
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic and computational studies of nitrile hydratase: insights into geometric and electronic structure and the mechanism of amide synthesis†
Kenneth M.
Light‡
a,
Yasuaki
Yamanaka§
b,
Masafumi
Odaka
 *b and
Edward I.
Solomon
*a
*b and
Edward I.
Solomon
*a
aDepartment of Chemistry, Stanford University, Stanford, CA 94305, USA. E-mail: edward.solomon@stanford.edu
bDepartment of Biotechnology and Life Science, Tokyo University of Agriculture and Technology, 2-24-16 Naka-cho, Koganei, Tokyo, Japan. E-mail: modaka@cc.tuat.ac.jp
First published on 30th July 2015
Abstract
Nitrile hydratases (NHases) are mononuclear nonheme enzymes that catalyze the hydration of nitriles to amides. NHase is unusual in that it utilizes a low-spin (LS) FeIII center and a unique ligand set comprised of two deprotonated backbone amides, cysteine-based sulfenic acid (RSO(H)) and sulfinic acid (RSO2−), and an unmodified cysteine trans to an exogenous ligand site. Electron paramagnetic resonance (EPR), magnetic circular dichroism (MCD) and low-temperature absorption (LT-Abs) spectroscopies are used to determine the geometric and electronic structures of butyrate-bound (NHaseBA) and active (NHaseAq) NHase. These data calibrate DFT models, which are then extended to explore the mechanism of nitrile hydration by NHase. In particular, the nitrile is activated by coordination to the LS FeIII and the sulfenate group is found to be deprotonated and a significantly better nucleophile than water that can attack the coordinated nitrile to form a cyclic species. Attack at the sulfenate S atom of the cyclic species is favorable and leads to a lower kinetic barrier than attack by water on coordinated, uncyclized nitrile, while attack at the C of the cyclic species is unfavorable. The roles of the unique ligand set and low-spin nature of the NHase active site in function are also explored. It is found that the oxidized thiolate ligands are crucial to maintaining the LS state, which is important in the binding and activation of nitrile susbtrates. The dominant role of the backbone amidate ligands appears to be as a chelate in keeping the sulfenate properly oriented for nucleophilic attack on the coordinated substrate.
Introduction
Nitriles produced by plants and animals are a source of carbon and nitrogen for some microorganisms. Nitrile hydratases (NHases) are enzymes found in bacteria that catalyze the hydrolysis of nitriles to amides as part of the nitrile degradation pathway.1 NHases have been used industrially as catalysts for the production of methacrylonitrile and nicotinamide,2 and have also been used in the synthesis of chiral amides3 and possess the potential to treat industrial wastewater.4 As shown in Fig. 1A, NHases possess an active site that uses either low-spin (LS) FeIII or LS CoIII complexed to a very unusual ligand set.5,6 This set is comprised of two deprotonated backbone amides or amidates, a cysteine thiolate, cysteine-derived post-translationally modified sulfenic/sulfenate (Cys-SO(H)) and sulfinate (Cys-SO2−) groups, and an exogenous ligand (X). The protonation state of the Cys-SO(H) group in the active form of the enzyme has not been unambiguously determined, with conflicting spectroscopic evidence for the sulfenate and sulfenic acid forms in the literature.7,8 | ||
| Fig. 1 The active site structure of NHase. (A) Diagram of the NHase active site, depicting the thiolate (green), amidate (red), sulfinate (blue), sulfenate/sulfenic acid (orange) and water-derived (denoted X) ligands. (B) The Fe(III) active site of Rhodococcus erythropolis N771 NHase after photolysis (PDB ID 2CYZ). Heavy atom distances showing H bonding are marked with dashed lines. All distances are in Å. | ||
NHases are αβ heterodimers with the metal ligand residues residing in the α subunit. The Cys-SO2− and Cys-SO(H) residues H-bond with two arginines on the β subunit, as shown in Fig. 1B.5,6 For the FeIII NHase of Rhodococcus erythropolis N771 βArg56 (Fig. 1B) was found to be essential for catalysis. In CoIII NHases the sixth ligand (X in Fig. 1A) is derived from water,6 whereas FeIII NHase produced in the dark has a NO bound to Fe that is photolytically cleaved to produce the active form containing a water-derived ligand (Fig. 2A).9–11 If left exposed to air for a sufficient period of time, the Cys-SO(H) ligand is oxidized to Cys-SO2− and the enzyme becomes inactive (Fig. 2B).12 However, butyric acid may be added to act as a protecting agent, binding to Fe and inhibiting further oxidation of the Cys-SO(H) group (Fig. 2C).13–15 Butyric acid has also been found to be a competitive inhibitor, which becomes more strongly inhibiting with decreasing pH.16 This indicates that it is the protonated form of the acid that stabilizes the enzyme (although from EPR data and DFT calculations (vide infra) the proton transfers to the sulfenate group upon butyric acid coordination to NHase FeIII).
 | ||
| Fig. 2 NHase active site forms. (A) Photolytic cleavage of Fe-bound NO to produce the active form. (B) Oxidation of the active form sulfenate/sulfenic acid group in aerobic conditions. (C) Protection of the active site through complexation with butyrate. | ||
The catalytic mechanism by which these enzymes operate has not yet been fully elucidated. The fact that the coordinated butyrate acts as a competitive inhibitor suggests that nitrile is activated for nucleophilic attack by coordination to metal, which has provided support for the mechanism of a water attacking this coordinated nitrile with a base accepting a proton as shown in Fig. 3A.17–20 Recently, a crystallographic study by Holz and coworkers21 showing that alkyl boronic acids, also competitive inhibitors, bind to the active site metal of NHase and are nucleophilically attacked by the sulfenate oxygen. This led to the proposal that the RSO(H) itself is the nucleophile that attacks the coordinated nitrile C as shown in Fig. 3B, activating it for nucleophilic attack by water on either the C or S atoms (Fig. 3C and D, respectively).21 A related mechanism has been proposed22 that involves the axial thiolate acting as the initial nucleophile and the subsequent formation of a disulfide bond, as shown in Fig. 3E. The protonation states of the water-derived and Cys-SO(H) ligands in the active form of the enzyme are not well defined, and their determination is important in understanding the mechanism of nitrile hydrolysis.
 | ||
| Fig. 3 Potential mechanisms for NHase catalysis. (A) Activation of coordinated nitrile for nucleophilic attack by water. (B) Nucleophilic attack by RSO(H) and subsequent activation for attack by water on either (C) the nitrile carbon, or (D) the RSO(H) sulfur. The oxygen atom originating from water is shown in red. (E) Nucleophilic attack on the cyclic intermediate by the axial thiolate. | ||
It is also important to note that a second-sphere mechanism involving nucleophilic attack on an uncoordinated nitrile in the active site pocket by a coordinated hydroxide has been proposed and found to have a theoretically similar barrier to coordinated nitrile activation via DFT calculations.20,23 However, such a mechanism is not in agreement with the crystallographic results involving boronic acids described above.21
In this study we use electronic paramagnetic resonance (EPR), absorption, and magnetic circular dichroism (MCD) spectroscopies to determine the geometric and electronic structures of the paramagnetic LS FeIII NHase from Rhodococcus erythropolis N771 in its butyrate-bound (NHaseBA) and active (NHaseAq) forms. EPR spectra of the oxidized, inactive form of NHase (NHaseOX) further allow us to characterize the protonated and deprotonated forms of NHaseAq. Due to the relatively complex nature of the NHase ligand set with regard to possible ligand-to-metal charge transfer (LMCT) transitions, band assignment is assisted through a density functional theory (DFT) computational investigation of a series of LS CoIII complexes [(en)2Co(XCH2CH2NH2)–N,S]2+/3+ (Fig. 4) and their hypothetical LS FeIII counterparts, where X is a thiolate, sulfenate, sulfenic acid, or sulfinate group.24–26 These results provide insight into the relative energy ordering of the LMCT transitions for the different sulfur ligands. We use the subsequently assigned experimental data to calibrate DFT models of the butyrate bound and active forms of NHase and to extend these computational models to examine potential mechanisms for nitrile hydrolysis. These results provide insight into the electronic structure of the unusual active site of NHases and the mechanistic strategy developed by this class of enzymes.
 | ||
| Fig. 4 General structure of the [(en)2M(XCH2CH2NH2)–N,S]2+/3+ complexes. | ||
Results and discussion
EPR spectroscopy of NHaseBA, NHaseAq, and NHaseOx
EPR powder pattern spectra of LS FeIII systems generally exhibit 3 features around geff ≈ 2.0.27 The g values are sensitive to the ligand environment, and can allow the determination of the number of species in a given sample, as well as their relative abundance and a quantitative analysis of the ground state. The EPR spectra of NHaseBA at pH 7.5 and NHaseAq at pH 6.5, 7.5, and 8.5 are shown in Fig. 5. NHaseBA (Fig. 5, blue) is a clean single species with g values of 2.28, 2.14 and 1.97. NHaseAq at pH 7.5, on the other hand, is a mixture of 2 species (Fig. 5, green). At pH 8.5 the minor species component has increased in relative contribution (Fig. 5, purple). Simulations of the spectra (see ESI†) indicate that at pH 7.5 NHaseAq is a 74%/26% ± 5% mixture, with the major species having g values of 2.20, 2.13, and 1.99, and the minor species having g values of 2.22, 2.14 and 1.98. These are indicated in Fig. 5. On going from pH 7.5 to 8.5 the ratio changes to 52%/48% ± 5%. Buffer exchanging the sample back to pH 7.5 leads to the original EPR spectrum (data not shown). These results indicate that the NHaseAq active site possesses a deprotonatable ligand with a pKa ≈ 8.5. On going from pH 7.5 to 6.5 (Fig. 5, orange), the EPR spectrum of NHaseAq shows that the high-pH minor species (above) has disappeared, and has been replaced by a third species with g values of 2.28, 2.14 and 1.97. The pH 6.5 sample is comprised of approximately 28% of this low-pH minor form and 72% of the major form, indicating a second deprotonatable ligand with a pKa ≈ 6.1. This value is identical to the pKa determined for the sulfenate of the alkyl hydroperoxide reductase AhpC.28 Again, increasing the pH back to 7.5 restores the pH 7.5 spectrum, showing that the process is reversible (data now shown). | ||
| Fig. 5 77 K X band EPR spectra of NHaseBA (blue) and NHaseAq at pH 6.5 (orange), 7.5 (green) and 8.5 (purple). | ||
EPR spectra of NHaseOx at pH 6.5, 7.5 and 8.5 are shown in Fig. 6. Spectra taken at 7.5 (red) and 8.5 (purple) indicate there is an acid–base equilibrium present for NHaseOx similar to that of NHaseAq with a pKa > 7 (a shoulder to the low field side of g = 2.2 (arrow) increases in intensity on going from pH 7.5 to 8.5). However, on going from pH 7.5 to 6.5 (cyan), no third species is observed. This implies that the moiety in NHaseAq with a pKa of 6.1 is the Cys-SO(H) group, and that at the active form present under functional conditions possesses a deprotonated sulfenate ligand. As sulfinic acids generally have pKa values of approximately 2,29 this means that the moiety in NHaseAq with a pKa of 8.5 is the water-derived ligand, and that under functional conditions NHaseAq also possess a coordinated water ligand (X in Fig. 1A).
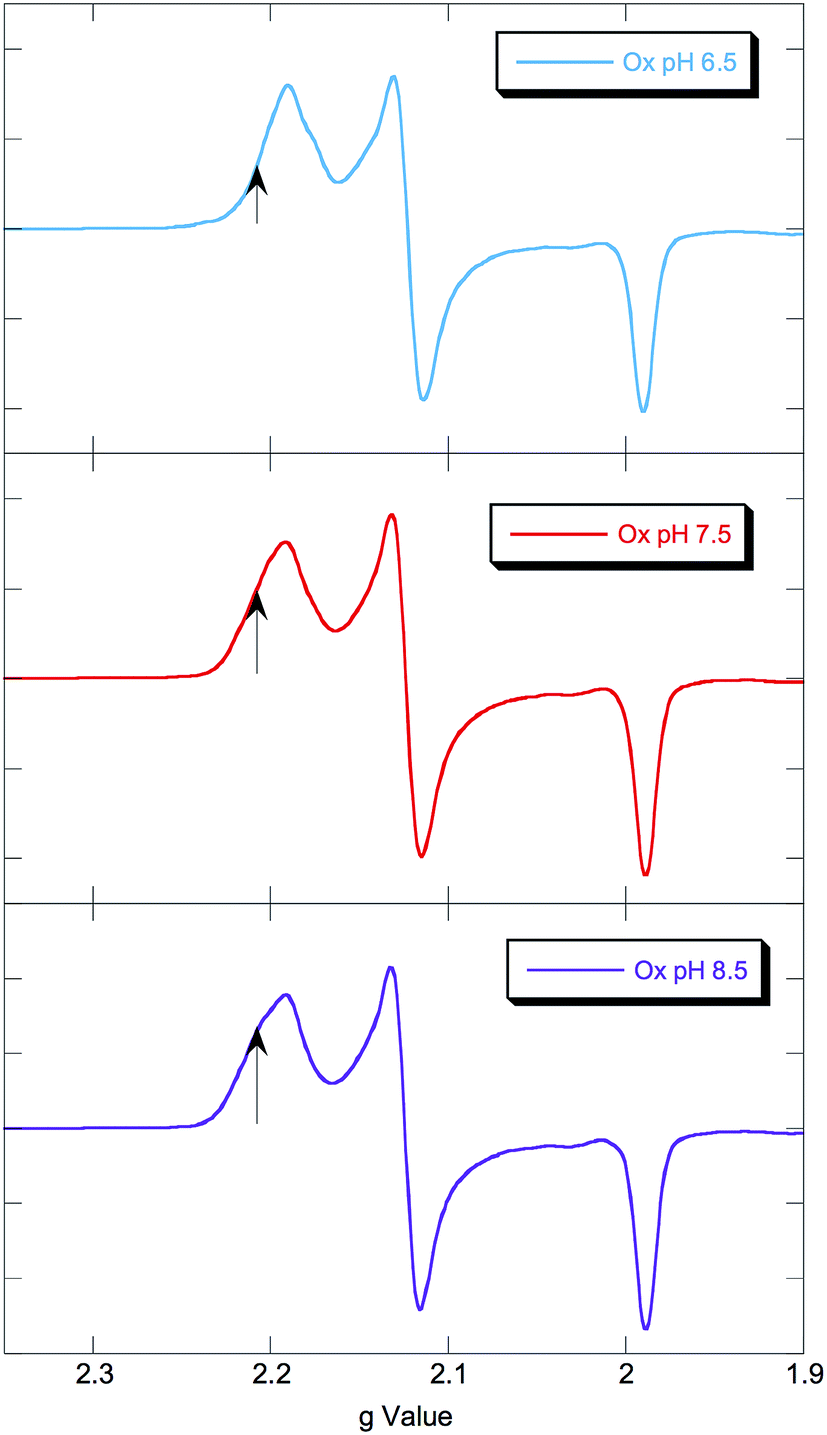 | ||
| Fig. 6 77 K X band EPR spectra of NHaseOx at pH 6.5 (cyan), 7.5 (red), and 8.5 (purple). Signals from minor species at low field marked with arrows. | ||
NIR MCD spectra of NHaseBA and NHaseAq: low energy d–d transitions
Octahedral LS FeIII species possess a (t2g)5 ground configuration giving a threefold orbitally degenerate 2T2g ground state, which in the rhombic ligand environment of a protein site splits in energy and leads to two t2g → t2g (dπ → dπ) transitions at ≤5000 cm−1. The orbitally degenerate doublet excited states also split to produce a manifold of t2g → eg (dπ → dσ) transitions from ≈15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 30000 cm−1, although the higher-energy ligand field (LF) transitions are frequently obscured by the intense LMCT transitions and may be difficult to detect.27 The 1.5 K, 7 T NIR MCD spectra of NHaseBA and NHaseAq at pD 7.5 are shown in Fig. 7A and B, respectively. The spectrum of NHaseBA has a band at ≈5600 cm−1 corresponding to the highest energy dπ–dπ transition, as well as a band at ≈10600 cm−1, which corresponds to the lowest energy dπ–dσ transition.30 The shoulder of the ≈13600 cm−1 Cys-Sπ → dπ LMCT is also visible as a tail in the higher energy region of this spectrum (vide infra). The spectrum of NHaseAq shows two bands at low energy, one with a
000 to 30000 cm−1, although the higher-energy ligand field (LF) transitions are frequently obscured by the intense LMCT transitions and may be difficult to detect.27 The 1.5 K, 7 T NIR MCD spectra of NHaseBA and NHaseAq at pD 7.5 are shown in Fig. 7A and B, respectively. The spectrum of NHaseBA has a band at ≈5600 cm−1 corresponding to the highest energy dπ–dπ transition, as well as a band at ≈10600 cm−1, which corresponds to the lowest energy dπ–dσ transition.30 The shoulder of the ≈13600 cm−1 Cys-Sπ → dπ LMCT is also visible as a tail in the higher energy region of this spectrum (vide infra). The spectrum of NHaseAq shows two bands at low energy, one with a ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) max < 5000 cm−1 (Gaussian fit at ≈4700 cm−1) and another with max ≈ 6300 cm−1. Thus the dπ–dπ transitions of NHaseAq are raised in energy relative to those of NHaseBA. The lowest energy dπ → dσ transition of NHaseAq is also higher than that of NHaseBA at ≈11000 cm−1.
max < 5000 cm−1 (Gaussian fit at ≈4700 cm−1) and another with max ≈ 6300 cm−1. Thus the dπ–dπ transitions of NHaseAq are raised in energy relative to those of NHaseBA. The lowest energy dπ → dσ transition of NHaseAq is also higher than that of NHaseBA at ≈11000 cm−1.
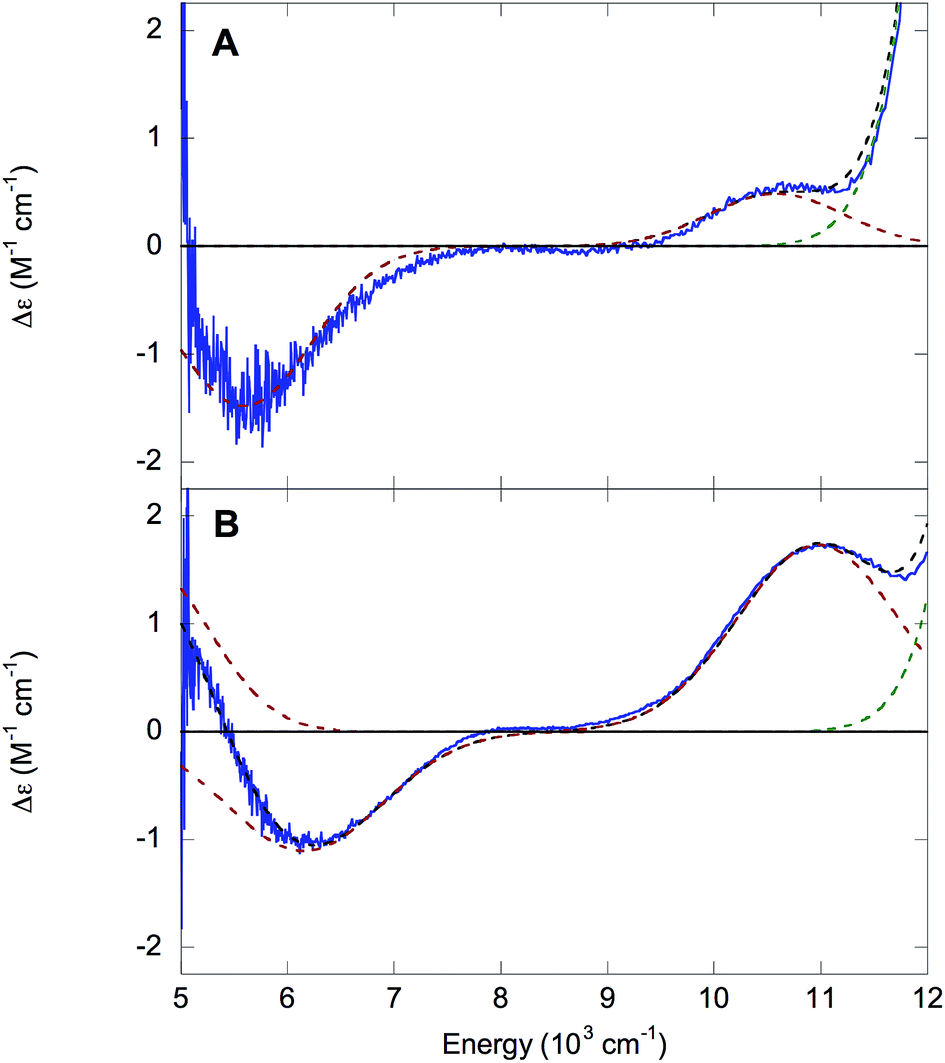 | ||
| Fig. 7 NIR MCD of NHase. 1.5 K, 7 T NIR MCD spectra of NHaseBA (A) and NHaseAq (B) at pD 7.5. Experimental data are in blue, the fit is in black dashes, red dashed curves denote d–d transitions and the green dashed curve denotes the thiolate-to-dπ LMCT. | ||
Assignment of t2g orbital splittings
As the ground states of six-coordinate LS FeIII active sites are derived from the 2T2g states of Oh, in addition to the low symmetry splitting of the dπ orbitals considered above they have in-state orbital angular momentum and thus undergo in-state spin–orbit coupling (SOC). This in-state coupling has a significant effect on the experimental EPR g values of a complex. A system of equations developed by Taylor includes this SOC and allows for the determination of the energy splitting of the t2g orbitals as well as the order and sign of the experimental g values.31 The t2g orbital energies of a given site are defined by the tetragonal splitting, Δ, of the dxy orbital from the {dxz, dyz} pair, and the rhombic splitting, V, of the dxz and dyz orbitals. The quantity |V/2Δ| is a measure of the rhombicity of the LS FeIII site, with a value of zero indicating a purely axial system and a value of 1/3 being the rhombic limit in which the t2g orbital splittings are equal. The assigned g values, energy splitting parameters, and predicted transitions for NHaseBA and NHaseAq are given in Table 1. The values of a, b, and c are the coefficients of the dyz, dxz, and dxy orbitals, respectively, in the half-occupied dπ ground state. The predicted dπ–dπ transition energies of NHaseBA (≈2800 cm−1 and 5300 cm−1) and NHaseAq (≈3900 cm−1 and 6200 cm−1) compare well with the experimental values (≈5600 cm−1 for NHaseBA, the lowest energy dπ → dπ transition being below the detection limit of the instrumentation, and ≈4700 cm−1 and 6200 cm−1 for NHaseAq), indicating that the Taylor method provides an accurate description of the t2g dπ orbital energies. The negative values of Δ for NHaseBA and NHaseAq indicate that dxy is the half-occupied orbital for both of these forms of the enzyme. This is reflected in the coefficients where c is dominant. The |V/2Δ| value for NHaseBA indicates that this form has a strongly rhombic FeIII site, whereas the value of NHaseAq indicates that its site is closer to axial.| Form | g x | g y | g z | a | b | c | Δ | |V| | |V/2Δ| | Trans. 1 | Trans. 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BA | −2.14 | 2.29 | −1.97 | 0.07 | 0.04 | 1.00 | −4030 | 2560 | 0.32 | 2750 | 5300 |
| Aq | −2.13 | 2.21 | −1.99 | 0.05 | 0.03 | 1.00 | −5010 | 2320 | 0.23 | 3850 | 6170 |
LS CoIII and FeIII models: bonding interactions and their associated LMCT transitions
The unusual ligation of NHase allows a number of different possible CT transitions, whose nature and energy order need to be clarified. One may initially anticipate thiolate π → dπ*, amidate π → dπ*, and thiolate/sulfenate/sulfenic/sulfinate σ → dσ* CT transitions as potentially occurring within the accessible spectral range for UV-Vis absorption (≈11000–33000 cm−1). It has been previously shown through resonance Raman spectroscopy32 that the thiolate π → dπ* CT transition for butyrate-free Fe NHases occurs at ≈14700 cm−1 (680 nm) and shifts to ≈14100 cm−1 (710 nm) upon butyrate binding.16 The LS FeIII cyanide-bound form of superoxide reductase (SOR) also exhibits a thiolate π → dπ at ∼15200 cm−1.33 Resonance Raman spectroscopy identified amidate π → dπ CT transitions in ferric bleomycin (FeBLM) and activated bleomycin (ABLM) at ≈26400 cm−1 and ≈27300 cm−1, respectively.30 NHase has two amidates, whose proximity leads to in-phase (+) and out-of-phase (−) combinations of ligand donor orbitals π+ and π− as shown in Fig. 8. Therefore, it is reasonable for two amidate π–dπ* CT transitions to contribute to the spectrum of NHase in the ≈26000 cm−1 energy region.
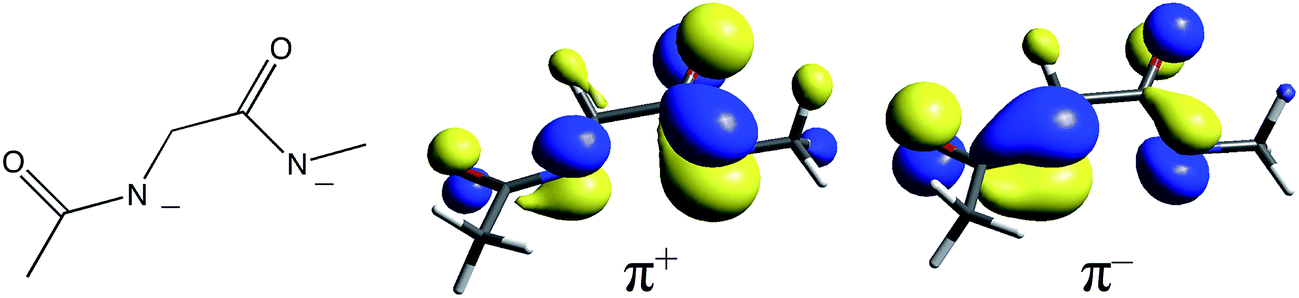 | ||
| Fig. 8 The diamidate moiety in NHase (left) and its in-phase (center) and out-of-phase (right) π MO combinations. | ||
There has been an interesting study of the UV-Vis absorption spectra of a series of LS CoIII complexes [(en)2Co(XCH2CH2NH2)–N,S]2+/3+, where X is S−, S-coordinated SO−, S-coordinated SOH, and S-coordinated SO2−.24–26 This series provides a complete set of spectroscopic data for the thiolate-based ligands of interest for which the computational extension to the hypothetical LS FeIII forms is straightforward. The observed LMCT's are given in Table 2. For LS CoIII (d6) these can only be Lσ → dσ* CT transitions as there is no electron hole in the dπ manifold. The thiolate and sulfinate complexes exhibit LMCT transitions at ≈35000 cm−1, with the latter lower in energy by 800 cm−1. The sulfenate complex, however, exhibits a lower energy LMCT transition at 27400 cm−1, in addition to a transition at 35200 cm−1. Addition of protons diminishes the intensity of the lower energy transition, while addition of BF3 eliminates it entirely.26,34
| Ligand | Expt'l trans. (ε) | CoIII TD-DFT | FeIII TD-DFT |
|---|---|---|---|
| a From ref. 26. b Due to the instability of the sulfenato complex the uncertainty on the reported ε values is high. c No ε value given. d From ref. 25. | |||
| Thiolatea | 35500 (13800) |
39300 (RSσ–dσ) |
19600 (RSπ–dπ), |
| 40600 (RSσ–dσ) |
|||
| Sulfenatea,b | 27400 (6700) |
29800 (RSOσIP–dσ) |
32700 (RSOσIP–dσ) |
| 35200 (3700) |
34900 (RSOσOOP–dσ) |
38000 (RSOσOOP–dσ) |
|
| Prot. sulfenatea | ≈35200c |
39200 (RSOHσIP–dσ) |
42300 (RSOHσIP–dσ) |
| 42800 (RSOHσOOP–dσ) |
49500 (RSOHσOOP–dσ) |
||
| Sulfinated | 34700 (14200) |
36600 (RSO2σ–dσ) |
37800 (RSO2σ–dσ) |
TD-DFT calculations were performed on the LS CoIII complexes and their LS FeIII counterparts, with the predicted transition energies listed in Table 2. Contours of the ligand donor molecular orbitals involved in these transitions are shown in Fig. 9. The thiolate ligand has two S 3p orbitals for bonding to metal (the third 3p orbital is involved in bonding to C), one aligned along the metal–S bond axis (RS− σ) positioned for σ bonding and one orthogonal to the bond axis (RS− π) oriented for π bonding. The RSO− and RSOH ligands are S-bonded to the metal and have occupied S–O π* orbitals aligned in the Fe–S–O plane (RSO(H) σIP) and out of the Fe–S–O plane (RSO(H) σOOP) that are oriented for σ overlap with the metal. The sulfinate ligand has an occupied S–(O2−) π* orbital which is oriented to interact with the metal in a σ fashion (RSO2− σ). The calculated CoIII transition energies agree qualitatively with the experimental results. The thiolate σ → dσ* transition predicted by TD-DFT is higher in energy than the corresponding sulfinate by 2700 cm−1, vs. 800 cm−1 in the experimental data. The TD-DFT sulfenate σIP → dσ* transition is lower in energy than the thiolate σ and sulfinate transitions at 29800 cm−1, comparable to the experimental value of 27400 cm−1.
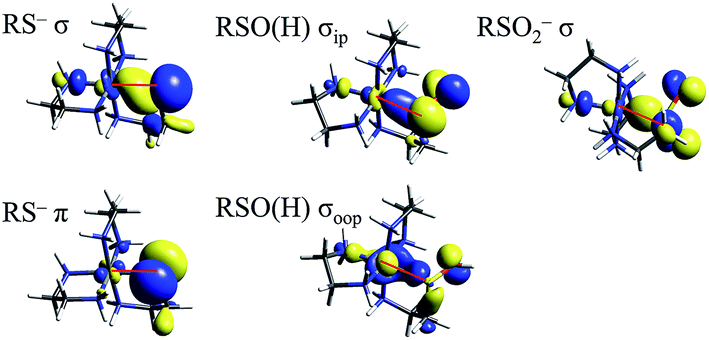 | ||
| Fig. 9 Molecular orbitals of thiolate, sulfenate, and sulfinate ligands involved in model complex LMCT transitions. Red lines mark the Fe–S bond in each picture. | ||
A sulfenate σOOP → dσ* transition is predicted at 34900 cm−1, but this transition will not be observed in an NHase active site with a deprotonated sulfenate group. The protonated sulfenate complex possesses a calculated RSOH σIP → dσ* at 39200 cm−1, comparable to the analogous transition in the thiolate complex. A calculated RSOH σOOP → dσ* transition occurs at a significantly higher energy of 42800 cm−1, which is beyond the protein cutoff for spectroscopy on an enzyme (≈33000 cm−1).
In order to extend the above findings to the LS FeIII site of NHase, FeIII was computationally substituted for CoIII in the starting structures of the 4 model complexes above and re-optimized (with an S = 1/2 ground state). These hypothetical FeIII complexes have TD-DFT transitions that are comparable to their CoIII counterparts, with the FeIII transitions at higher energy due to the lower Zeff of FeIII leading to a higher energy d orbital manifold relative to CoIII. The FeIII thiolate complex also has a dπ* hole due to the LS d5 configuration leading to a RS− π → dπ* transition at 19600 cm−1 from TD-DFT, or about 5000 cm−1 higher in energy than the corresponding transitions of NHaseBA, NHaseAq and cyanide-bound SOR. Scaling the other FeIII TD-DFT calculated transition energies by a similar amount gives predicted deprotonated sulfenate σIP → dσ* transition at ≈28000 cm−1 and sulfenate/thiolate/sulfinate σ transitions predicted at 33000–36000 cm−1 for NHaseBA and NHaseAq. The relative energy order for the LMCT transitions in NHase is therefore predicted to be Cys-S− π → dπ* (experimentally assigned by resonance Raman at ≈15000 cm−1) < amidate π → dπ* ≈ Cys-SO− σIP → dσ* < Cys-SO2− σIP → dσ* < Cys-S− σIP → dσ* < Cys-SOH σIP → dσ*. It should be noted that the Cys-SO− σIP MO is the highest in energy among the occupied S-based ligands, and has considerable O character (48% O vs. 32% S in the FeIII model), indicating that it has the potential to be a good nucleophile (vide infra).
UV-Vis low-temperature absorption and MCD spectroscopy of NHaseBA and NHaseAq
The 5 K absorption spectrum and 5 K, 7 T MCD spectrum of NHaseBA are given in Fig. 10A and B, respectively. A list of band energies, ε and Δε values, and C/D ratios is given in Table 3. Temperature dependence indicates the MCD transitions are all C terms of a paramagnetic complex. The C/D ratio of a transition is proportional to its Δε/ε value, and is generally higher for d–d than CT transitions.35 The Cys-S− π → dπ* LMCT transition occurs at 13600 cm−1 (band a) from resonance Raman spectroscopy.32 The transitions at 15700, 17500, 19500 and 21800 cm−1 (bands b, c, d, and e, respectively) are determined by their relative lower ε values (and higher Δε in MCD) as dπ → dσ* ligand field transitions. As NHaseBA has a protonated sulfenate group, the intense in absorption, and therefore CT in nature, transitions at 24700 and 27000 cm−1 (bands f and g) are ascribed to amidate π → dπ* transitions, while the 30000 cm−1 (band h) transition is assigned to the Cys-SO2− σ → dσ* CT transition.
 | ||
| Fig. 10 UV-Vis spectra of NHaseBA. (A) Absorption spectrum of NHaseBA at 5 K. (B) 5 K 7 T MCD spectrum of NHase BA. Experimental data are in black, the total fit is in dashed gray, and dashed Gaussian curves denote Cys-S− π → dπ (a, green), dπ → dσ (b–e, black), amidate π → dπ (f–g, red) and Cys-SO2− σ → dσ (h, blue) transitions. | ||
| Band | Energy (cm−1) | ε (M−1 cm−1) | Δε (M−1 cm−1) | C/D × 104 |
|---|---|---|---|---|
| a | 13600 |
1250 | 36 | 306 |
| b | 15700 |
500 | 10 | 213 |
| c | 17500 |
350 | 2 | 61 |
| d | 19500 |
760 | 38 | 532 |
| e | 21800 |
1060 | 14 | 140 |
| f | 24700 |
2050 | −59 | 306 |
| g | 27000 |
2200 | −3 | 15 |
| h | 30000 |
3350 | −14 | 44 |
The LS nature of the FeIII site implies strong ligand bonds and relatively high covalency, which can lead to significant ligand character in the d-based molecular orbitals and increased absorption intensity for the d–d transitions through LF excited state mixing with CT transitions. The CT transitions of NHaseBA generally have low C/D ratios, except for the Cys-S− π → dπ* (band a in Fig. 10) and amidate π+ → dπ* (band f in Fig. 10) transitions which have significant MCD intensities of opposite sign. This can be explained by the two transitions interacting through SOC to form a pseudo-A term in MCD. The pseudo-A term requires 2 perpendicularly polarized transitions to SOC in a third mutually orthogonal direction.36 From the DFT results given in the ESI,† the Cys-S− π → dπ* CT transition is y-polarized with the Cys-S− π donor MO having dxz character, while the amidate π+ → dπ* transition is x-polarized with the amidate π+ donor MO having dyz character. The dxz and dyz characters of these two donor MOs couple by the Lz component of the angular momentum operator and therefore spin–orbit couple to produce the pseudo-A-term.
The absorption and MCD spectra of NHaseAq are given in Fig. 11. A list of band energies, ε and Δε values, and C/D ratios is given in Table 4. The spectra in Fig. 11 are similar to those in Fig. 10, but with more absorption intensity in the 25000–30000 cm−1 region associated with the butyrate being replaced by H2O and Cys-SOH deprotonated to Cys-SO−. NHaseAq possesses a Cys-S− π → dπ* LMCT transition at 14300 cm−1 (band a) which is shifted up from that of NHaseBA by 700 cm−1 and is similar to the results previously found in the room temperature absorption spectra for other NHases.14 The dπ → dσ* LF transitions of NHaseAq (bands b–e) are also shifted up in energy relative to that of NHaseBA, consistent with the LF effects on the dπ splitting. The amidate π → dπ* CT transitions (band f) are also shifted higher in energy relative to NHaseBA at ≈27500 cm−1. Instead of two features, however, one broad feature is observed with higher intensity than the two amidate π → dπ* CT transitions of NHaseBA (≈2100 M−1 cm−1 for each NHaseBA transition vs. 3200 M−1 cm−1 for the NHaseAq transition). It is possible that the Cys-SO− σ → dσ* transition (predicted to be in this energy region from the model complex DFT calculations, vide supra) overlaps with the two amidate π → dπ* transitions to form this broad, intense feature. The transition in the NHaseAq spectra at ≈30600 cm−1 (band g) is similar in energy and intensity to band h in NHaseBA and is reasonably assigned to the Cys-SO2− σ → dσ* CT transition.
 | ||
| Fig. 11 UV-Vis spectra of NHaseAq. (A) Absorption spectrum of NHaseAq at 5 K. (B) 5 K 7 T MCD spectrum of NHaseAq. Experimental data are in black, the total fit is in dashed gray, and dashed Gaussian curves denote Cys-S− π → dπ (a, green), dπ → dσ (b–e, black), amidate π → dπ/Cys-SO− σ → dσ (f, orange) and Cys-SO2− σ → dσ (g, blue) transitions. | ||
| Band | Energy (cm−1) | ε (M−1 cm−1) | Δε (M−1 cm−1) | C/D × 104 |
|---|---|---|---|---|
| a | 14300 |
1200 | 54 | 479 |
| b | 16900 |
310 | −4 | 137 |
| c | 20100 |
450 | 16 | 378 |
| d | 21700 |
780 | 16 | 218 |
| e | 23900 |
1000 | −9 | 96 |
| f | 27500 |
3180 | −32 | 107 |
| b | 30600 |
2890 | −13 | 48 |
DFT and TD-DFT calculations: EPR parameters, dπ orbital splittings and ground state wave functions
The DFT optimized structures of NHaseBA and NHaseAq are shown in Fig. 12 (note that both are based on crystal structures). Optimization of an NHaseBA structure with a sulfenate and protonated butyric acid ligand leads to the proton moving to the sulfenate group. NHaseAq contains an exogenous water that is H bonding with both the sulfenate oxygen and the coordinated water ligand. The B3LYP, BP86, and BP86 with 10% Hartree–Fock (HF) functionals were tested by optimizing the NHaseBA and NHaseAq structures in the S = 1/2, S = 3/2, and S = 5/2 states to determine if each functional correctly predicted the ground state spin (S = 1/2). The values of ΔH(1/2 → 3/2) and ΔH(1/2 → 5/2) for the different functionals are given in Table 5. The B3LYP functional predicts the S = 1/2 and S = 3/2 states are virtually isoenthalpic for both NHaseBA and NHaseAq, rendering this functional unsuitable for modeling the NHase active site. While the BP86 with 10% HF functional predicts an S = 1/2 ground state for both NHaseBA and NHaseAq, it predicts Cys-S− π → dπ CT transitions of 16400 cm−1 for both NHaseBA and NHaseAq, whereas the pure BP86 functional predicts the NHaseAq transition to be 500 cm−1 higher in energy than the NHaseBA transition (vide infra), which is very close to the 700 cm−1 difference observed experimentally. The pure GGA BP86 functional was therefore used for all subsequent calculations.
 | ||
| Fig. 12 Geometry-optimized structures of NHaseBA (left) and NHaseAq (right). | ||
| Form | Functional | ΔH(1/2 → 3/2) | ΔH(1/2 → 5/2) |
|---|---|---|---|
| NHaseBA | BP86 | 8.9 | 24.7 |
| BP86 + 10% HF | 5.4 | 16.1 | |
| B3LYP | 0.9 | 4.0 | |
| NHaseAq | BP86 | 11.4 | 28.5 |
| BP86 + 10% HF | 8.7 | 20.3 | |
| B3LYP | 1.2 | 6.7 |
The ORCA program was used to calculate the EPR g values for the NHaseBA and NHaseAq models as listed in Table 6. Significant deviations from the largest experimental g value for each form of NHase are calculated consistent with past results that DFT tends to underestimate the largest g value.37 The calculated g tensor directions for both the NHaseBA and NHaseAq models indicate that the x direction is approximately along the amidate N–Fe–sulfenate S bond axis, the y direction is pointing along the thiolate S–Fe–butyrate/water O bond axis and the z direction is effectively pointing along the amidate N–Fe–sulfinate S bond axis (see ESI†).
| Form | |gx| | |gy| | |gz| | |
|---|---|---|---|---|
| NHaseBA | Exp. | 2.14 | 2.29 | 1.97 |
| ORCA | 2.07 | 2.11 | 2.01 | |
| NHaseAq | Exp. | 2.13 | 2.21 | 1.99 |
| ORCA | 2.07 | 2.10 | 2.01 | |
The minority spin β molecular orbital energy diagrams for NHaseBA and NHaseAq are given in Fig. 13 left and right, respectively. Contour plots for all MOs in Fig. 13 are supplied in the ESI.† For both forms of the enzyme the calculated half-occupied t2g orbital is dxy (perpendicular to the amidate N–sulfenate S axis) 206β for NHaseBA and 200β for NHaseAq in Fig. 14 (left and right, respectively). For both MOs dxy interacts most strongly with the Cys-S− π orbital, but also interacts with the amidate π orbitals trans to the sulfenate group. The thiolate ligand of NHase is therefore the strongest π donor ligand in this enzyme. The implications of this strong donor bonding interaction will be considered further in the Discussion. From Fig. 13, the unoccupied σ* orbitals are dx2−y2 and dz2, with the dz2 orbital highest in energy. These two dσ orbitals are split by ≈6000 cm−1 in NHaseBA and ≈6300 cm−1 in NHaseAq. The sulfinate and its trans amidate both overlap with dz2 and are therefore the strongest σ donors in the active site.
 | ||
| Fig. 13 β MO energy diagram for NHaseBA (left) and NHaseAq (right). Metal d MOs are in black, thiolate MOs in green, sulfinate MOs in blue, sulfenate MOs in orange, and amidate MOs in red. | ||
 | ||
| Fig. 14 NHase β LUMOs. Isosurface contour plots of the DFT-calculated, dxy β MOs of NHaseBA (center) and NHaseAq (right), along with the coordinate system for both models (left). | ||
DFT and TD-DFT calculations: dπ → dσ* LF and CT transitions
From Fig. 13, closest in energy to the Fe d orbital manifold for both active site models is the Cys-S− π orbital, as would be expected from its π bonding nature and the relatively low electronegativity of the S valence 3p orbitals. For the NHaseBA model, the next highest-energy MO is the diamidate in-phase, π+, combination, with the out-of-phase, π−, combination at lower energy. The Cys-SO2− σ orbital (Fig. 9) is sandwiched between the π+ and π− MOs. For NHaseBA, below π− lies the Cys-SOH σ and the Cys-S− σ MOs are lowest in energy.For the NHaseAq model, the next MO below the thiolate π is the Cys-SO− σ donor orbital. The removal of the proton from the coordinated sulfenic acid significantly raises the energy of this orbital, reflected in the low energy Cys-SO− σ → dσ* CT transition of the CoIII sulfenate complex that was eliminated upon protonation of the sulfenate group (vide supra). The high energy of this Cys-SO− MO has implications for this ligand's capacity as a nucleophile (vide infra). Below the Cys-SO− σ lie the amidate π+ and π− MOs followed by the Cys-SO2− σ and Cys-S− σ MOs. Thus the calculated energy ordering of the different S ligands reasonably parallels the order determined experimentally for the model complexes.
The experimentally-determined and TD-DFT-predicted transitions for NHaseBA and NHaseAq are given in Fig. 15 left and right, respectively. The predicted dπ → dπ* transitions for NHaseBA are at 3600 and 6900 cm−1, similar in energy to the experimentally determined (from Taylor analysis and NIR MCD data) values of 2800 and 5300 cm−1. The same is true for NHaseAq, with calculated values of 4300 and 7200 cm−1 compared to experimental values of 3900 and 6200 cm−1. The calculations correctly predict the increase in the dπ → dπ LF transition energies upon replacing the butyrate ligand with water, and the values of |V/2Δ| determined from these TD-DFT transitions (0.31 for NHaseBA and 0.25 for NHaseAq) agree well with the experimental values (0.32 and 0.23, respectively). The calculated dπ → dσ* LF energies for NHaseBA range from 10200 to 18900 cm−1, which compares reasonably well to the experimental range of 10600 to 21800 cm−1. The corresponding dπ → dσ* transition energies for the NHaseAq model range from 10900 to 19400 cm−1, similar to the experimental transitions of 11000 to 23900 cm−1. The calculated dπ → dσ* transitions shift up in energy upon replacing butyrate with water, as is observed experimentally, which indicates an increase in 10Dq, reflecting replacement of the π donor butyrate in NHaseBA by water.
 | ||
| Fig. 15 Experimental (top) and TD-DFT (bottom) transitions for NHaseBA (left) and NHaseAq (right). Lines are colored purple for dπ → dπ, black for dπ → dσ, green for Cys-S− π → dπ, red for amidate π → dπ, orange for Cys-SOH/-SO− σ → dσ and blue for Cys-SO2− σ → dσ. The transition at ≈27000 cm−1 in the upper-right plot is marked half red, half orange to denote the overlap of the amidate dπ → dπ and Cys-SO− σ → dσ transitions. | ||
The calculated Cys-S− π → dπ CT transitions are at 11800 cm−1 for NHaseBA and 12300 cm−1 for NHaseAq. These values are in the range of those determined experimentally (13600 cm−1 for NHaseBA and 14300 cm−1 for NHaseAq), and the increase in transition energy upon substituting a water ligand for butyrate is correctly predicted. The calculated amidate π → dπ* transition energies for NHaseBA (16200 and 19700 cm−1) and NHaseAq (18800 and 20600 cm−1) are significantly lower than their experimental counterparts (≈26000 cm−1 for NHaseBA and ≈27000 cm−1 NHaseAq), perhaps reflecting a large self-interaction error for the anionic ligands.38 The models do, however, correctly predict that the amidate-based transitions of NHaseAq are shifted up in energy relative to those of NHaseBA. For NHaseBA the calculated Cys-SOH σ → dσ and Cys-SO2− σ → dσ CT transitions are 31900 and 33600 cm−1, respectively. From the model studies it was predicted that the Cys-SOH-based transition would lie to higher energy, but in this system the Cys-SO2− transition is to the higher energy dz2 orbital while the Cys-SOH transition is to the lower energy dx2−y2 orbital. In the TD-DFT calculation of NHaseAq, the Cys-SO−-based transition at 25200 cm−1 is 6000 cm−1 lower in energy relative to that of Cys-SOH in NHaseBA. This is consistent with the experimental Cys-SOH σ → dσ* CT transition of NHaseBA being unobserved and the broad, intense transition of NHaseAq at 27500 cm−1 arising from the overlap of the amidate π → dπ* and Cys-SO− σ → dσ* CT transitions. The lower energy CT transition for sulfenate vs. sulfenic acid indicates an increase in the sulfenate frontier MO energy with deprotonation and a consequent increase in its nucleophilic character. While TD-DFT calculations of the CT excited states are shifted relative to experiment, their ordering and energy shifts between enzyme forms are consistent with experiment. Also, the functional used predicts the correct LS ground state and from the Taylor analysis above the ground state parameters are well described. The functional (BP86) and basis set employed above were thus used to evaluate possible mechanisms of nitrile hydration by this unusual LS FeIII active site.
Reaction coordinate calculations
Nucleophilic attack on coordinated acetonitrile by water with the sulfenate group acting as a proton acceptor was explored as described previously.20 This is shown in blue in the energy diagram in Fig. 16, with corresponding structures in Fig. 17, top. At the transition state, the proton is approximately equidistant between the water and sulfenate oxygens. The electronic energy of the transition state lies 16.7 kcal mol−1 above the ground state, similar to the value of 20.2 kcal mol−1 obtained previously.20 Complete transfer of the proton to the sulfenate group leads to a coordinated amidate tautomer structure 11.3 kcal mol−1 above the starting structure, labeled as ‘SOH’ in Fig. 16. Rotation of the amidate tautomer around the Fe–N axis leads to the abstraction of the proton of sulfenic acid by amidate tautomer N, generating the coordinated amide tautomer at −14.4 kcal mol−1 relative to the starting coordinated nitrile structure. Tautomerization/dissociation of the product amide and regeneration of the active form of NHase leads to a net energy change of −10.4 kcal mol−1. For the reaction of two water molecules with free acetonitrile, one water acting as nucleophile and the other as proton acceptor,20 the electronic energy of the transition state is 27.4 kcal mol−1, or approximately 11 kcal mol−1 higher than for water attack on NHase LS FeIII-coordinated nitrile. Thus the combination of activation of nitrile by coordination to metal and the better nucleophilic character of sulfenate relative to water lowers the energy barrier by ≈11 kcal mol−1.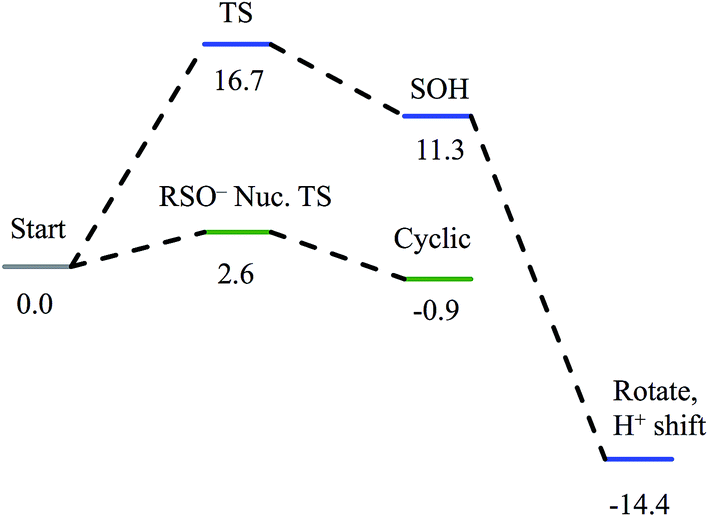 | ||
| Fig. 16 Reaction coordinates for attack on coordinated nitrile by water (blue) and attack on coordinated nitrile by sulfenate (green). All energies are in kcal mol−1. | ||
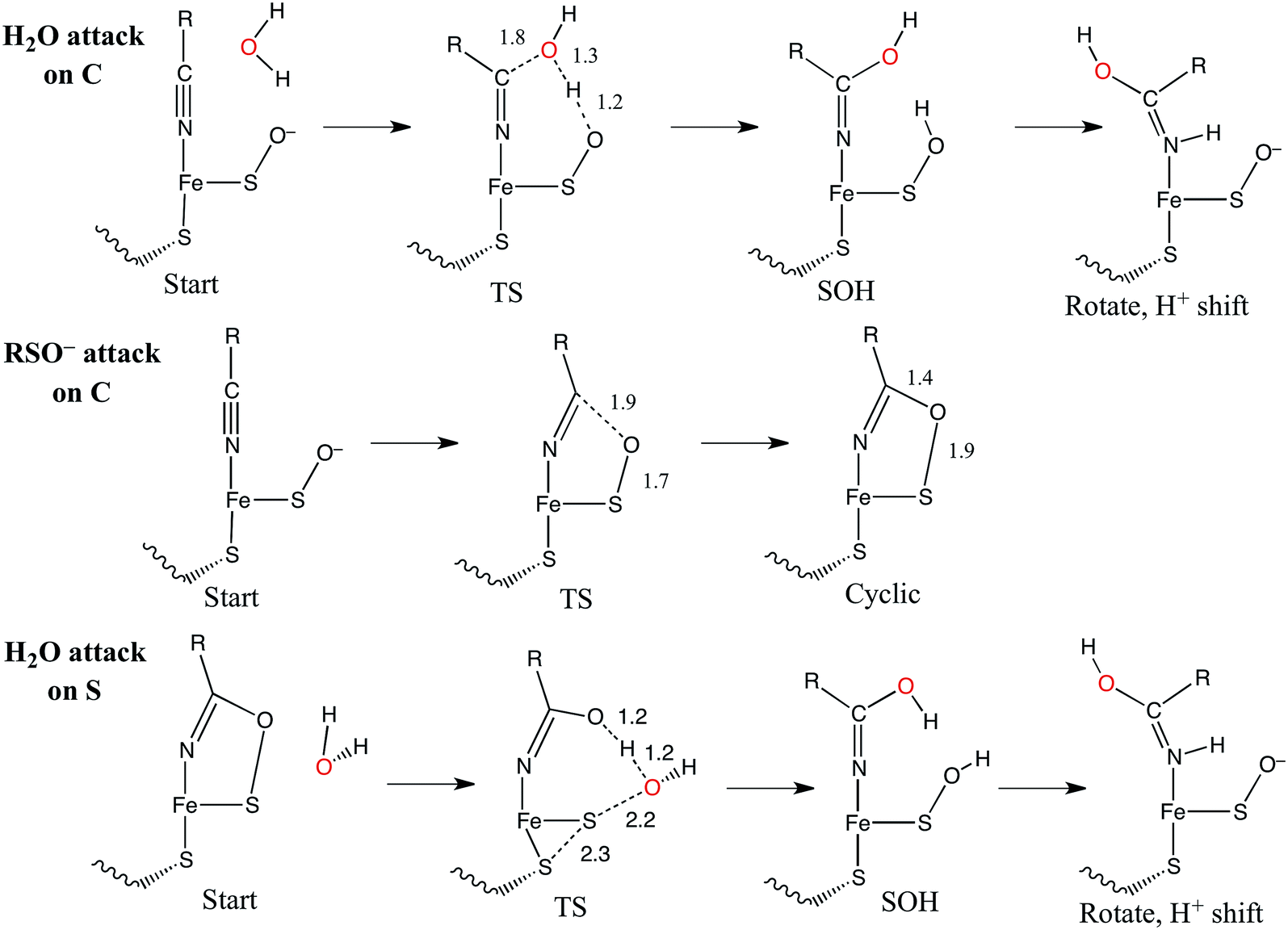
Alternatively, the sulfenate group can act as the nucleophile towards the coordinated nitrile to form a cyclic intermediate.21 The reaction coordinate and associated structures for this process are also shown in Fig. 16 and 17, center, respectively. The process of direct attack by sulfenate on nitrile (shown in green in Fig. 16) has a barrier of only 2.6 kcal mol−1, significantly lower than the barrier for nucleophilic attack on nitrile by water (16.7 kcal mol−1, blue in Fig. 16). The cyclic species formed by attack of the sulfenate is effectively isoenergetic with the starting structure.
Subsequent attack by water on the cyclic species can occur at the (formerly) nitrile carbon or at the (formerly) sulfenate sulfur. Attempts to produce a transition state for attack at the nitrile carbon led only to cleavage of the sulfenate–O bond, with no transfer of a proton from attacking water to the O now bound to C. Also, the energy was approximately 28 kcal mol−1 higher than the starting structure for a water O–nitrile C distance of 1.8 Å. Nucleophilic attack by water on the C of the cyclic species was therefore deemed unfeasible. However, nucleophilic attack by water on the S of the cyclic structure did lead to cleavage of the sulfenate S–O bond and generation of a coordinated amidate tautomer (SOH) similar to the nucleophilic attack by water on (uncyclized) coordinated nitrile. The reaction coordinate for this attack at S is given in Fig. 18, and structures of the transition state and end point before and after amidate tautomer rotation are given in Fig. 17, bottom. Sulfenate S–O bond-breaking occurs relatively early in the reaction coordinate: from a linear transit study, at a water O–cyclic intermediate S distance of 2.6 Å the cyclic intermediate O–cyclic intermediate S distance has increased to 2.5 Å, and at the transition state this latter distance has increased to 3.6 Å, as the attacking water is forming a bond to the cyclic intermediate S and transferring a proton to the cyclic intermediate O. Points along the IRC (compared with the SOH product and the structure for a water O–cyclic intermediate S distance of 2.4 Å from the linear transit, Fig. 19) indicate that the transition state proceeds towards the reactants and products. The barrier for attack at the S of the cyclized species is 14.4 kcal mol−1, or 2.3 kcal mol−1 lower than that of attack at coordinated, uncyclized C in Fig. 16. At the transition state (one imaginary frequency, confirmed to be on the IRC), the proton transfer is similar to that of an attack on uncyclized nitrile. Interestingly, in the transition state for attack on the S of the cyclized species, the sulfenate S–axial thiolate S distance has shortened to 2.3 Å, indicating some bonding between these atoms (a Mayer bond order of 0.53 between the sulfenate S–axial thiolate S was calculated, whereas the S–S bond in dimethyl disulfide has a calculated Mayer bond order of 1.61). This is similar to a recent computational study proposing a mechanism involving nucleophilic attack by the axial thiolate upon the sulfur of the cyclic intermediate, resulting in the formation of a disulfide bond.22 In the computed mechanism at the bottom of Fig. 17 and 18 the S–S interaction aids in stabilizing the transition state and lowering the barrier for this reaction step involving H2O attack on the cyclic S. After amidate tautomer rotation and proton shifting the S–S distance increases to 3.1 Å and the bonding interaction is eliminated. The nature of the activation of the coordinated nitrile and the cyclic structure are explored below.
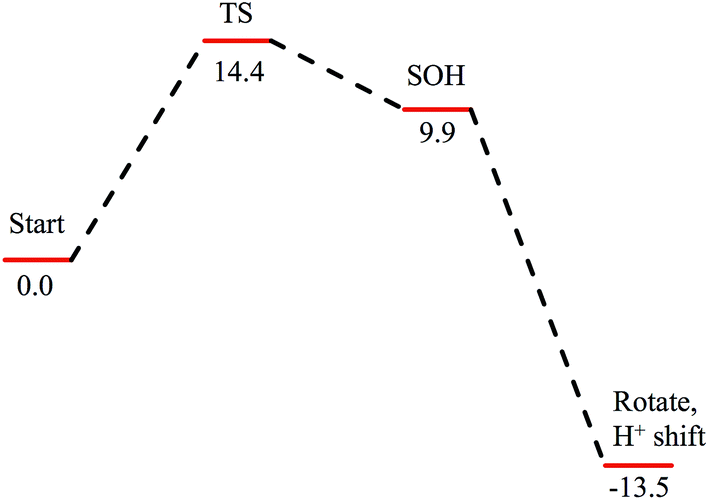 | ||
| Fig. 18 Reaction coordinate for nucleophilic attack by water on the S atom of the cyclic structure. Species defined at bottom of Fig. 17, with zero energy referenced to the cyclic intermediate at −0/9 kcal mol−1 in Fig. 16. All energies are given in kcal mol−1. | ||
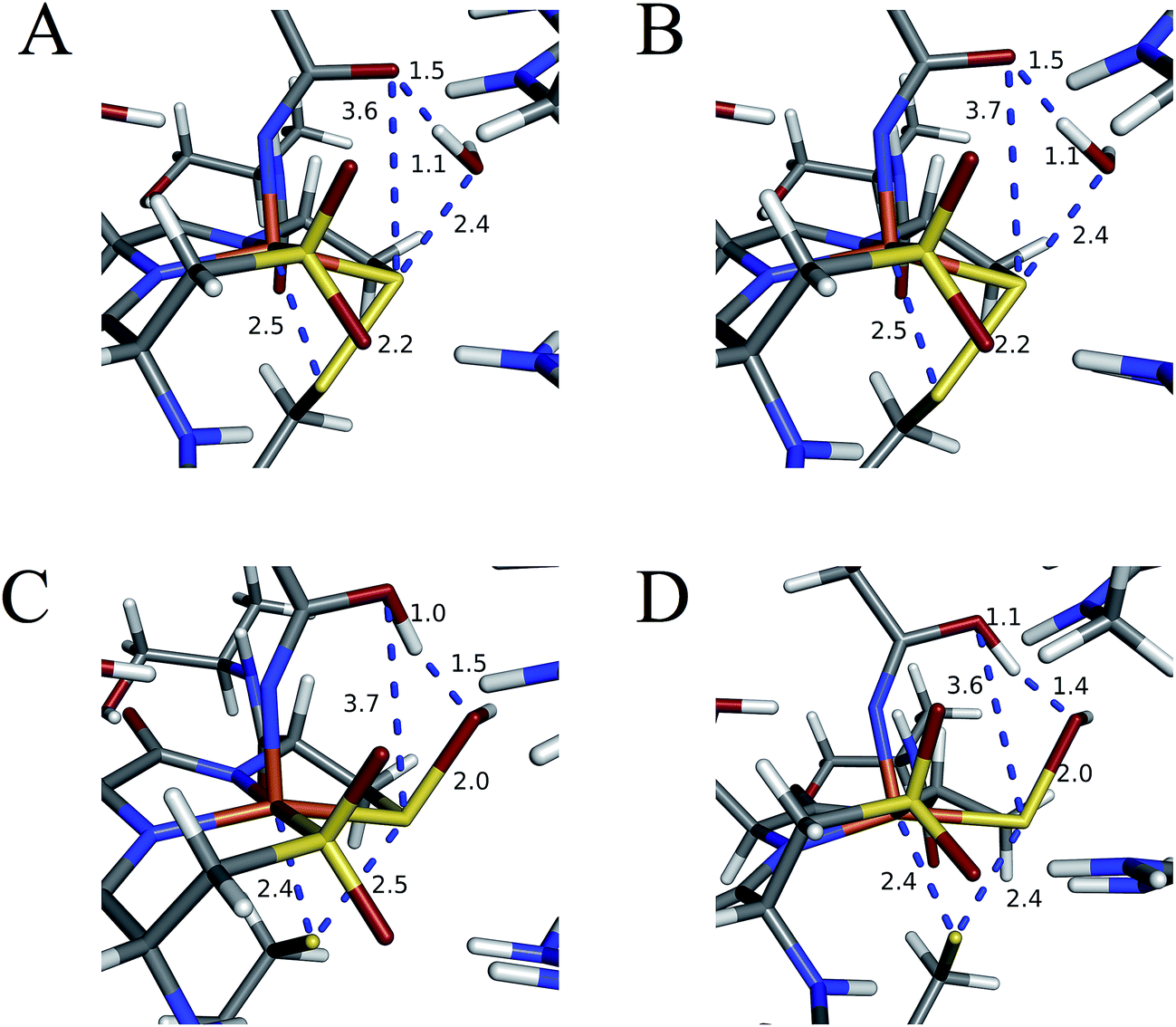 | ||
| Fig. 19 Structures of (A) linear transit species with a water O–cyclic intermediate S distance of 2.4 Å, (B) IRC step towards reactants with this same distance, (C) SOH species, and (D) IRC step towards products. All distances are in Å. | ||
Conclusions
The EPR and UV-Vis MCD spectra of NHaseBA and NHaseAq in this study have shown that the LS FeIII site of NHase, which has a protonated sulfenate group when butyrate is coordinated to metal, has a deprotonated sulfenate group and coordinated water ligand in the active form as shown in Fig. 20. For both forms, the active site (including the positively charged βArg56 and βArg141 that are H-bonding to the sulfenate and sulfinate ligands) has zero net charge therefore the NHase active site maintains charge neutrality and the coordinated sulfenate can easily change protonation state at functional pHs (here determined to have a pKa of 6.1 and deprotonated in the active form of the site). From the EPR, UV-Vis LT Abs and UV-Vis/NIR MCD spectroscopic data coupled to DFT calculations the highest energy, half-occupied dπ orbital for both NHase forms is strongly π bonding to the cysteine thiolate ligand, and the sulfenate ligand possesses a high-energy occupied σIP MO with significant O character (Fig. 9), implying significant nucleophilic capability for this ligand. These spectrally evaluated structural and electronic properties of the NHase active site provide insight into its reactivity.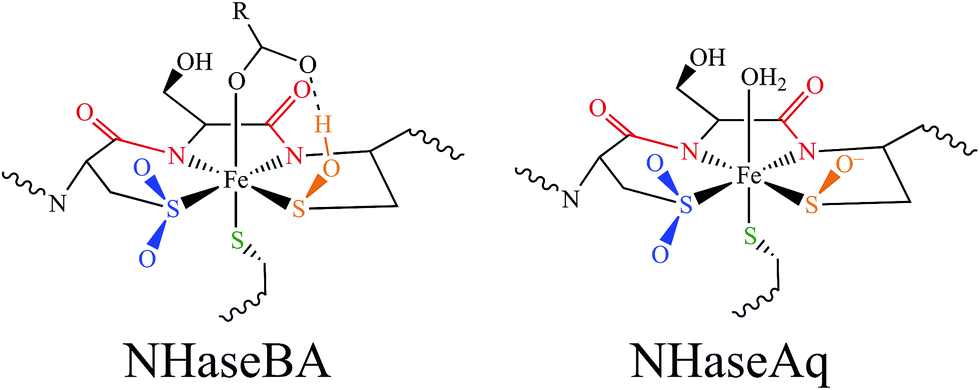 | ||
| Fig. 20 Spectroscopically determined active site structures of NHaseBA (left) and NHaseAq (right). | ||
Using EPR, NIR MCD, and UV-Vis LT-Abs and MCD spectroscopic data, computational models of NHaseBA and NHaseAq were developed which qualitatively reproduce the spectroscopic features and geometric and electronic structures of the two enzyme forms. These findings show that the computational models provide a reasonable basis for evaluating possible NHase catalytic mechanisms. The five negatively charged protein-derived ligands combined with the nearby positively charged βArg56 and βArg141 residues lead to a relatively high pKa for the bound H2O, allowing its displacement and nitrile substitution. Upon coordination to the LS FeIII the nitrile is activated for nucleophilic attack: (a) the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond becomes more polarized such that the C atom has an increased electrostatic interaction with the nucleophile, (b) the nitrile frontier π* LUMO has more C character, leading to better overlap with the HOMO of the nucleophile, and (c) the energy of the nitrile π* MO is decreased, leading to better covalent interaction. DFT calculations on free acetonitrile and acetonitrile bound to the FeIII of NHase show that upon coordination to LS FeIII the positive charge on the nitrile C increases from +0.12 to +0.21, the C character in the acetonitrile π* LUMO increases from 53% to 59% and the energy decreases by ≈0.2 eV. This activation is shown in Fig. 21, left and center.
N bond becomes more polarized such that the C atom has an increased electrostatic interaction with the nucleophile, (b) the nitrile frontier π* LUMO has more C character, leading to better overlap with the HOMO of the nucleophile, and (c) the energy of the nitrile π* MO is decreased, leading to better covalent interaction. DFT calculations on free acetonitrile and acetonitrile bound to the FeIII of NHase show that upon coordination to LS FeIII the positive charge on the nitrile C increases from +0.12 to +0.21, the C character in the acetonitrile π* LUMO increases from 53% to 59% and the energy decreases by ≈0.2 eV. This activation is shown in Fig. 21, left and center.
 | ||
| Fig. 21 Donor/acceptor orbitals for free nitrile and water (left), coordinated nitrile and water (center) and the coordinated cyclic species and water (right). | ||
The NHase active site also possesses a good internal nucleophile in the coordinated sulfenate ligand. This is shown in Fig. 21, center. The occupied sulfenate S–O σIP MO may act as a donor and is higher in energy than the water MO by ≈1.6 eV. It is negatively charged, and is well-oriented for overlap with the nitrile π* acceptor orbital. Sulfenate can therefore attack the nitrile C to form the cyclic species shown in Fig. 21, right. This cyclic species has an unoccupied sulfenate S–O σ* FMO which is ≈3 eV lower than the π* orbital of the uncyclized coordinated nitrile, leading to favorable attack by water at S and a lower reaction barrier than for attack by water on the coordinated, uncyclized nitrile.
Alternatively, the axial thiolate could act as a nucleophile as previously predicted.22 In this reported mechanism, the barriers for formation of the cyclic intermediate and the disulfide intermediate are the largest and of similar energy, indicating that both should be observed over the course of enzyme turnover. Holz and coworkers reported stopped flow data for an Fe NHase enzyme with substoichiometric nitrile reactant, which showed intermediates with a blue shift in the Cys-S− π → dπ CT transition of approximately 1100 cm−1.39 TD-DFT calculations on the cyclic intermediate both in this work and on the structure in ref. 22 showed Cys-S− π → dπ CT transitions approximately 4000 cm−1 higher in energy than NHaseAq, whereas the disulfide intermediate of ref. 22 showed no transition in the ≈12000–18000 cm−1 region. As the ≈14300–15400 cm−1 band does not disappear over the course of the stopped-flow experiment, the presence of a disulfide intermediate does not appear likely.
The ligand set and spin state of NHase are unusual relative to the other mononuclear non-heme iron enzymes. Most are ferrous enzymes that activate O2 and utilize histidine, glutamate, and aspartate residues to coordinate the metal and facilitate the redox reaction. In the intradiol dioxygenases an FeIII with Tyr ligands is active, but this is high spin and this spin state and its change along the reaction coordinate are important in activating the singlet substrate for the spin-forbidden reaction with 3O2.40 In order to determine the contribution of the low spin state on the nitrile-bound form of NHase, geometry optimizations were performed on models of this complex with S = 3/2 and 5/2 ground states. Whereas the nitrile N–Fe bond length in the S = 1/2 spin state is ≈1.9 Å, this length is ≈2.6 Å in the S = 3/2 state and ≈2.4 Å in the S = 5/2 state, indicating that the bonds are very weak to nonexistent. Indeed, the nitrile N–Fe bond dissociation energies for the S = 1/2, S = 3/2, and S = 5/2 forms are +6.6, −2.3, and −7.0 kcal mol−1 respectively, indicating that the low spin state of NHase is required to assist in the coordination and activation of nitrile substrates.
In order to explore the effects of the NHase ligands on the spin state and activity of the enzyme, DFT calculations were performed on active site models with unoxidized Cys residues, as well as with two His or two acetate ligands replacing the backbone amidates of the WT site. The active site model with all unoxidized thiolates was found to not have an S = 1/2 ground state; the S = 3/2 ground state was lower in energy by 14.2 kcal mol−1. Geometry optimization of this S = 3/2, structure leads to dissociation of the exogenous ligand and a 5C form that would not be catalytic. Alternatively, the amidate ligands of NHase are not critical to maintaining the low-spin active site. The in silico results with the amidates replaced with weaker donors indicate better activation of coordinated nitriles for nucleophilic attack (i.e. these form stronger FeIII–nitrile bonds). Nature may have selected this deprotonated amide ligand set for its rigidity and chelate ring for orienting the sulfenate ligand for attack on the C of the coordinated nitrile.
Finally, consistent with the EPR g value analysis and LF MCD, the DFT calculations indicate that the axial Cys thiolate is the strongest π donor in the NHase coordination sphere and controls the orientation of the half-occupied dπ orbital. This strong π donor trans to coordinated nitrile would not assist in its activation, but may serve to increase both H2O and product lability from the LS FeIII site, as was observed by Kovacs et al. for NHase model complexes.41 This increased lability would be especially important for NHase forms that utilize low-spin CoIII, which generally undergoes very slow ligand exchange. The presence of a weak interaction between the thiolate and sulfenate S atoms in the transition state for nucleophilic attack on the S of the cyclic species (Fig. 17, bottom, TS) also indicates that the axial thiolate may serve to lower the energy barrier for nitrile hydrolysis.
In summary, our spectroscopic results have provided new insight into the geometric and electronic structure of NHase, which activates nitriles by coordination to a LS FeIII and contains a sulfenate group that acts as a good nucleophile oriented well for this attack. These spectroscopically-calibrated computational results show that the cyclic intermediate that would be formed in this reaction is activated for nucleophilic attack by water at the S atom leading to formation of the amide product and regeneration of the active site sulfenate. Nature has selected an unusual set of ligands for this enzyme to ensure that the low-spin state necessary for nitrile binding is maintained, and that a rigid chelate ring is present, which properly orients the frontier MO of the sulfenate group for nucleophilic attack on the bound substrate.
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM040392 (E. I. S.) and by a Grant-in-Aid for Scientific Research from the Scientific Research (B) KAKENHI 24350082 (M. O.). K. M. L. was also supported by the Althouse Stanford Graduate Fellowship.Notes and references
- N. D'Antona and R. Morrone, Biocatalysis: Green Transformations of Nitrile Function, in Green Chemistry for Environmental Sustainability, ed. S. Sharma and A. Mudhoo, CRC Press, Boca Raton, FL, 2011, pp. 357–415 Search PubMed.
- Y. Ashina, M. Suto and T. Endo, Nitrile Hydratase, in Encyclopedia of Industrial Biotechnology, ed. M. C. Flickinger, John Wiley & Sons, Hoboken, NJ, 2010, vol. 6, pp. 3671–3676 Search PubMed.
- A. Yanenko and S. Osswald, Hydrolysis of Nitriles to Amides, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Groeger and O. May, Wiley, Weinheim, Germany, 3rd edn, 2012, vol. 2, pp. 533–544 Search PubMed.
- L. Martinkova, B. Uhnakova, M. Patek, J. Nesvera and V. Kren, Environ. Int., 2009, 35, 162–177 CrossRef CAS PubMed.
- S. Nagashima, M. Nakasako, N. Dohmae, M. Tsujimura, K. Takio, M. Odaka, M. Yohda, N. Kamiya and I. Endo, Nat. Struct. Mol. Biol., 1998, 5, 347–351 CAS.
- A. Miyanaga, S. Fushinobu, K. Ito and T. Wakagi, Biochem. Biophys. Res. Commun., 2001, 288, 1169–1174 CrossRef CAS PubMed.
- A. Dey, M. Chow, K. Taniguchi, P. Lugo-Mas, S. Davin, M. Maeda, J. Kovacs, M. Odaka, K. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2006, 128, 533–541 CrossRef CAS PubMed.
- T. Noguchi, M. Nojiri, K. Takei, M. Odaka and N. Kamiya, Biochemistry, 2003, 42, 11642–11650 CrossRef CAS PubMed.
- T. Noguchi, J. Honda, T. Nagamune, H. Sasabe, Y. Inoue and I. Endo, FEBS Lett., 1995, 358, 9–12 CrossRef CAS.
- H. Takarada, Y. Kawano, K. Hashimoto, H. Nakayama, S. Ueda, M. Yohda, N. Kamiya, N. Dohmae, M. Maeda and M. Odaka, Biosci., Biotechnol., Biochem., 2006, 70, 881–889 CrossRef CAS.
- The crystal structure of WT NHase with a bound water-derived ligand is deposited in the Protein Databank (2CYZ), but unpublished. However, the structure near the active site is virtually identical to that of the αQ90N as published in ref. 8.
- M. Taku, M. Nojiri, H. Nakayama, M. Odaka, M. Yohda, M. Dohmae, K. Takio, T. Nagamune and I. Endo, Protein Sci., 2000, 9, 1024–1030 CrossRef PubMed.
- T. Nagasawa, K. Ryuno and Y. Hideaki, Biochem. Biophys. Res. Commun., 1986, 139, 1305–1312 CrossRef CAS.
- The crystal structure of WT NHase with a bound butyrate ligand is deposited in the Protein Databank (2CZ1), but unpublished. However, the structure of a Co NHase PDB ID 1UGP does have the butyrate directly bound to metal. See ref. 13.
- A. Miyanaga, S. Fushinobu, K. Ito, H. Shoun and T. Wakagi, Eur. J. Biochem., 2004, 271, 429–438 CrossRef CAS.
- M. A. Kopf, D. Bonnet, I. Artaud, D. Pétré and D. Mansuy, Eur. J. Biochem., 1996, 240, 239–244 CrossRef CAS PubMed.
- W. Huang, J. Jia, J. Cummings, M. Nelson, G. Schneider and Y. Lindqvist, Structure, 1997, 5, 691–699 CrossRef CAS.
- Y. Yamanaka, K. Hashimoto, A. Ohtaki, K. Noguchi, M. Yohda and M. Odaka, JBIC, J. Biol. Inorg. Chem., 2010, 15, 655–665 CrossRef CAS PubMed.
- J. A. Kovacs, Chem. Rev., 2004, 104, 825–848 CrossRef CAS PubMed.
- K. H. Hopmann, J.-D. Guo and F. Himo, Inorg. Chem., 2007, 46, 4850–4856 CrossRef CAS PubMed.
- S. Martinez, R. Wu, R. Sanishvili, D. Liu and R. C. Holz, J. Am. Chem. Soc., 2014, 136, 1186–1189 CrossRef CAS PubMed.
- K. H. Hopmann, Inorg. Chem., 2014, 53, 2760–2762 CrossRef CAS PubMed.
- K. H. Hopmann and F. Himo, Eur. J. Inorg. Chem., 2008, 1406–1412 CrossRef CAS PubMed.
- R. C. Elder, L. R. Floria, R. E. Lake and A. M. Yacynych, Inorg. Chem., 1973, 12, 2690–2699 CrossRef CAS.
- B. A. Lange, K. Libson, E. Deutsch and R. C. Elder, Inorg. Chem., 1976, 15, 2985–2989 CrossRef CAS.
- I. K. Adzamli, K. Libson, J. D. Lydon, R. C. Elder and E. Deutsch, Inorg. Chem., 1979, 18, 303–311 CrossRef CAS.
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y.-S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235–349 CrossRef CAS PubMed.
- L. B. Poole and H. R. Ellis, Identification of Cysteine Sulfenic Acid in AhpC of Alkyl Hydroperoxide Reductase, in Methods in Enzymology, ed. H. Sies and L. Packer, Academic Press, Amsterdam, 2002, vol. 238, pp. 122–136 Search PubMed.
- R. K. Burkhard, D. E. Sellers, F. DeCou and J. L. Lambert, J. Org. Chem., 1959, 24, 767–769 CrossRef CAS.
- F. Neese, J. Zaleski, K. L. Zaleski and E. I. Solomon, J. Am. Chem. Soc., 2000, 122, 11703–11724 CrossRef CAS.
- C. P. S. Taylor, Biochim. Biophys. Acta, 1977, 491, 137–149 CrossRef CAS.
- B. A. Brennan, J. G. Cummings, D. B. Chase, I. M. Turner Jr and M. J. Nelson, Biochemistry, 1996, 35, 10068–10077 CrossRef CAS PubMed.
- M. D. Clay, F. E. Jenney, P. L. Hagedoorn, G. N. George, M. W. W. Adams and M. K. Johnson, J. Am. Chem. Soc., 2002, 124, 788–805 CrossRef CAS PubMed.
- J. D. Lydon and E. Deutsch, Inorg. Chem., 1982, 21, 3180–3185 CrossRef CAS.
- A. A. Gewirth and E. I. Solomon, J. Am. Chem. Soc., 1988, 110, 3811–3819 CrossRef CAS.
- F. Neese and E. I. Solomon, Inorg. Chem., 1999, 38, 1847–1865 CrossRef CAS PubMed.
- F. Neese, JBIC, J. Biol. Inorg. Chem., 2006, 11, 702–711 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, J. Am. Chem. Soc., 2004, 126, 4007–4016 CrossRef CAS PubMed.
- N. Gumataotao, M. L. Kuhn, N. Hajnas and R. C. Holz, J. Biol. Chem., 2013, 288, 15532–15536 CrossRef CAS PubMed.
- M. Y. Pau, J. D. Lipscomb and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18355–18362 CrossRef CAS PubMed.
- J. Shearer, I. Y. Kung, S. Lovell, W. Kaminsky and J. A. Kovacs, J. Am. Chem. Soc., 2001, 123, 463–468 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: DFT geometry optimization procedure, simulation of EPR spectra, g tensor coordinate axes, MO isosurface contours for NHaseBA and NHaseAq, and optimized coordinates for computational models. See DOI: 10.1039/c5sc02012c |
| ‡ Currently at the Department of Chemistry, University of Utah, Salt Lake City, UT 84112. |
| § Currently at the Department of Cell Biology, Graduate School of Medicine, Kyoto University Yoshida-Konoe-cho, Sakyo-ku, Kyoto 606-8501, Japan. |
| This journal is © The Royal Society of Chemistry 2015 |