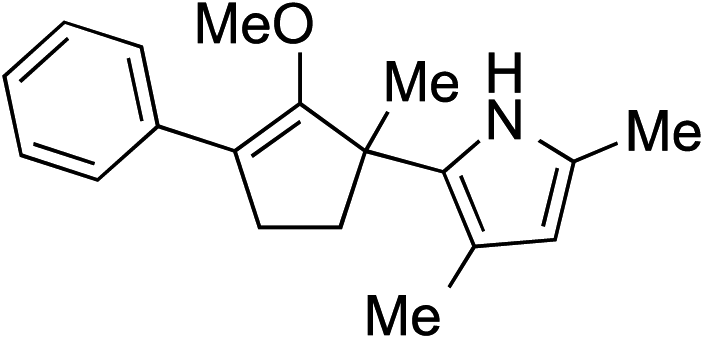
|
27b 83% (0.5 h) |
| 3 |

|
|
27c 62%c, (0.5 h), 28c 39%b,c,d (39 h) |
| 4 |
|

|
27d 62%, 20:1 dr (72 h) |
| 5 |

|

|
27e 69%e (23 h) |
| 6 |
|
|
27f 39% (1 h), 28f 94% (4 h) |
| 7 |

|

|
27g 78% (1.5 h), 28g 93% (20 h) |

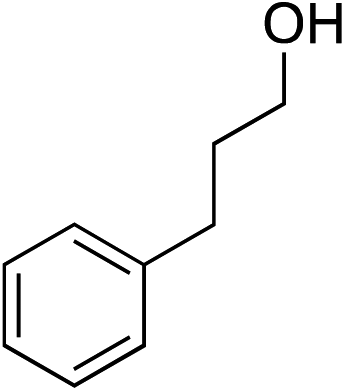
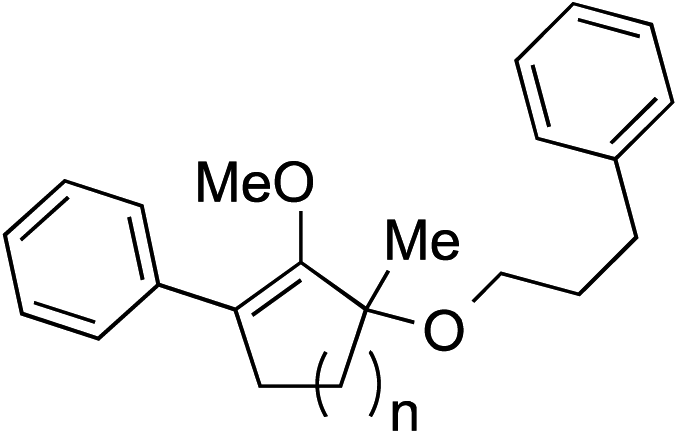
We recognized the challenge of using heteroatom-centered nucleophiles to capture carbocations in Brønsted acid catalyzed reactions due to the potentially reversible ionization of the newly generated carbon-heteroatom bonds promoted by the catalyst.12,14 We explored the applicability of such nucleophiles by initially focusing on alcohols.15 Interestingly, a reaction between α-hydroxy enol ether 19 and methanol produced the corresponding methanol adduct 27e in 69% yield as a single regioisomer. While the use of 3-phenyl-1-propanol afforded methylenol ether 27f in modest 39% yield, the addition of this primary alcohol to six-membered substrate 20 formed quaternary center 28f in near quantitative yield. An attempt to incorporate thiophenol was also successful to produce the respective mercaptan adducts 27g and 28g in high yields as a single regioisomer.
Construction of these highly functionalized enol ethers provided a unique venue to access various structurally formidable quaternary center-containing molecular architectures (Scheme 3). For example, treatment of methylenol ether 21a to stoichiometric toluenesulfonic acid monohydrate in THF at −20 °C readily produced the corresponding stereochemically elaborate ketone 29 in 73% yield with 5.5:1 diastereoselection. Our ability to install two aromatic substituents, each at the α- and α′-positions of ketone, while simultaneously introducing an α-quaternary center, clearly demonstrated the strength of our methodology. Methylenol ether 21a could also be subjected to palladium-catalyzed hydrogenation. These conditions introduced a stereotriad in methylated cyclopentanol 30 in a good yield with a decent control of diastereoselectivity.
 | ||
| Scheme 3 Synthetic applications of methylenol ethers. | ||
The indole ring in compound 21a could also be easily protected without compromising the methylenol ether functionality via treatment with LDA and acetic anhydride. The resulting acetate-protected product 31 could be further subjected to another diastereoselective transformation, such as oxidation of the carbon–carbon double bond using m-CPBA.16 Interestingly, this reaction cleanly produced structurally intricate epoxide 32 with three contiguous quaternary centers, in 80% yield as a 5:1 mixture of diastereomers. Similarly, the stereochemical identity of compounds 29, 30, and 32 was confirmed by single crystal X-ray diffraction.11 We were also able to oxidatively cleave the carbon–carbon double bond in methylenol ether 31 using RuCl3/NaIO4 to furnish 1,5-ketoester 33.17 In addition to the generation an sp3–sp2 connectivity, chemoselective α-quaternarization of ester in the presence ketone are challenging due to the subtle acidity differences between the relevant α-hydrogens.8
Conclusions
In conclusion, we detailed a new method to generate benzylic–oxyallylic stabilized carbocations under mild Brønsted acid catalysis and discovered that the reactivity of these novel intermediates could be harnessed towards a regioselective construction of quaternary centers through a direct capture with indoles and other high value nucleophiles. Overall, this chemistry efficiently furnished highly functionalized enol ethers that could be conveniently derivatized to other complex molecular architectures. Detailed mechanistic investigations on the origin of this unprecedented control of regioselectivity are ongoing in our laboratory, and the results will be reported in due course.Acknowledgements
This material is based upon work supported by the National Science Foundation under CHE-1464788. We gratefully acknowledge generous financial support from Louisiana State University (LSU) College of Science and the Louisiana Board of Regents through the PFUND Award (LEQSF-EPS(2015)-PFUND-395-PFUND-395). C.E.A. thanks the Louisiana Board of Regents for the Graduate Fellowship (LEQSF(2011-16)-GF-03). J.R.S. thanks the Louisiana Board of Reagents for the Graduate Fellowship (LEQSF(2014-19)-GF-02).Notes and references
- (a) A. J. Frontier and C. Collison, Tetrahedron, 2005, 61, 7577–7606 CrossRef CAS PubMed; (b) T. N. Grant, C. J. Rieder and F. G. West, Chem. Commun., 2009, 5676–5688 RSC; (c) W. Nakanishi and F. G. West, Curr. Opin. Drug Discovery Dev., 2009, 12, 732–751 CAS; (d) N. Shimada, C. Stewart and M. A. Tius, Tetrahedron, 2011, 67, 5851–5870 CrossRef CAS PubMed; (e) T. Vaidya, R. Eisenberg and A. J. Frontier, ChemCatChem, 2011, 3, 1531–1548 CrossRef CAS PubMed.
- (a) A. G. Lohse and R. P. Hsung, Chem.–Eur. J., 2011, 17, 3812–3822 CrossRef CAS PubMed; (b) M. Harmata, Chem. Commun., 2010, 46, 8904–8922 RSC; (c) M. Harmata, Chem. Commun., 2010, 46, 8886–8903 RSC; (d) D. A. Foley and A. R. Maguire, Tetrahedron, 2010, 66, 1131–1175 CrossRef CAS PubMed; (e) M. Harmata, Adv. Synth. Catal., 2006, 348, 2297–2306 CrossRef CAS PubMed; (f) M. A. Battiste, P. M. Pelphrey and D. L. Wrightthl, Chem.–Eur. J., 2006, 12, 3438–3447 CrossRef CAS PubMed; (g) B. Niess and H. M. R. Hoffmann, Angew. Chem., Int. Ed., 2005, 44, 26–29 CrossRef CAS PubMed; (h) I. V. Hartung and H. M. R. Hoffmann, Angew. Chem., Int. Ed., 2004, 43, 1934–1949 CrossRef CAS PubMed; (i) M. Harmata and P. Rashatasakhon, Tetrahedron, 2003, 59, 2371–2395 CrossRef CAS; (j) M. Harmata, Acc. Chem. Res., 2001, 34, 595–605 CrossRef CAS PubMed.
- (a) E. H. Krenske, S. Z. He, J. Huang, Y. F. Du, K. N. Houk and R. P. Hsung, J. Am. Chem. Soc., 2013, 135, 5242–5245 CrossRef CAS PubMed; (b) H. Li, R. P. Hughes and J. Wu, J. Am. Chem. Soc., 2014, 136, 6288–6296 CrossRef CAS PubMed; (c) H. Li and J. Wu, Synthesis, 2015, 47, 22–33 CAS.
- (a) A. W. Fort, J. Am. Chem. Soc., 1962, 84, 2620–2625 CrossRef CAS; (b) K. Freter, Liebigs Ann. Chem., 1978, 1357–1364 CrossRef CAS PubMed; (c) B. Föhlisch and R. Joachimi, Chem. Ber., 1987, 120, 1951–1960 CrossRef PubMed; (d) J. Leitich and I. Heise, Eur. J. Org. Chem., 2001, 2707–2718 CrossRef CAS; (e) M. Harmata, C. Huang, P. Rooshenas and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 8696–8699 CrossRef CAS PubMed; (f) Q. Tang, X. Chen, B. Tiwari and Y. R. Chi, Org. Lett., 2012, 14, 1922–1925 CrossRef CAS PubMed; (g) M. N. V. Wal, A. K. Dilger and D. W. C. MacMillan, Chem. Sci., 2013, 4, 3075–3079 RSC; (h) J. Luo, H. Zhou, J. W. Hu, R. Wang and Q. Tang, RSC Adv., 2014, 4, 17370–17377 RSC.
- C. E. Ayala, N. S. Dange, F. R. Fronczek and R. Kartika, Angew. Chem., Int. Ed., 2015, 54, 4641–4645 CrossRef CAS PubMed.
- (a) Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed; (b) C. M. Reeves, D. C. Behenna and B. M. Stoltz, Org. Lett., 2014, 16, 2314–2317 CrossRef CAS PubMed; (c) P. Zhang, H. Le, R. E. Kyne and J. P. Morken, J. Am. Chem. Soc., 2011, 133, 9716–9719 CrossRef CAS PubMed; (d) J. T. Mohr, D. C. Behenna, A. M. Harned and B. M. Stoltz, Angew. Chem., Int. Ed., 2005, 44, 6924–6927 CrossRef CAS PubMed; (e) W.-B. Liu, C. M. Reeves, S. C. Virgil and B. M. Stoltz, J. Am. Chem. Soc., 2013, 135, 10626–10629 CrossRef CAS PubMed; (f) J. Li and D. Lee, Chem. Sci., 2012, 3, 3296–3301 RSC; (g) A. Y. Hong, M. R. Krout, T. Jensen, N. B. Bennett, A. M. Harned and B. M. Stoltz, Angew. Chem., Int. Ed., 2011, 50, 2756–2760 CrossRef CAS PubMed; (h) D. C. Behenna, Y. Liu, T. Yurino, J. Kim, D. E. White, S. C. Virgil and B. M. Stoltz, Nat. Chem., 2012, 4, 130–133 CrossRef CAS PubMed.
- (a) Y.-K. Wu and F. G. West, Org. Lett., 2014, 16, 2534–2537 CrossRef CAS PubMed; (b) Y.-K. Wu, C. R. Dunbar, R. McDonald, M. J. Ferguson and F. G. West, J. Am. Chem. Soc., 2014, 136, 14903–14911 CrossRef CAS PubMed; (c) Y. Kwon, O. Scadeng, R. McDonald and F. G. West, Chem. Commun., 2014, 50, 5558–5560 RSC; (d) Y. Kwon, R. McDonald and F. G. West, Angew. Chem., Int. Ed., 2013, 52, 8616–8619 CrossRef CAS PubMed; (e) Y.-K. Wu, R. McDonald and F. G. West, Org. Lett., 2011, 13, 3584–3587 CrossRef CAS PubMed; (f) K. Yaji and M. Shindo, Synlett, 2009, 2524–2528 CAS; (g) D. Song, A. Rostami and F. G. West, J. Am. Chem. Soc., 2007, 129, 12019–12022 CrossRef CAS PubMed; (h) F. Dhoro, T. E. Kristensen, V. Stockmann, G. P. A. Yap and M. A. Tius, J. Am. Chem. Soc., 2007, 129, 7256–7257 CrossRef CAS PubMed; (i) T. D. White and F. G. West, Tetrahedron Lett., 2005, 46, 5629–5632 CrossRef CAS PubMed; (j) F. Dhoro and M. A. Tius, J. Am. Chem. Soc., 2005, 127, 12472–12473 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- The use of 1.1 equivalents of indole resulted in a slightly lower yield and longer reaction time.
-
(a) F. Johnson, Chem. Rev., 1968, 68, 375–413 CrossRef CAS;
(b) R. W. Hoffmann, Chem. Rev., 1989, 89, 1841–1860 CrossRef CAS . We believed that the lower reactivity of the six-membered substrates in this methodology was most likely attributed by destabilization of their corresponding six-membered oxyallyl cations due to the more severe allylic strain that was readily introduced by the smaller angles among the participating subsituents than those from the analogous five-membered systems.
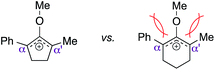 .
. - ESI†.
- R. Sanz, A. Martinez, D. Miguel, J. M. Alvarez-Gutierrez and F. Rodriguez, Adv. Synth. Catal., 2006, 348, 1841–1845 CrossRef CAS PubMed . The extent of regioselectivity in the addition of nucleophiles to unsymmetrical α,α′-disubstituted allylic cations under Brønsted acid catalysis was unpredictable.
- (a) H. Mayr and A. R. Ofial, J. Phys. Org. Chem., 2008, 21, 584–595 CrossRef CAS PubMed; (b) H. Mayr and A. R. Ofial, Pure Appl. Chem., 2005, 77, 1807–1821 CrossRef CAS; (c) H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77 CrossRef CAS PubMed; (d) S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 9088–9095 CrossRef CAS PubMed; (e) B. Kempf, N. Hampel, A. R. Ofial and H. Mayr, Chem.–Eur. J., 2003, 9, 2209–2218 CrossRef CAS PubMed; (f) T. A. Nigst, M. Westermaier, A. R. Ofial and H. Mayr, Eur. J. Org. Chem., 2008, 2369–2374 CrossRef CAS PubMed; (g) J. Ammer, C. Nolte and H. Mayr, J. Am. Chem. Soc., 2012, 134, 13902–13911 CrossRef CAS PubMed; (h) M. Kedziorek, P. Mayer and H. Mayr, Eur. J. Org. Chem., 2009, 1202–1206 CrossRef CAS PubMed.
- (a) S. E. Larson, J. C. Baso, G. L. Li and J. C. Antilla, Org. Lett., 2009, 11, 5186–5189 CrossRef CAS PubMed; (b) G. L. Li, F. R. Fronczek and J. C. Antilla, J. Am. Chem. Soc., 2008, 130, 12216–12217 CrossRef CAS PubMed; (c) Y. Liang, E. B. Rowland, G. B. Rowland, J. A. Perman and J. C. Antilla, Chem. Commun., 2007, 4477–4479 RSC; (d) E. B. Rowland, G. B. Rowland, E. Rivera-Otero and J. C. Antilla, J. Am. Chem. Soc., 2007, 129, 12084–12085 CrossRef CAS PubMed; (e) G. B. Rowland, H. L. Zhang, E. B. Rowland, S. Chennamadhavuni, Y. Wang and J. C. Antilla, J. Am. Chem. Soc., 2005, 127, 15696–15697 CrossRef CAS PubMed; (f) M. Senatore, A. Lattanzi, S. Santoro, C. Santi and G. Della Sala, Org. Biomol. Chem., 2011, 9, 6205–6207 RSC.
- Sterically bulky alcohols and nitrogen-centered nucleophiles were not tolerated in this methodology..
- J. Carreras, M. Livendahl, P. R. McGonigal and A. M. Echavarren, Angew. Chem., Int. Ed., 2014, 53, 4896–4899 CrossRef CAS PubMed.
- (a) L. F. Silva Jr, R. S. Vasconcelos and M. A. Nogueira, Org. Lett., 2008, 10, 1017–1020 CrossRef PubMed; (b) M. C. Pierre, A. Tenaglia and M. Santelli, Tetrahedron, 1998, 54, 14803–14810 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data of new compounds. CCDC 1057455–1057460. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01914a |
| ‡ These authors contributed equally. |
| § X-ray crystallographer. |
| This journal is © The Royal Society of Chemistry 2015 |