
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective reductive multicomponent coupling reactions between isatins and aldehydes†
Matthew A.
Horwitz‡
a,
Naoya
Tanaka‡
b,
Takuya
Yokosaka
a,
Daisuke
Uraguchi
b,
Jeffrey S.
Johnson
*a and
Takashi
Ooi
*bc
aDepartment of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA. E-mail: jsj@unc.edu
bInstitute of Transformative Bio-Molecules (WPI-ITbM) and Department of Applied Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho D2-1, Chikusa, Nagoya 464-8602, Japan. E-mail: tooi@apchem.nagoya-u.ac.jp
cCREST, Japan Science and Technology Agency (JST), Nagoya University, Nagoya 464-8603, Japan
First published on 30th July 2015
Abstract
A metal-free stereoselective reductive coupling reaction between isatins and aldehydes is reported. The reaction relies on commercial diethyl phosphite (∼€70 kg−1) as the stoichiometric reductant. Base-catalyzed Pudovik addition and phosphonate/phosphate rearrangement achieved polarity inversion on the isatin, and the derived carbanions were trapped by aldehydes with subsequent dialkoxyphosphinyl migration. Chiral iminophosphoranes were used as basic catalysts to achieve high diastereo- and enantioselectivities with excellent yields.
The reductive coupling of π-unsaturation is a powerful method for the construction of carbon–carbon bonds. When the two coupling partners are prochiral, there exists the opportunity to establish multiple stereogenic centers concurrent with C–C bond formation. In the specific case of two carbonyl reactants, reductive coupling offers an attractive and straightforward method for the synthesis of vicinal diols, valuable building blocks in organic chemistry. A generic carbonyl reductive coupling manifold encompasses many mechanistic subtypes,1 but the pinacol reaction is preeminent among them. The traditional pinacol coupling entails single-electron reduction of the carbonyl functionality to generate the corresponding ketyl radical and subsequent dimerization between two radical species. The reaction has been studied extensively using low-valent metals in this single-electron transfer manifold.2–6 Despite numerous advances, however, myriad challenges remain: a stoichiometric or superstoichiometric amount of metal agents is often required and there are sparse examples that use catalytic conditions.4n–r Moreover, the nature of the mechanism can render it difficult to control both chemoselectivity (homo- versus cross-coupling) and stereoselectivity, and the lack of differentiation of the nascent alcohols can be nettlesome. These precedents collectively informed our interest in developing an alternative, potentially generalizable reductive coupling strategy that utilizes a polar two-electron reaction mechanism for addressing the aforementioned issues. The purpose of this communication is to detail a new base-catalyzed cross coupling of carbonyls mediated by an economical organic reductant, diethyl phosphite; the stereochemical outcome of this multicomponent process is precisely controlled by a chiral triaminoiminophosphorane (Figure 1a).7,8
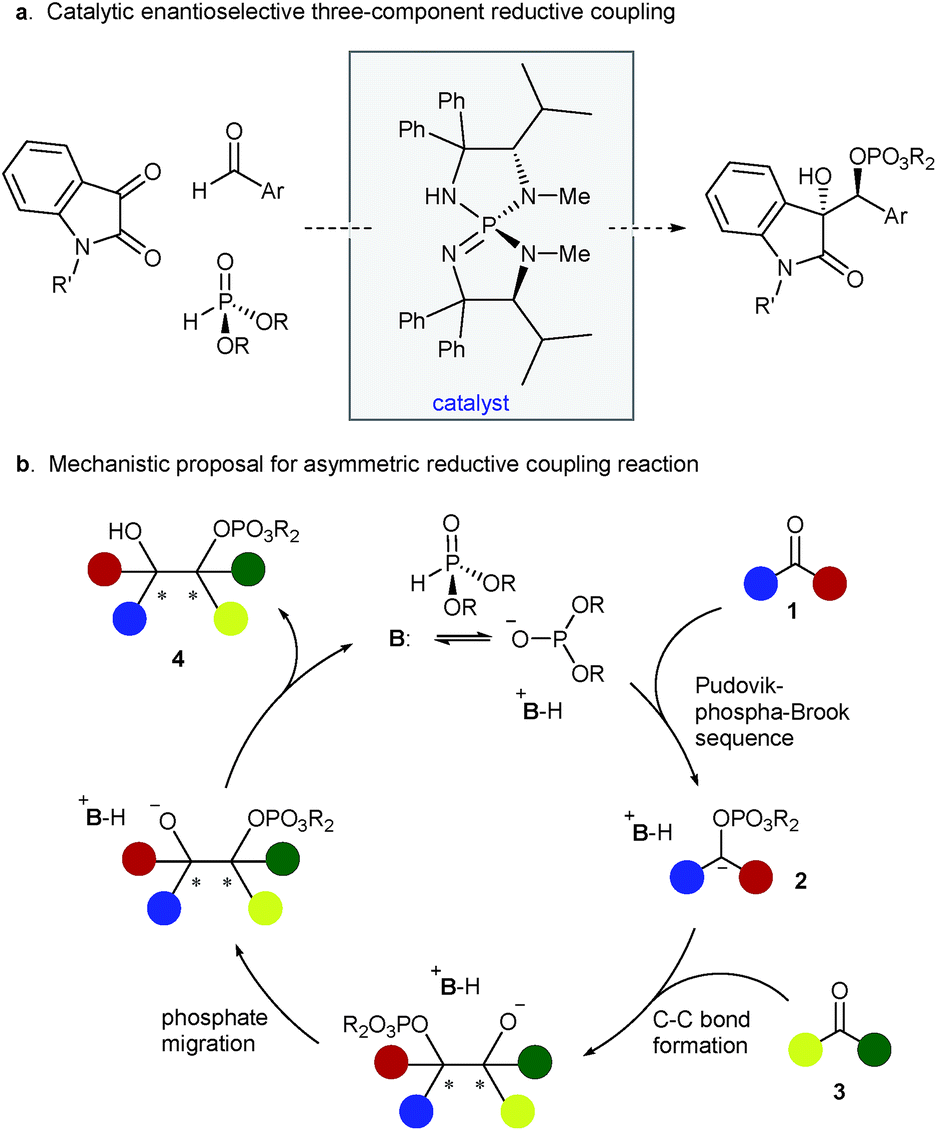 | ||
| Fig. 1 Stereoselective reductive coupling reactions. | ||
At the outset, we envisaged the possibility of catalytic generation of an α-oxycarbanion from a carbonyl substrate and its rapid and selective trapping with another carbonyl compound to form 1,2-diols. For substantiating this hypothesis, polarity reversal of a particular carbonyl group is of critical importance and we sought to take advantage of the phosphonate–phosphate (phospha-Brook) rearrangement to achieve this requisite process. Thus, a base-catalyzed sequence of Pudovik addition and phosphonate–phosphate rearrangement between ketone 1 and dialkyl phosphite was projected to lead to carbanion 2. The interception of this key intermediate by aldehyde 3 would afford mono-protected diol 4 through dialkoxyphosphinyl migration (Figure 1b).9 A crucial departure from prior art is the fully intermolecular nature of the coupling and the need for the phosphite to exhibit complete selectivity between the two carbonyl reactants. We reasoned that the crucial chemoselectivity issue underlying this mechanistic framework, viz. the selective generation of α-oxycarbanion 2 from ketone 1, would be ensured by the inherent reversibility of Pudovik reaction and the reluctance of the aldehyde Pudovik product to undergo phospha-Brook rearrangement. In addition, absolute stereochemical guidance in the C–C bond-forming event could be provided by the conjugate acid of a suitable chiral base. In providing the conceptual blueprint for this scenario, we focused our attention on the exceptional electrophilicity and utility of α-dicarbonyls.9d–g,10
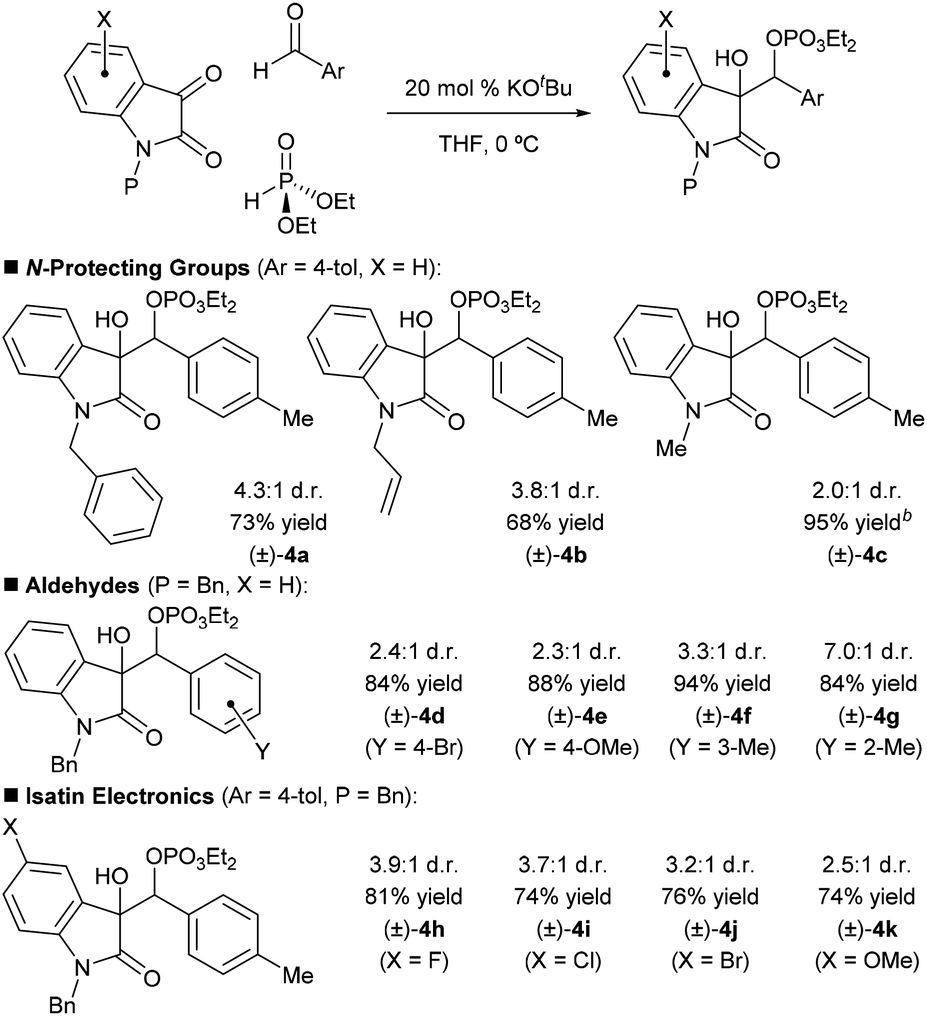
Steps were initially taken to assess the feasibility of the proposed reaction in a racemic sense using achiral bases such as potassium tert-butoxide (KOtBu). Initial trials with diethyl phosphite as the stoichiometric reductant indicated that the reaction proceeds most cleanly and efficiently when a protecting group is used on the isatin. Benzyl, allyl, and methyl protecting groups were examined using 20 mol% KOtBu in THF at 0 °C (Table 1, (±)-4a–(±)-4c). Under these conditions, the reactions were complete in minutes with no observable intermediates (if the aldehyde is omitted from the reaction, the Pudovik-phospha-Brook product can be observed, however).9f These experiments revealed that the benzyl protecting group provided the highest isolated yield and diastereoselectivity. We subsequently verified that para-tolualdehyde is not capable of phospha-Brook rearrangement when treated with diethyl phosphite and 20 mol% KOtBu: only the Pudovik adduct was observed, implying that it is the isatin that is undergoing polarity reversal as we expected.
| a All reactions were run on 0.2 mmol scale, using 1.1 equiv. of dialkylphosphite and 5.0 equiv. of aldehyde. % Yields refer to isolated yields. All d.r. and % yield values are the averages of two trials. Reactions were run until complete as adjudged by TLC. b % Yield determined by crude 1H NMR using mesitylene as an internal standard. Products derived from apparent retro-reaction significantly diminished the isolated yield; therefore, this substrate was not selected for further study. |
|---|
|
We then briefly studied the scope of the racemic reaction. The reaction gives consistently good yields for various aryl aldehydes incorporating substituents of different electronic properties (Table 1, (±)-4d–(±)-4g). At the current level of optimization, alkyl aldehydes and Boc-protected imine electrophiles were not well tolerated and only provided messy reactions.11 The substitution pattern of the isatin was also examined; we found that the racemic reaction is reasonably flexible in terms of isatin electronics ((±)-4h–(±)-4k).
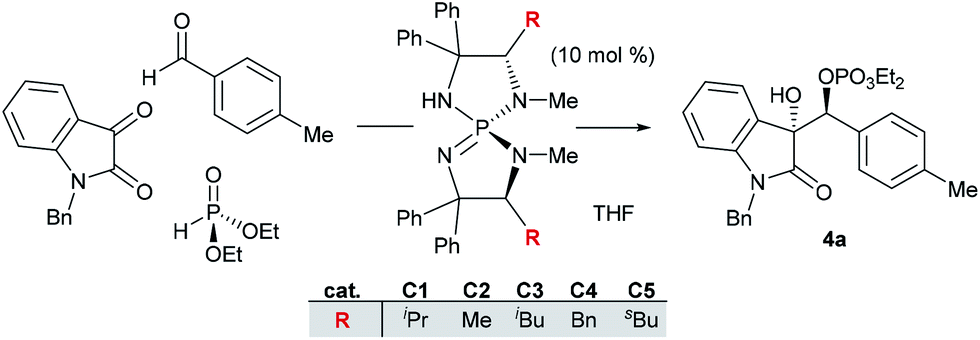
Efforts were next directed to the development of the enantioselective variant.12 We were encouraged to find that when we used the chiral iminophosphorane (C1), we obtained the secondary phosphate 4a with appreciable enantioenrichment (er 89.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.5), although the diastereoselectivity was poor (Table 2, entry 1). Gratifyingly, we found that upon lowering the temperature to −78 °C, phosphate 4a was obtained in 82% yield, 15:1 diastereoselectivity and an er of 96.5:3.5 (entry 2). Using the same temperature, we proceeded to evaluate the effect of the catalyst structure (entries 3 to 6), but ultimately concluded that α-branching in ligand substituent R is essential for promoting the desired transformations and the valine-derived iminophosphorane C1 was optimal in terms of stereoselectivity and chemical yield.
:10.5), although the diastereoselectivity was poor (Table 2, entry 1). Gratifyingly, we found that upon lowering the temperature to −78 °C, phosphate 4a was obtained in 82% yield, 15:1 diastereoselectivity and an er of 96.5:3.5 (entry 2). Using the same temperature, we proceeded to evaluate the effect of the catalyst structure (entries 3 to 6), but ultimately concluded that α-branching in ligand substituent R is essential for promoting the desired transformations and the valine-derived iminophosphorane C1 was optimal in terms of stereoselectivity and chemical yield.
|
|
|||||
|---|---|---|---|---|---|
| Entry | T (°C) | Catalyst | d.r. | e.r. | % Conv. |
| a All reactions were conducted on a 0.1 mmol scale, using 1.1 equiv. of dialkylphosphite and 5.0 equiv. of 4-tolualdehyde. Argon was used to purge the reaction flasks. All d.r., e.r., and % conversion values are the average of two trials. n.a. = not analyzed. | |||||
| 1 | 0 | C1 | 3.4:1 |
89.5:10.5 |
96 |
| 2 | −78 | C1 | 15:1 |
96.5:3.5 |
82 |
| 3 | −78 | C2 | n.a. | n.a. | 18 |
| 4 | −78 | C3 | n.a. | n.a. | 15 |
| 5 | −78 | C4 | n.a. | n.a. | 12 |
| 6 | −78 | C5 | 7.9:1 |
86:14 |
80 |
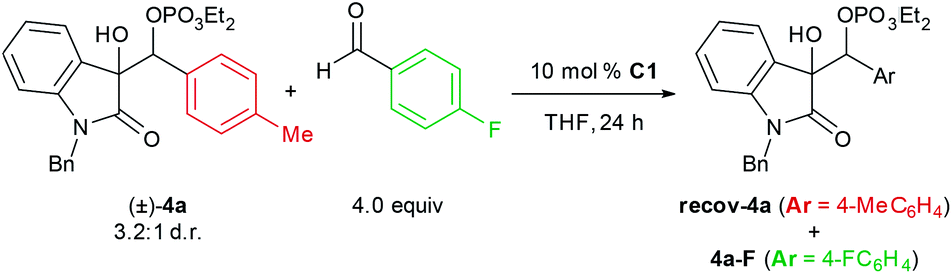
The disparity between the stereoselectivities at 0 °C and −78 °C prompted us to investigate the reversibility of the carbon–carbon bond formation via crossover experiments in that temperature range (Table 3). When racemic phosphate (±)-4a was subjected to standard conditions in the presence of 4-fluorobenzaldehyde, significant incorporation of that component in the form of phosphate 4a–F was observed at 0 °C and −40 °C, but no crossover was observed at −78 °C. These data support the hypothesis that the increase in enantioselectivity at −78 °C is not only a consequence of more rigorous facial discrimination of both substrates but also shutting down a stereoablative retro-aldol process that is operative at higher temperatures.
|
|
||
|---|---|---|
| Entry | T (°C) |
4a:4a–F |
| a Product distributions were determined by 1H NMR analysis (800 MHz) of the crude mixture. | ||
| 1 | 0 | 1.0:1.5 |
| 2 | −40 | 1.0:1.1 |
| 3 | −78 | Only 4a |
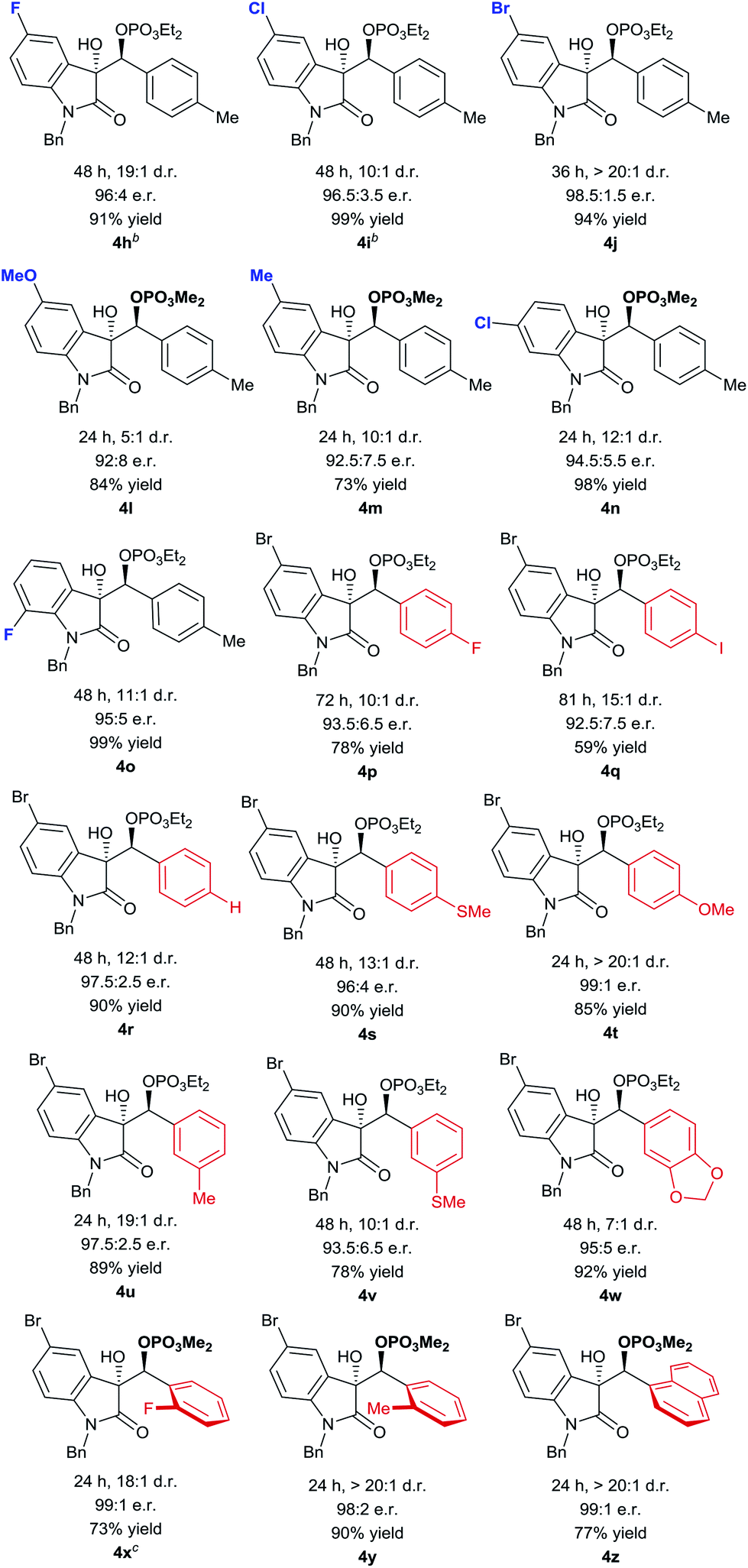
Using the optimized conditions, we evaluated the scope of the asymmetric reaction by initially looking at various isatins. While electron-deficient 5-halogenated isatins were well accommodated under the optimized conditions, use of dimethyl phosphite was indispensable for completion of the reactions with 5-methyl and methoxy isatins probably because of the slow phospha-Brook rearrangement (Table 4, 4h–4m).13 6-Chloro and 7-fluoro isatins were also smoothly converted into the reductive coupling products of high stereochemical purity using appropriate phosphite (4n and 4o). The absolute stereochemistry was determined at this stage by an X-ray diffraction study of phosphate 4j (Fig. 2).14
|
|
|---|
| a All reactions were conducted on a 0.1 mmol scale, using 1.1 equiv. of dialkylphosphite and 5.0 equiv. of ArCHO. Argon was used to purge the reaction flasks. % Yields refer to isolated yields. All d.r., e.r., and % yield values are the average of two trials. b 15 mol% of catalyst was used. c 2.2 equiv. of dialkylphosphite was used. |
|
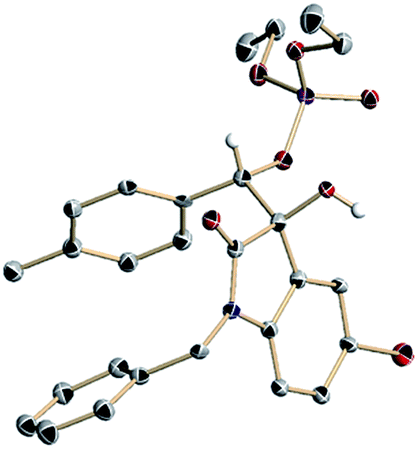 | ||
| Fig. 2 ORTEP diagram of 4j (ellipsoids displayed at 50% probability. Calculated hydrogen atoms except for that attached to the stereogenic carbon atom are omitted for clarity. Black: carbon, red: oxygen, purple: phosphorous, blue: nitrogen, vermilion: bromine, white: hydrogen). | ||
For exploration of aldehyde generality, we selected 5-bromo isatin as a coupling partner in consideration of its high reactivity and advantage of having an additional functional handle at the aromatic nuclei. As included in Table 4, various para-substituted aromatic aldehydes were tolerated and relatively electron rich aldehydes exhibited higher reactivity and selectivity (4p–4t). Hetero-substituents at the meta-position slightly affected the stereochemical outcome (4u–4w). For sterically demanding ortho-substituted aldehydes, dimethyl phosphite was needed to accelerate the reaction and virtually complete stereocontrol could be achieved (4x–4z).
In summary, we have developed a highly stereoselective, fully organic multicomponent coupling reaction between isatins and aldehydes with dialkyl phosphite as an economical reductant. The advantages of extending the reductive coupling into a two-electron manifold are manifest, and the mechanistic framework established herein may be applicable to other stereoselective reductive carbon–carbon bond constructions. Efforts to exploit this reaction paradigm in other systems are ongoing in our laboratories.
Acknowledgements
Financial support to TO, DU, and NT was provided by CREST-JST, a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” from MEXT, Program for Leading Graduate Schools “Integrative Graduate Education and Research Program in Green Natural Sciences” in Nagoya University, and Grants of JSPS for Scientific Research. NT acknowledges JSPS for financial support. Financial support to JSJ, MH, and TY was provided by Award R01 GM103855 from the National Institute of General Medical Sciences and a JSPS Research Fellowship for Young Scientists (TY).Notes and references
- For example: (a) F. Ramirez, N. B. Desai and N. Ramanathan, Tetrahedron Lett., 1963, 4, 323–328 CrossRef; (b) T. Mukaiyama, H. Sugimura, T. Ohno and S. Kobayashi, Chem. Lett., 1989, 18, 1401–1404 CrossRef; (c) S. N. Greszler and J. S. Johnson, Org. Lett., 2009, 11, 827–830 CrossRef CAS PubMed.
- Reviews: (a) B. S. Terra and F. Macedo Jr, Arkivoc, 2012, 134–151 CAS; (b) A. Chatterjee and N. N. Joshi, Tetrahedron, 2006, 62, 12137–12158 CrossRef CAS PubMed.
- Seminal papers on cross pinacol reactions with low-valent metal reagents: (a) J. E. McMurry and L. R. Krepski, J. Org. Chem., 1976, 41, 3929–3930 CrossRef CAS; (b) J. E. McMurry, Chem. Rev., 1989, 89, 1513–1524 CrossRef CAS.
- Developments toward using aldehydes as coupling partners in stereoselective reductive coupling reactions: (a) P. M. Takahara, J. H. Freudenberger, A. W. Konradi and S. F. Pedersen, Tetrahedron Lett., 1989, 30, 7177–7180 CrossRef CAS; (b) J. H. Freudenberger, A. W. Konradi and S. F. Pedersen, J. Am. Chem. Soc., 1989, 111, 8014–8016 CrossRef CAS; (c) R. Annunziata, M. Cinquini, F. Cozzi and P. Giaroni, Tetrahedron: Asymmetry, 1990, 1, 355–358 CrossRef CAS; (d) R. Annunziata, M. Cinquini, F. Cozzi, P. Giaroni and M. Benaglia, Tetrahedron, 1991, 47, 5737–5758 CrossRef CAS; (e) R. Annunziata, M. Benaglia, M. Cinquini, F. Cozzi and P. Giaroni, J. Org. Chem., 1992, 57, 782–784 CrossRef CAS; (f) A. W. Konradi and S. F. Pedersen, J. Org. Chem., 1990, 55, 4506–4508 CrossRef CAS; (g) A. W. Konradi, S. J. Kemp and S. F. Pedersen, J. Am. Chem. Soc., 1994, 116, 1316–1323 CrossRef CAS; (h) J. Park and S. F. Pedersen, J. Org. Chem., 1990, 55, 5924–5926 CrossRef CAS; (i) F. R. Askham and K. M. Carroll, J. Org. Chem., 1993, 58, 7328–7329 CrossRef CAS; (j) H. Yoda, K. Matsuda, H. Nomura and K. Takabe, Tetrahedron Lett., 2000, 41, 1775–1779 CrossRef CAS; (k) K. Takai, K. Nitta and K. Utimoto, Tetrahedron Lett., 1988, 29, 5263–5266 CrossRef CAS; (l) K. Takai, R. Morita and C. Toratsu, Angew. Chem., Int. Ed., 2001, 40, 1116–1119 CrossRef CAS; (m) K. Takai, R. Morita, H. Matsushita and C. Toratsu, Chirality, 2003, 15, 17–23 CrossRef CAS PubMed; (n) R. K. Boeckman Jr and R. A. Hudack Jr, J. Org. Chem., 1998, 63, 3524–3525 CrossRef; (o) M. Jung and U. Groth, Synlett, 2002, 2015–2018 CAS; (p) U. Groth, M. Jung and T. Vogel, Chem.–Eur. J., 2005, 11, 3127–3135 CrossRef CAS PubMed; (q) S. Fischer, U. Groth, M. Jung, M. Lindenmaier and T. Vogel, Tetrahedron Lett., 2005, 46, 6679–6682 CrossRef CAS PubMed; (r) N. Miyoshi, T. Fukuma and M. Wada, Chem. Lett., 1995, 24, 999–1000 CrossRef; (s) H. Maekawa, Y. Yamamoto, H. Shimada, K. Yonemura and I. Nishiguchi, Tetrahedron Lett., 2004, 45, 3869–3872 CrossRef CAS PubMed; (t) V. Nair, S. Ros, C. N. Jayan and N. P. Rath, Tetrahedron Lett., 2002, 43, 8967–8969 CrossRef CAS; (u) Y.-S. Yang, Z.-L. Shen and T.-P. Loh, Org. Lett., 2009, 11, 2213–2215 CrossRef CAS PubMed; (v) N. Kise, Y. Shiozawa and N. Ueda, Tetrahedron, 2007, 63, 5415–5426 CrossRef CAS PubMed; (w) N. Takenaka, G. Xia and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 13198–13199 CrossRef CAS PubMed; (x) X.-F. Duan, J.-X. Feng, G.-F. Zi and Z.-B. Zhang, Synthesis, 2009, 277–282 CrossRef CAS.
- R. Fittig, Justus Liebigs Ann. Chem., 1859, 110, 23–45 CrossRef PubMed.
- Recent developments in using ketones in pinacol reactions: (a) S. Matsukawa and Y. Hinakubo, Org. Lett., 2003, 5, 1221–1223 CrossRef CAS PubMed; (b) L. Shi, C.-A. Fan, Y.-Q. Tu, M. Wang and F.-M. Zhang, Tetrahedron, 2004, 60, 2851–2855 CrossRef CAS PubMed; (c) H. C. Aspinall, N. Greeves and C. Valla, Org. Lett., 2005, 7, 1919–1922 CrossRef CAS PubMed.
- (a) D. Uraguchi, T. Ito and T. Ooi, J. Am. Chem. Soc., 2009, 131, 3836–3837 CrossRef CAS PubMed; (b) D. Uraguchi, T. Ito, S. Nakamura and T. Ooi, Chem. Sci., 2010, 1, 488–490 RSC.
- (a) D. Uraguchi and T. Ooi, J. Synth. Org. Chem., Jpn., 2010, 68, 1185–1194 CrossRef CAS; (b) D. Uraguchi, K. Yoshioka, Y. Ueki and T. Ooi, J. Am. Chem. Soc., 2012, 134, 19370–19373 CrossRef CAS PubMed; (c) D. Uraguchi, R. Tsutsumi and T. Ooi, J. Am. Chem. Soc., 2013, 135, 8161–8164 CrossRef CAS PubMed; (d) D. Uraguchi, S. Nakamura, H. Sasaki, Y. Konakade and T. Ooi, Chem. Commun., 2014, 50, 3491–3493 RSC; (e) H. Krawczyk, M. Dzięgielewski, D. Deredas, A. Albrecht and Ł. Albrecht, Chem.–Eur. J., 2015, 21, 10268–10277 CrossRef CAS PubMed.
- Phosphonate–phosphate rearrangement from acyl phosphonates: (a) C. C. Bausch and J. S. Johnson, Adv. Synth. Catal., 2005, 347, 1207–1211 CrossRef CAS PubMed; (b) A. S. Demir, Ö. Reis, A. Ç. İğdir, İ. Esiringü and S. Eymur, J. Org. Chem., 2005, 70, 10584–10587 CrossRef CAS PubMed ; Phosphonate–phosphate rearrangements from α-hydroxy trialkyl phosphonoacetates; (c) M. T. Corbett, D. Uraguchi, T. Ooi and J. S. Johnson, Angew. Chem., Int. Ed., 2012, 51, 4685–4689 CrossRef CAS PubMed ; Addition of phosphite to α-keto amides followed by phosphonate–phosphate rearrangement and intramolecular trapping; (d) A. Kondoh, T. Aoki and M. Terada, Org. Lett., 2014, 16, 3528–3531 CrossRef CAS PubMed ; Phosphite addition and rearrangement with protonation; (e) M. Hayashi and S. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 2249–2252 CrossRef CAS PubMed; (f) L. Wang, Z. Yao, F. Xu and Q. Shen, Heteroat. Chem., 2012, 23, 449–456 CrossRef CAS PubMed; (g) W. Fang, G.-G. Liu, X.-F. Huang, J. Jia and X.-W. Wang, Chin. J. Org. Chem., 2014, 34, 1177–1182 CrossRef CAS.
- For example: (a) J. K. Whitesell, A. Bhattacharya, D. A. Aguilar and K. Henke, J. Chem. Soc., Chem. Commun., 1982, 989–990 RSC; (b) D. A. Evans, C. S. Burgey, M. C. Kozlowski and S. W. Tregay, J. Am. Chem. Soc., 1999, 121, 686–699 CrossRef CAS; (c) L. Peng, L.-L. Wang, J.-F. Bai, L.-N. Jia, Q.-C. Yang, Q.-C. Huang, X.-Y. Xu and L.-X. Wang, Tetrahedron Lett., 2011, 52, 1157–1160 CrossRef CAS PubMed; (d) C. G. Goodman, M. M. Walker and J. S. Johnson, J. Am. Chem. Soc., 2015, 137, 122–125 CrossRef CAS PubMed.
- With respect to aldehyde electrophiles, we examined linear and alpha-branched alkyl aldehydes and observed slow conversion affording a mixture of unidentified products, together with a certain amount of the aldehyde Pudovik product and the desired mono-protected diol.
- We studied a number of cinchona alkaloid-derived catalysts, as well as hydrogen-bonding catalysts of other types (ureas, thioureas, cyclopropyl imines) and found low levels of stereoselectivity and unacceptable isolated yields.
- We were able to detect the formation of the Pudovik adduct with larger phosphites, implicating steric effects as being important in determining the rate of phosphonate–phosphate rearrangement.
- Crystallographic data (excluding structure factors) for phosphate 4j have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-1055582†.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterizations of compounds. CCDC 1055582. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02170g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |