
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper-catalyzed condensation of imines and α-diazo-β-dicarbonyl compounds: modular and regiocontrolled synthesis of multisubstituted pyrroles†
Wei Wen
Tan
and
Naohiko
Yoshikai
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: nyoshikai@ntu.edu.sg.
First published on 3rd August 2015
Abstract
In the presence of a copper(II) catalyst, enolizable imines bearing various N-substituents and α-diazo-β-ketoesters undergo denitrogenative and dehydrative condensation to afford highly substituted pyrroles in moderate to good yields with exclusive regioselectivity. The reaction likely involves nucleophilic addition of the imine nitrogen to a copper carbenoid, tautomerization of the resulting azomethine ylide to an α-enaminoketone, and a subsequent enamine–ketone cyclocondensation. With Yb(OTf)3 as a unique cocatalyst, α-diazo-β-diketones also participate in the same condensation reaction. The present reaction is applicable to acyclic, exocyclic, and endocyclic imines with tolerance of a broad range of functional groups and heterocyclic moieties, thus opening a new convenient route for the synthesis of the lamellarin family of natural products.
Introduction
Transition metal carbenoids, generated through the decomposition of diazocarbonyl compounds, have been proven to serve as extremely versatile electrophilic intermediates for organic synthesis.1 Among the various carbenoid transformations, reactions using aldimines bearing N-aryl or N-alkyl substituents as nucleophilic reaction partners have been extensively explored as they offer useful methods for the synthesis of nitrogen-containing heterocycles (Scheme 1a). In a prototypical reaction pattern, the nucleophilic addition of the aldimine nitrogen atom to the carbenoid gives rise to a metal-bound or a free azomethine ylide.2 The azomethine ylide then undergoes intramolecular cyclization to afford an aziridine derivative—a process that can be made enantioselective using a chiral metal catalyst.3 The azomethine ylide can also be intercepted by an appropriate dipolarophile, such as an electron-deficient alkene or alkyne, thus affording a pyrrolidine or related heterocycle through [3 + 2] cycloaddition.4,5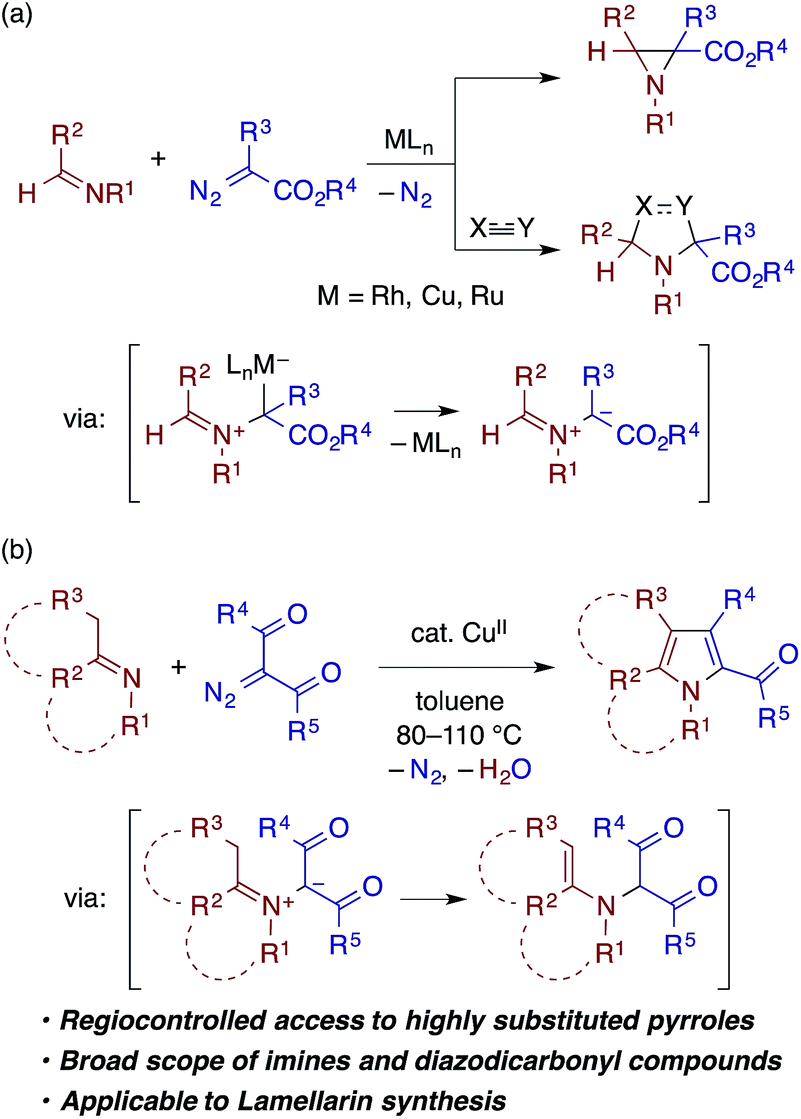 | ||
| Scheme 1 Transition metal-catalyzed condensation of imines and diazocarbonyl compounds. | ||
In contrast to the extensive studies on the carbenoid reactions with aldimines, reports on transition metal-catalyzed reactions of ketimines with diazocarbonyl compounds are very rare. While isatin-derived ketimines were reported to react with a rhodium carbenoid derived from diazomalonate to generate azomethine ylides for [3 + 2] cycloaddition,6 in other examples, ketimines do not directly react with a metal carbenoid but undergo [2 + 2] cycloaddition with a ketene generated through a Wolff rearrangement of the carbenoid.7 To our knowledge, reactions of other types of ketimines, including those bearing α-protons, with metal carbenoids have not been documented in the literature. In pursuit of heterocycle synthesis through the transition metal catalysis of ketimines,8 we have found that a ketimine derived from an enolizable ketone participates in the reaction with an α-diazo-β-ketoester (or diketone) under copper catalysis to afford a multisubstituted pyrrole with the concomitant loss of dinitrogen and water (Scheme 1b), which is reported herein. The reaction is considered to involve the nucleophilic addition of the ketimine nitrogen to a copper carbenoid and the tautomerization of the resulting azomethine ylide to an α-enamino-β-dicarbonyl intermediate, which then undergoes dehydrative cyclocondensation to give the pyrrole product. The reaction is applicable to ketimines with various skeletons and N-substituents, and features a simple catalytic system and operation.
Multisubstituted pyrroles are present in many natural products, pharmaceutically relevant compounds, and other functional molecules (Chart 1).9 While a number of new synthetic methods for multisubstituted pyrroles, those catalyzed by transition metals in particular, have been developed in the last decades,10,11 the demand for simple, robust, and sustainable methods remains high. Notably, in many of the recent transition metal-catalyzed methods, the catalyst plays a key role in the formation of linear intermediates such as α-enaminoketone,12 γ-ketoimine (or its tautomers),13 and 1,4-diimine,14 which undergo dehydrative or deaminative cyclocondensation to afford the pyrrole products. In this context, the present reaction represents a useful addition to the synthetic repertoire for pyrroles, because the substitution patterns of the key α-enamino-β-dicarbonyl intermediates are otherwise not readily accessible.15 As such, the present reaction enables the modular and expeditious preparation of dozens of new multisubstituted pyrroles and also opens a new alternative route for the synthesis of the lamellarin family of natural products.
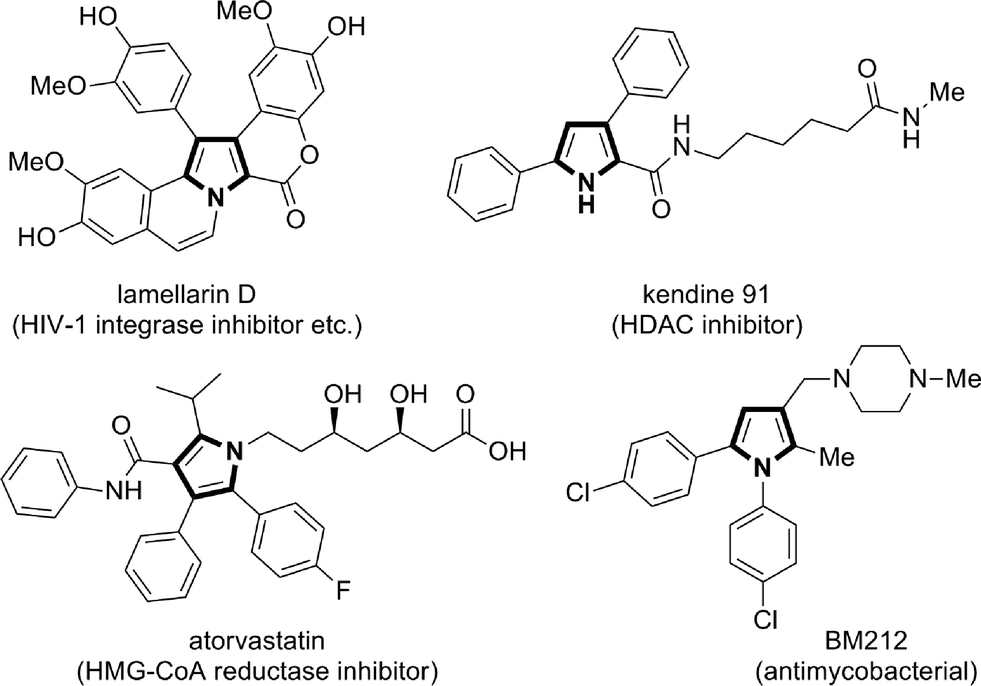 | ||
| Chart 1 Examples of pyrrole-containing bioactive molecules. | ||
Results and discussion
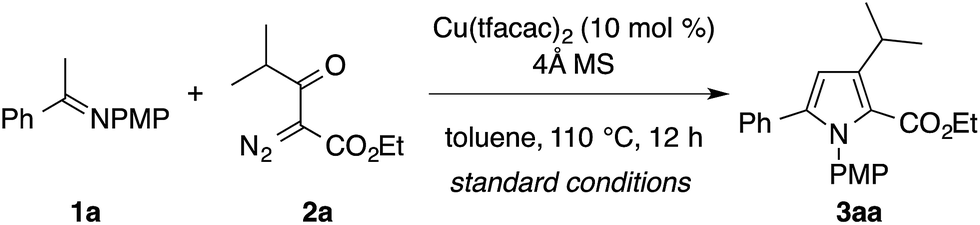
The present study commenced with attempts on the condensation of imine 1a, derived from acetophenone and p-anisidine, with α-diazo-β-ketoester 2a, derived from ethyl isobutyrylacetate (Table 1). Upon the screening of various reaction conditions, the desired condensation was found to proceed efficiently in the presence of catalytic copper(II) trifluoroacetylacetonate (Cu(tfacac)2, 10 mol%) and 4 Å molecular sieves (MS) in toluene at 110 °C to afford the tetrasubstituted pyrrole 3aa as the exclusive regioisomer in 88% yield (entry 1). The regiochemistry of 3aa was confirmed by two-dimensional NMR (HMQC and HMBC) analysis. While copper(II) hexafluoroacetylacetonate (Cu(hfacac)2) showed a comparable catalytic activity to Cu(tfacac)2 (entry 2), other copper salts such as Cu(acac)2 and Cu(OAc)2 were much less effective (entries 3 and 4). The use of Rh2(OAc)4 resulted in the formation of an intractable mixture of products, in which the desired product 3aa was not detected (entry 5). The yield of 3aa diminished substantially in the absence of the 4 Å MS (entry 6). The reduction of the catalyst loading (to 5 mol%) or the reaction temperature (to 90 °C) resulted in slightly lower yields (entries 7 and 8). 1,2-Dichloroethane (DCE) can be used as an alternative solvent (entry 9), while the reaction was completely shut down in DMSO (entry 10). Note that the formation of an aziridine product was not observed during the optimization study.3a–e|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yieldb (%) |
| a The reaction was performed using 0.2 mmol of 1a and 0.3 mmol of 2a. PMP = p-methoxyphenyl. Cu(tfacac)2 = copper(II) trifluoroacetylacetonate. Cu(hfacac)2 = copper(II) hexafluoroacetylacetonate. b Determined by GC. c Isolated yield. | ||
| 1 | None | 88c |
| 2 | Cu(hfacac)2 instead of Cu(tfacac)2 | 87 |
| 3 | Cu(acac)2 instead of Cu(tfacac)2 | 7 |
| 4 | Cu(OAc)2 instead of Cu(tfacac)2 | 21 |
| 5 | Rh2(OAc)4 instead of Cu(tfacac)2 | 0 |
| 6 | Without 4 Å MS | 52 |
| 7 | 5 mol% of Cu(tfacac)2 | 76 |
| 8 | T = 90 °C | 82 |
| 9 | DCE instead of toluene | 81 |
| 10 | DMSO instead of toluene | 0 |
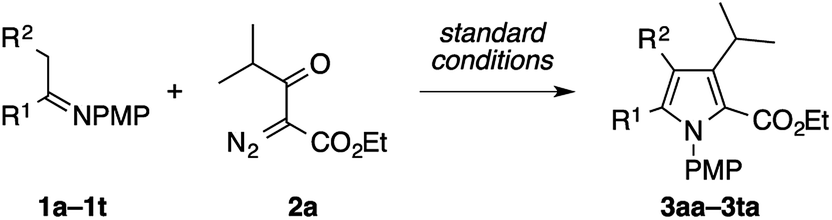
To explore the scope of the present pyrrole synthesis, we first subjected various N-PMP ketimines 1a–1u to the reaction with the diazoester 2a (Table 2). The imines 1a–1o derived from a wide variety of (hetero)aryl methyl ketones could be condensed with 2a, affording the tetrasubstituted pyrroles 3aa–3oa in moderate to good yields. The X-ray diffraction analysis of single crystals of 3ja unambiguously confirmed its regiochemistry. The reaction of the parent acetophenone imine 1a could be performed on a gram (5 mmol) scale without problem (see 3aa). Various functional groups, such as halogen (F, Cl, Br, I), trifluoromethyl, cyano, and nitro groups, as well as heteroaryl moieties such as furyl, benzofuryl, thienyl, and indolyl groups, were tolerated. The imines 1p–1t derived from propiophenone, 2-phenylacetophenone, cyclopropyl methyl ketone, and cycloalkanones also participated in the reaction with 2a to afford the corresponding penta- or tetrasubstituted pyrroles 3pa–3ta in moderate to good yields.
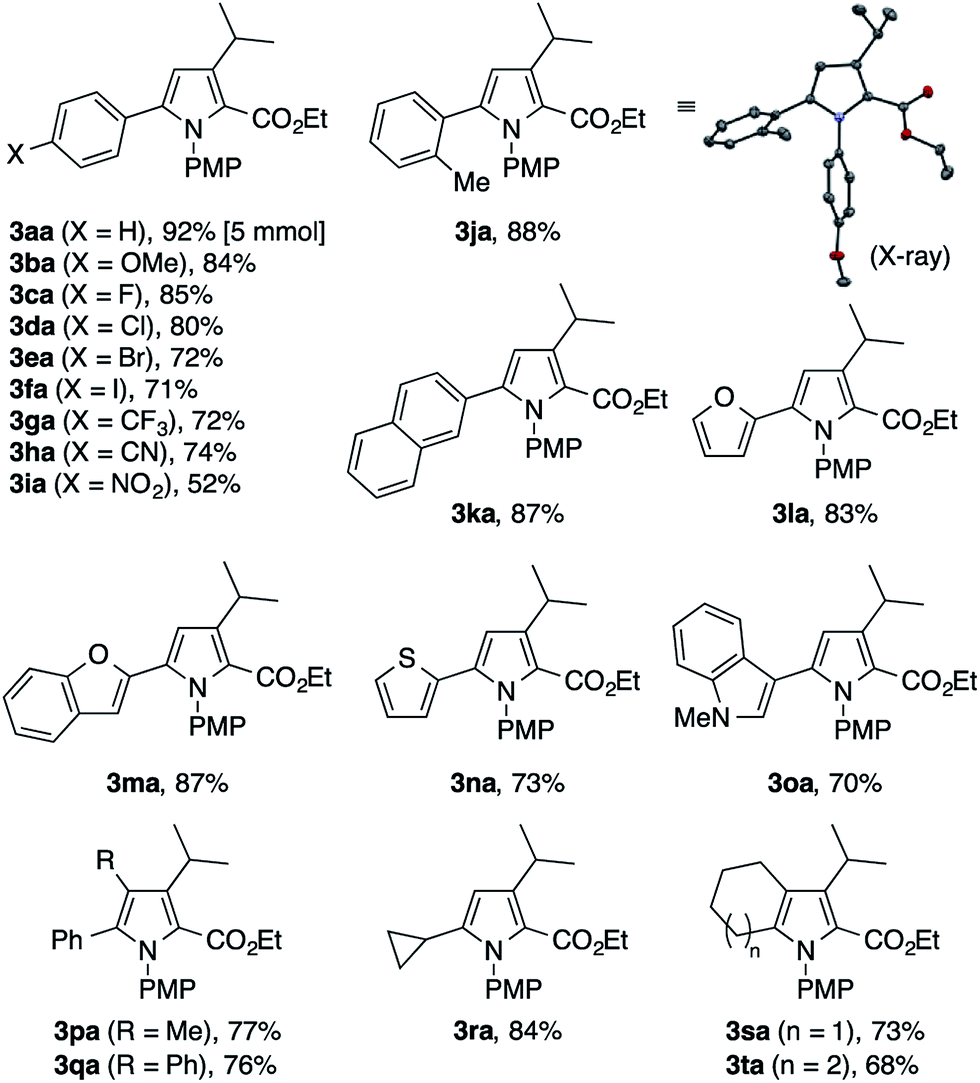
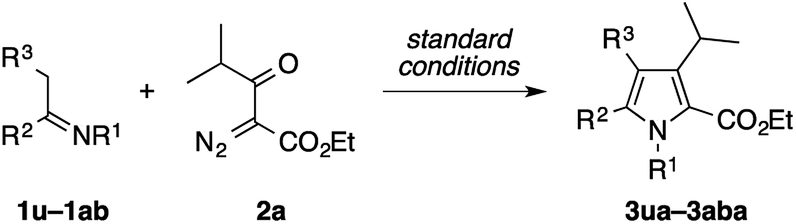
The present condensation reaction is applicable to imines bearing N-substituents other than the PMP group, as illustrated in Table 3. The reaction of the acetophenone imine bearing the N-4-chlorophenyl group (1u) with 2a afforded the desired product 3ua in 42% yield, which was markedly lower than that obtained with the N-PMP imine 1a. This suggests the relevance of the electron-richness of the nitrogen atom to the reactivity of the imine. The imines 1v–1z bearing removable benzyl, 4-methoxybenzyl (PMB), and allyl groups smoothly participated in the reaction with 2a to afford the pyrroles 3va–3za in respectable yields of 57–73%, thus making the preparation of N–H pyrroles feasible. For example, the removal of the PMB group of 3ya was achieved in 90% yield with the aid of trifluoroacetic acid and anisole. The methyl-substituted dihydroisoquinoline 1aa, readily prepared by the Bischler–Napieralski reaction, was also amenable to the condensation with 2a to afford a 5,6-dihydropyrrolo[2,1-a]isoquinoline derivative 3aaa, implying the potential applicability of the present method to the synthesis of the lamellarin family of natural products (vide infra).9b–c Interestingly, the benzyl-substituted dihydroisoquinoline 1ab afforded a mixture of 5,6-dihydropyrrolo[2,1-a]isoquinoline 3aba and an unexpected dihydrobenz[g]indolizinone derivative 3aba′.16 The latter product features the migration of the phenyl group of 1ab from the β-position of the nitrogen atom to the α-position, as well as the concomitant oxidation of the β-position. We consider that these structural changes occur on the reaction pathway leading to the pyrrole product 3aba, rather than after the formation of 3aba, because the ratio of 3aba and 3aba′ was not affected by the reaction time (see Scheme S1 in the ESI† for a possible mechanism).
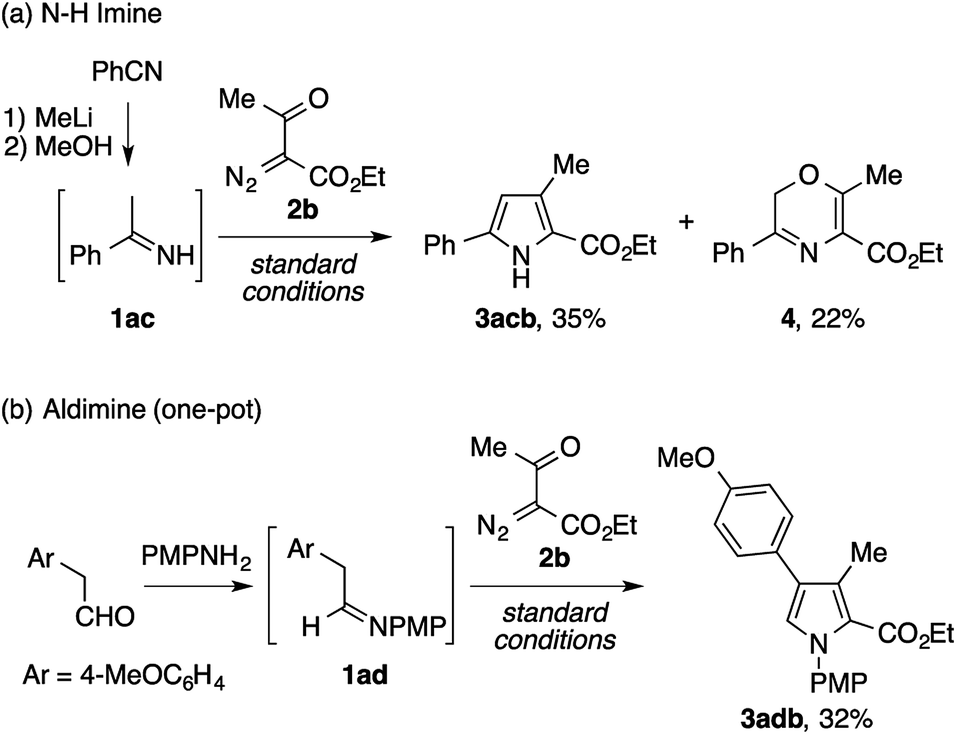
Besides the isolable imines described above, we also examined the viability of unstable imines as reactants for the present pyrrole synthesis (Scheme 2). First, an N–H imine of acetophenone 1ac, prepared from benzonitrile and methyllithium with minimal workup, was subjected to the reaction with ethyl acetyldiazoacetate 2b, which afforded the desired N–H pyrrole 3acb in a modest yield (Scheme 4a). Interestingly, the reaction was accompanied by the formation of a 2H-1,4-oxazine derivative 4 as a byproduct. Next, the condensation of 4-methoxyphenylacetaldehyde and p-anisidine and the subsequent reaction of the resulting aldimine 1ad and 2b were performed in a one-pot manner. Although the first condensation step was inevitably complicated by side reactions such as the enamine aldol reaction, the desired tetrasubstituted pyrrole 3adb was obtained in a modest yield (Scheme 4b).
 | ||
| Scheme 2 Condensation of unstable imines with diazoketoester 2b. | ||
During the exploration of the scope of the imines, we encountered a few cases of an unexpected mode of cyclization (Scheme 3). The reaction of the pinacolone-derived imine 1ae with 2a cleanly furnished a 2,3-dihydrooxazole derivative 5a, rather than the expected pyrrole. The same reaction was also observed using the trifluoroacetone-derived imine 1af.
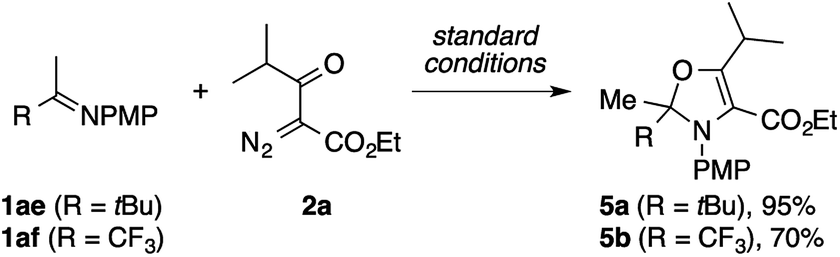 | ||
| Scheme 3 Formation of dihydrooxazole derivatives. | ||
We next explored the reaction of imine 1a with various α-diazo-β-ketoesters 2b–2p (Table 4). The reaction allowed the facile preparation of tetrasubstituted pyrroles bearing alkyl (entries 1–3), benzyl (entry 4), and (hetero)aryl (entries 5–12) groups in moderate to good yields, with tolerance of functional groups such as bromo and nitro groups (entries 7, 8, and 10). Diazo compounds derived from diethyl 2-oxosuccinate and ethyl 4,4,4-trifluoroacetoacetate could also be condensed with 1a, thus furnishing 2,3-diethoxycarbonylpyrrole 3an and 2-ethoxycarbonyl-3-trifluoromethylpyrrole 3ao, respectively (entries 13 and 14). In addition to the 1,2,3,5-tetrasubstituted pyrroles 3ab–3ao, the 1,2,5-trisubstituted pyrrole 3ap was also successfully prepared in a moderate yield using ethyl formyldiazoacetate 2p as the reactant (entry 15).
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yield (%) |
| a The reaction was performed on a 0.2 mmol scale according to the standard conditions described in Table 1. | ||||
| 1 | Me | Et | 3ab | 81 |
| 2 | c-C3H5 | Et | 3ac | 80 |
| 3 | c-C6H11 | Me | 3ad | 88 |
| 4 | Bn | Me | 3ae | 94 |
| 5 | Ph | Et | 3af | 77 |
| 6 | 4-MeOC6H4 | Et | 3ag | 77 |
| 7 | 4-BrC6H4 | Et | 3ah | 86 |
| 8 | 4-NO2C6H4 | Et | 3ai | 80 |
| 9 | 2-MeC6H4 | Et | 3aj | 67 |
| 10 | 2-BrC6H4 | Et | 3ak | 60 |
| 11 | 1-Naphthyl | Me | 3al | 74 |
| 12 | 2-Furyl | Me | 3am | 70 |
| 13 | CO2Et | Et | 3an | 79 |
| 14 | CF3 | Et | 3ao | 41 |
| 15 | H | Et | 3ap | 47 |
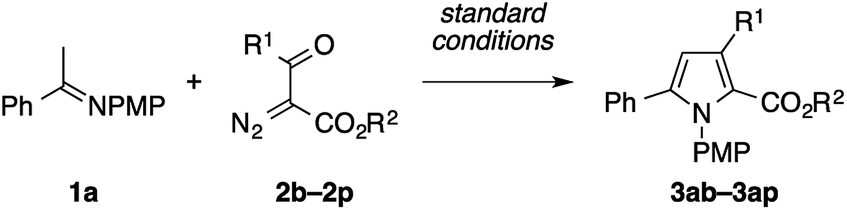
The reaction of 1a with 3-diazopentane-2,4-dione 2q under the standard conditions with Cu(tfacac)2 did not give the desired pyrrole product. The use of Cu(hfacac)2 instead of Cu(tfacac)2 promoted the reaction, but produced a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the expected pyrrole 3aq and its positional isomer 3aq′ in a low overall yield (Scheme 4a). The latter isomer would be formed through the formal C–H insertion of a copper carbenoid into the α-position of 1a. Interestingly, the efficiency and the chemoselectivity of this reaction were substantially improved by adding Yb(OTf)3 (10 mol%) and lowering the temperature to 80 °C.17 Thus, the 2-acetylpyrrole isomer 3aq was obtained almost exclusively in a respectable yield of 57%. The Cu/Yb cocatalytic system also effected the condensation of 1a with 1-diazo-1-benzoylacetone 2r in the same mode of cyclization, in favor of the 2-benzoylpyrrole isomer 3ar over the 2-acetylpyrrole isomer 3ar′ (Scheme 4b). Note that the same reaction in the absence of Yb(OTf)3 produced a mixture of four isomers, including 3ar and 3ar′ in a low overall yield, as suggested by the GC analysis of the crude product. While the role of Yb(OTf)3 is not clear, we speculate that it serves as a Lewis acid to the diketo moiety to enhance the electrophilicity of the copper carbenoid. Note that the α-diazoketones, such as 2-diazo-1,2-diphenylethanone, decomposed too quickly not only under the standard conditions, but also under conditions employing the less reactive Cu(acac)2 at a lower temperature, hence producing none of the desired pyrrole product.
:1 mixture of the expected pyrrole 3aq and its positional isomer 3aq′ in a low overall yield (Scheme 4a). The latter isomer would be formed through the formal C–H insertion of a copper carbenoid into the α-position of 1a. Interestingly, the efficiency and the chemoselectivity of this reaction were substantially improved by adding Yb(OTf)3 (10 mol%) and lowering the temperature to 80 °C.17 Thus, the 2-acetylpyrrole isomer 3aq was obtained almost exclusively in a respectable yield of 57%. The Cu/Yb cocatalytic system also effected the condensation of 1a with 1-diazo-1-benzoylacetone 2r in the same mode of cyclization, in favor of the 2-benzoylpyrrole isomer 3ar over the 2-acetylpyrrole isomer 3ar′ (Scheme 4b). Note that the same reaction in the absence of Yb(OTf)3 produced a mixture of four isomers, including 3ar and 3ar′ in a low overall yield, as suggested by the GC analysis of the crude product. While the role of Yb(OTf)3 is not clear, we speculate that it serves as a Lewis acid to the diketo moiety to enhance the electrophilicity of the copper carbenoid. Note that the α-diazoketones, such as 2-diazo-1,2-diphenylethanone, decomposed too quickly not only under the standard conditions, but also under conditions employing the less reactive Cu(acac)2 at a lower temperature, hence producing none of the desired pyrrole product.
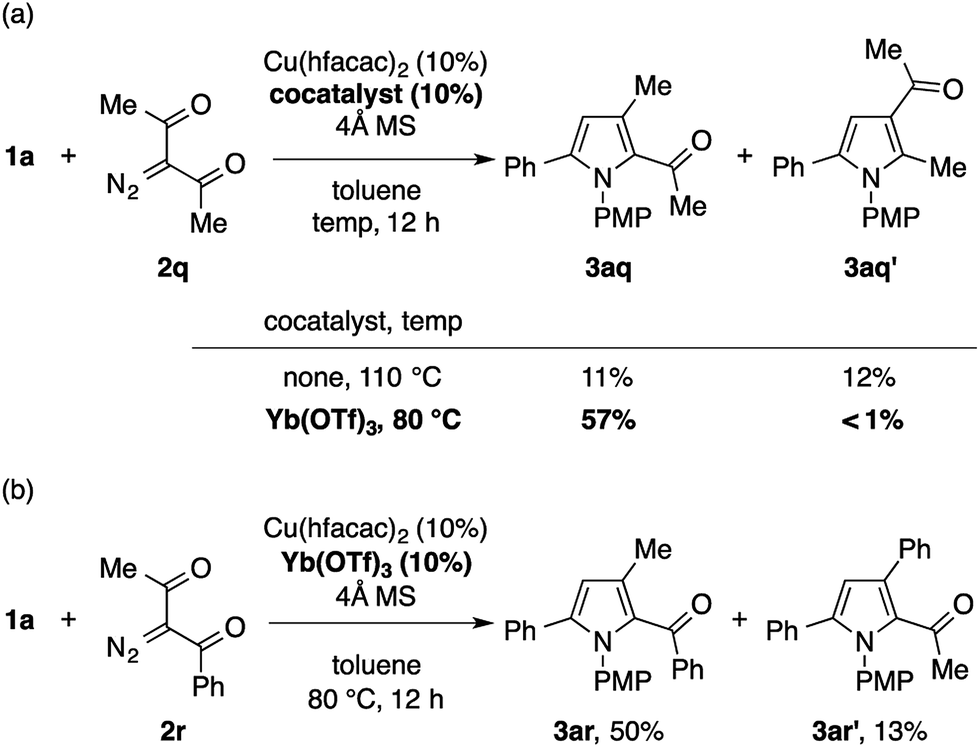 | ||
| Scheme 4 Condensation of 1a with α-diazo-β-diketones using a Yb(OTf)3 cocatalyst. | ||
Having established the scope and limitation with respect to imines and diazo compounds, we became interested in the applicability of the present condensation reaction to the synthesis of bioactive natural or unnatural products. In this connection, the successful reactions of the dihydroisoquinolines 1aa and 1ab (Table 3) turned our attention to the lamellarin family of natural products, many members of which contain a (5,6-dihydro)pyrrolo[2,1-a]isoquinoline skeleton. Since the early studies of Steglich, Ishibashi/Iwao, and Banwell,18 lamellarins have been popular synthetic targets due to the broad spectrum of their biological activities, and have also served as touchstones for new methods for the construction and the functionalization of pyrrole rings.9b–c,19 As some of the completed lamellarin syntheses involve 3-alkoxycarbonyl-5,6-dihydropyrrolo[2,1-a]isoquinoline derivatives as key intermediates, we wondered whether the present reaction offers an alternative and efficient route to such intermediates.
Aiming at a straightforward access to the polyarylated pyrrole structure of lamellarins, we first tested model reactions of methyl- and benzyldihydroisoquinolines 1aa and 1ab with ethyl benzoyldiazoacetate 2f (Scheme 5). The reaction of the former imine successfully furnished the desired pyrrole scaffold 3aaf in 62% yield (Scheme 5a). By contrast, the latter exclusively afforded a dihydrobenz[g]indolizinone derivative 6,16 the structure of which was unambiguously confirmed by X-ray crystallographic analysis (Scheme 5b).
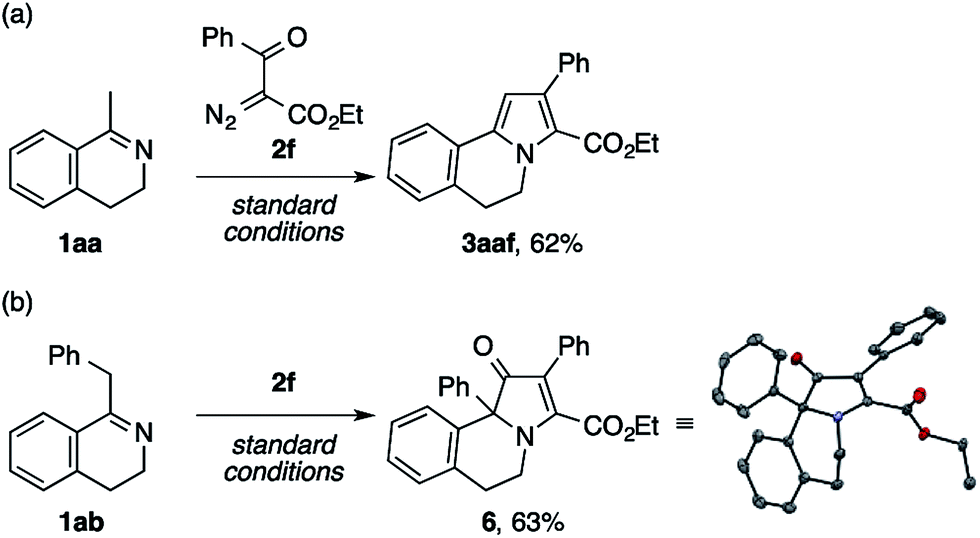 | ||
| Scheme 5 Model reactions of the dihydroisoquinolines 1aa and 1ab with ethyl benzoyldiazoacetate 2f. | ||
While the above results suggest that the present method may not be suitable for the direct assembly of the pentasubstituted pyrrole core of lamellarins, attempts using ethyl formyldiazoacetate 2p have proven its utility in the preparation of building blocks for the modular synthesis of lamellarins (Scheme 6). The reaction of methyldihydroisoquinoline with two methoxy groups (1ag) and 2p cleanly afforded the 3-ethoxycarbonyl-5,6-dihydropyrrolo[2,1-a]isoquinoline derivative 3agp (Scheme 6a). An analogous compound of 3agp was previously synthesized through a sequence of pyrrole N-alkylation and an intramolecular Heck reaction, and used as an intermediate to lamellarin D and its analogues.19b,20 Furthermore, benzyldihydroisoquinoline bearing four methoxy groups (1ah) also underwent condensation with 2p to afford the 1-aryl-3-ethoxycarbonyl-5,6-dihydropyrrolo[2,1-a]isoquinoline derivative 3ahp (Scheme 6b). In a previous study by Handy et al., 3ahp was prepared through the six-step manipulation of a pyrrole-based starting material, and was then converted to lamellarin G trimethyl ether in two steps.19a
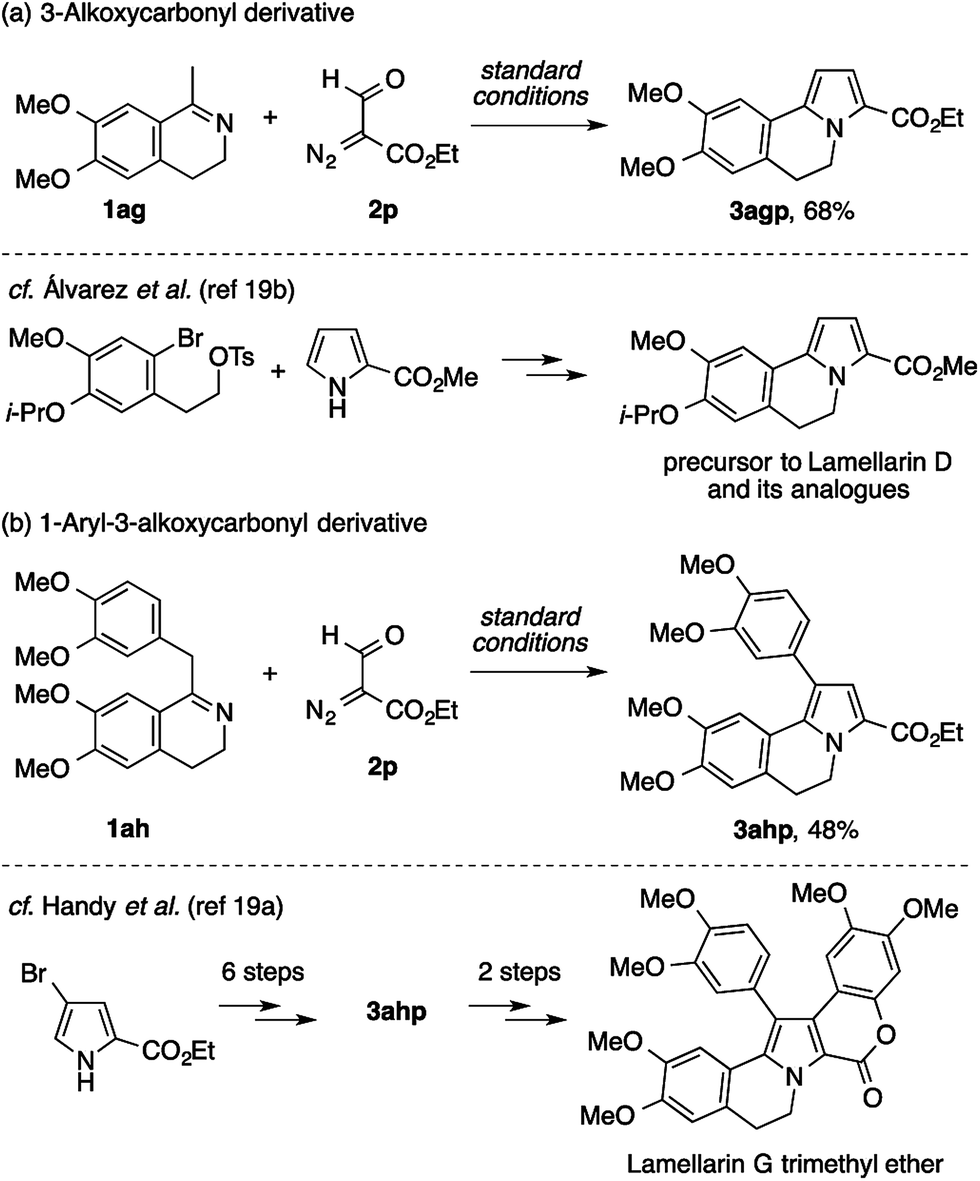 | ||
| Scheme 6 Construction of the 5,6-dihydropyrrolo[2,1-a]isoquinoline scaffolds for lamellarin synthesis. | ||
On the basis of the regiochemistry of the pyrrole products as well as common reaction patterns in the transition metal catalysis of diazo compounds, the present reaction is considered to involve the addition of the imine nitrogen to a copper carbenoid.2,3,4g To probe the nature of this putative step, competition experiments using imines bearing different substituents were performed. The reaction of a mixture of the imines 1b and 1h, derived from electron-rich and electron-poor acetophenones, respectively, with 2a afforded the product of the former (3ba) as the dominant product (Scheme 7a). Likewise, the reaction of a mixture of the imines 1a and 1u, derived from electron-rich and electron-poor anilines, respectively, preferentially produced the product of the former (Scheme 7b). Thus, the nucleophilicity of the imine nitrogen atom would play a critical role in its reaction with the copper carbenoid.
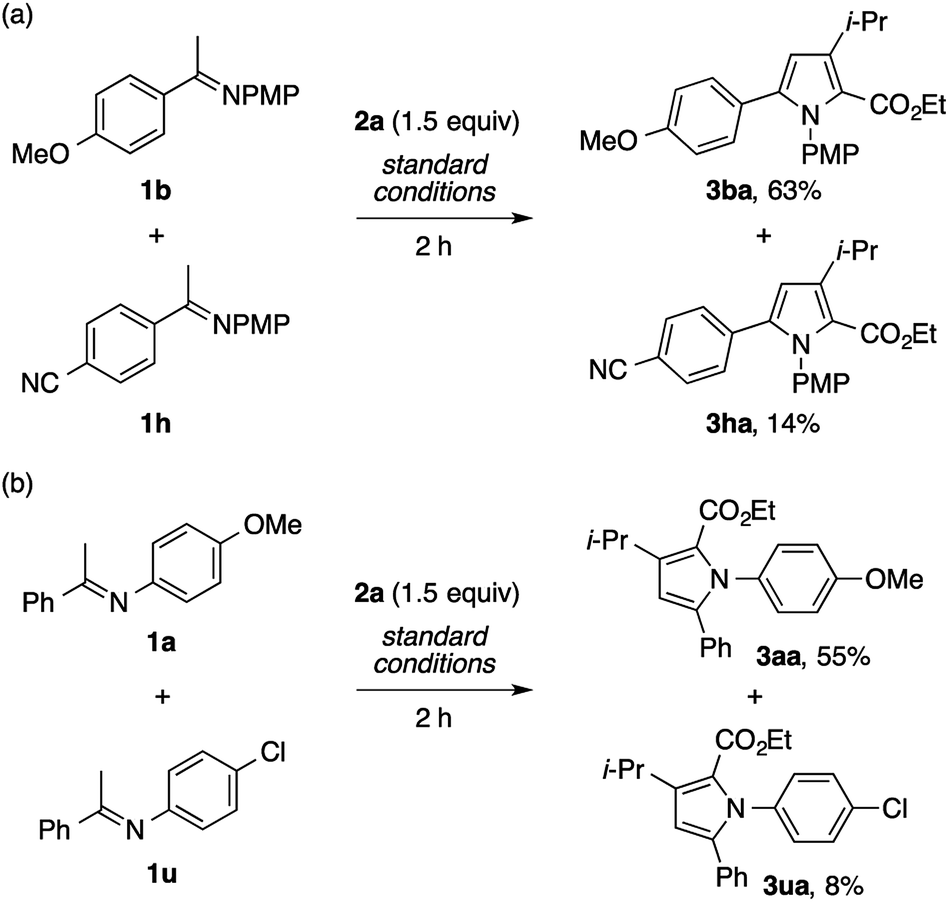 | ||
| Scheme 7 Competition experiments. | ||
Scheme 8 shows plausible reaction pathways of the present pyrrole synthesis. The decomposition of the diazo compound 2 with the copper catalyst generates an electrophilic copper carbenoid. Nucleophilic attack of the imine 1 to the carbenoid gives azomethine ylide I2–6 and regenerates the copper catalyst. The tautomerization of I to α-enaminoketone IIvia proton transfer is followed by cyclocondensation to afford the pyrrole product 3. Attempts to intercept the azomethine ylide I with typical dipolarophiles such as dimethyl acetylenedicarboxylate, dimethyl maleate, and N-phenylphthalimide failed to give the corresponding [3 + 2] cycloadducts, but exclusively afforded the pyrrole product, presumably because of the rapid tautomerization of I to II. Nevertheless, the intermediacy of I and II rationalizes not only the formation of the pyrrole 3, but also the side reactions observed in the present study. In the reaction of the N–H imine (Scheme 2), the intermediate II may be oxidized to the 1-acyl-2-azadiene III, which would then undergo a 6π electrocyclization to afford a 2H-1,4-oxazine derivative 4.21 The azomethine ylide I may also behave as an enolate/iminium bifunctional species I′, which undergoes an intramolecular attack of the enolate oxygen to the iminium moiety to afford the 2,3-dihydrooxazole 5. This side reaction would be operative in the cases shown in Scheme 3, possibly because of the lower acidity of the α-proton of the iminium moiety (for 1ae) or the increased electrophilicity of the iminium moiety (for 1af).
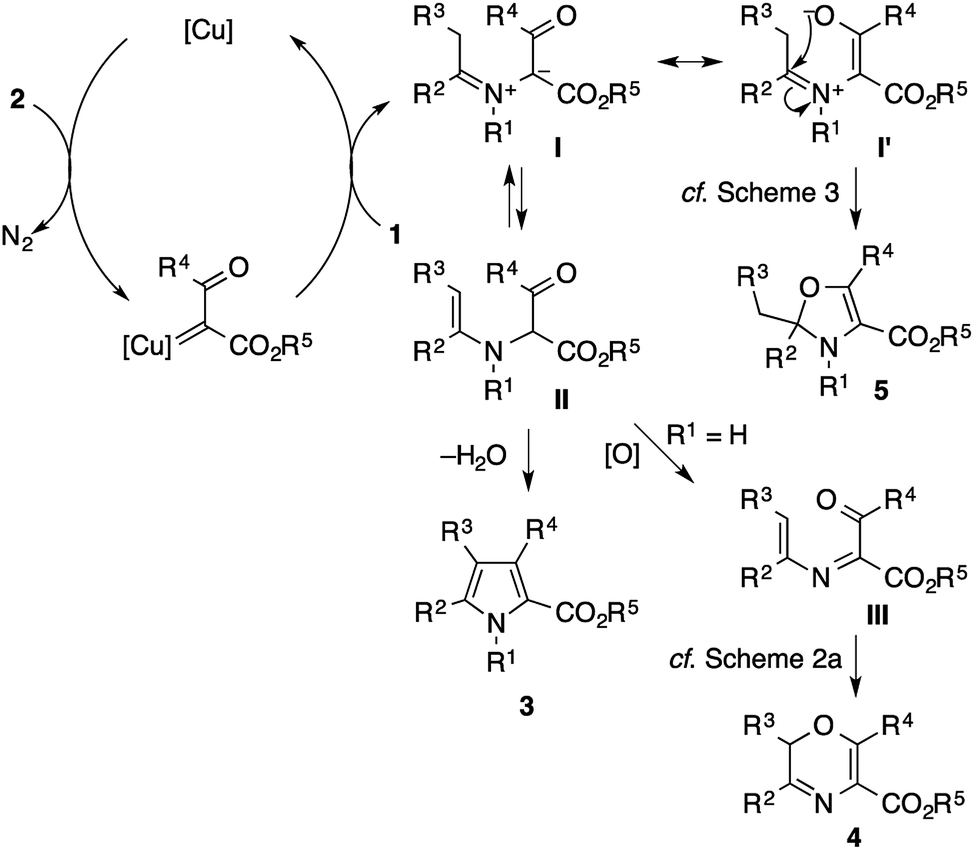 | ||
| Scheme 8 Plausible reaction pathways leading to pyrrole and other byproducts. | ||
Conclusions
In conclusion, we have developed a copper-catalyzed condensation reaction of imines and α-diazo-β-dicarbonyl compounds to afford multisubstituted pyrroles in a regiocontrolled manner. The reaction features a broad scope and the ready availability of starting materials and a simple operation, thus enabling the modular and quick preparation of a variety of densely functionalized pyrroles. Given the extensive previous studies on carbenoid reactions with imines,2–6 it is rather surprising that the present condensation of enolizable imines has not been documented. The reaction opens concise preparative routes to lamellarin scaffolds, and may find further applications in the synthesis of pyrrole alkaloids. The cooperative effect of the carbene transfer catalyst (Cu(hfacac)2) and the Lewis acid catalyst (Yb(OTf)3), observed in the reaction of α-diazo-β-diketones, also deserves mechanistic and synthetic explorations. Further studies on heterocycle synthesis using imines as starting materials8 and/or based on carbenoid chemistry are currently in progress in our laboratory.Acknowledgements
This work was supported by the Singapore National Research Foundation (NRF-RF2009-05) and Nanyang Technological University. We thank Dr Malleswara Rao Kuram for his early study and Drs Yongxin Li and Rakesh Ganguly for assistance with the X-ray crystallographic analysis.Notes and references
- (a) T. Ye and M. A. Mckervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS; (b) M. P. Doyle, M. A. Mckervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley-Interscience, New York, 1998 Search PubMed; (c) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (d) Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS PubMed.
- (a) A. Padwa and S. F. Hornbuckle, Chem. Rev., 1991, 91, 263 CrossRef CAS; (b) A. Padwa and M. D. Weingarten, Chem. Rev., 1996, 96, 223 CrossRef CAS PubMed.
- (a) K. G. Rasmussen and K. A. Jørgensen, J. Chem. Soc., Chem. Commun., 1995, 1401 RSC; (b) K. B. Hansen, N. S. Finney and E. N. Jacobsen, Angew. Chem., Int. Ed., 1995, 34, 676 CrossRef CAS PubMed; (c) M. Moran, G. Bernardinelli and P. Müller, Helv. Chim. Acta, 1995, 78, 2048 CrossRef CAS PubMed; (d) Y. Li, P. W. H. Chan, N.-Y. Zhu, C.-M. Che and H.-L. Kwong, Organometallics, 2004, 23, 54 CrossRef CAS; (e) X.-J. Zhang, M. Yan and D. Huang, Org. Biomol. Chem., 2009, 7, 187 RSC; (f) M. P. Doyle, W. H. Hu and D. J. Timmons, Org. Lett., 2001, 3, 933 CrossRef CAS PubMed.
- (a) A. Padwa, D. C. Dean, M. H. Osterhout, L. Precedo and M. A. Semones, J. Org. Chem., 1994, 59, 5347 CrossRef CAS; (b) G.-Y. Li, J. Chen, W.-Y. Yu, W. Hong and C.-M. Che, Org. Lett., 2003, 5, 2153 CrossRef CAS PubMed; (c) H.-W. Xu, G.-Y. Li, M.-K. Wong and C.-M. Che, Org. Lett., 2005, 7, 5349 CrossRef CAS PubMed; (d) M.-Z. Wang, H.-W. Xu, Y. Liu, M.-K. Wong and C.-M. Che, Adv. Synth. Catal., 2006, 348, 2391 CrossRef CAS PubMed; (e) C. V. Galliford and K. A. Scheidt, J. Org. Chem., 2007, 72, 1811 CrossRef CAS PubMed; (f) C. V. Galliford, J. S. Martenson, C. Stern and K. A. Scheidt, Chem. Commun., 2007, 631 RSC; (g) A. P. Kadina, A. F. Khlebnikov, M. S. Novikov, P. J. Perez and D. S. Yufit, Org. Biomol. Chem., 2012, 10, 5582 RSC.
- M. Yan, N. Jacobsen, W. Hu, L. S. Gronenberg, M. P. Doyle, J. T. Colyer and D. Bykowski, Angew. Chem., Int. Ed., 2004, 43, 6713 CrossRef CAS PubMed.
- T. Rajasekaran, G. Karthik, B. Sridhar and B. V. S. Reddy, Org. Lett., 2013, 15, 1512 CrossRef CAS PubMed.
- (a) B. V. S. Reddy, G. Karthik, T. Rajasekaran, A. Antony and B. Sridhar, Tetrahedron Lett., 2012, 53, 2396 CrossRef PubMed; (b) M. D. Mandler, P. M. Truong, P. Y. Zavalij and M. P. Doyle, Org. Lett., 2014, 16, 740 CrossRef CAS PubMed.
- (a) Y. Wei, I. Deb and N. Yoshikai, J. Am. Chem. Soc., 2012, 134, 9098 CrossRef CAS PubMed; (b) Z. Chen, B. Lu, Z. Ding, K. Gao and N. Yoshikai, Org. Lett., 2013, 15, 1966 CrossRef CAS PubMed; (c) B. Lu, J. Wu and N. Yoshikai, J. Am. Chem. Soc., 2014, 136, 11598 CrossRef CAS PubMed; (d) T. Yamakawa and N. Yoshikai, Org. Lett., 2013, 15, 196 CrossRef CAS PubMed.
- (a) M. d'Ischia, A. Napolitano and A. Pezzella, in Comprehensive Heterocyclic Chemistry III, Elsevier, Oxford, 2008, pp. 353–388 Search PubMed; (b) H. Fan, J. Peng, M. T. Hamann and J.-F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS PubMed; (c) T. Fukuda, F. Ishibashi and M. Iwao, Heterocycles, 2011, 83, 491 CrossRef CAS; (d) M. Biava, G. C. Porretta and F. Manetti, Mini-Rev. Med. Chem., 2007, 7, 65 CrossRef CAS; (e) D. Otaegui, A. Rodríguez-Gascón, A. Zubia, F. P. Cossío and J. L. Pedraz, Cancer Chemother. Pharmacol., 2009, 64, 153 CrossRef CAS PubMed; (f) I. S. Young, P. D. Thornton and A. Thompson, Nat. Prod. Rep., 2010, 27, 1801 RSC; (g) A. D. Yamaguchi, K. M. Chepiga, J. Yamaguchi, K. Itami and H. M. L. Davies, J. Am. Chem. Soc., 2015, 137, 644 CrossRef CAS PubMed.
- (a) J. Bergman and T. Janosik, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katrizky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, pp. 269–351 Search PubMed; (b) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2010, 39, 4402 RSC; (c) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2014, 43, 4633 RSC; (d) F. Bellina and R. Rossi, Tetrahedron, 2006, 62, 7213 CrossRef CAS PubMed; (e) C. Schmuck and D. Rupprecht, Synthesis, 2007, 3095 CrossRef CAS.
- (a) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed; (b) N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466 CrossRef CAS PubMed; (c) N. T. Patil and Y. Yamamoto, ARKIVOC, 2007, 121 CrossRef CAS; (d) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS PubMed.
- (a) Y. Aoyagi, T. Mizusaki, M. Shishikura, T. Komine, T. Yoshinaga, H. Inaba, A. Ohta and K. Takeya, Tetrahedron, 2006, 62, 8533 CrossRef CAS PubMed; (b) S. Michlik and R. Kempe, Nat. Chem., 2013, 5, 140 CrossRef CAS PubMed; (c) M. Zhang, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 597 CrossRef CAS PubMed; (d) M. Zhang, X. Fang, H. Neumann and M. Beller, J. Am. Chem. Soc., 2013, 135, 11384 CrossRef CAS PubMed; (e) D. Srimani, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 4012 CrossRef CAS PubMed; (f) X. Li, M. Chen, X. Xie, N. Sun, S. Li and Y. Liu, Org. Lett., 2015, 17, 2984 CrossRef CAS PubMed.
- (a) H. Takaya, S. Kojima and S.-i. Murahashi, Org. Lett., 2001, 3, 421 CrossRef CAS; (b) S. Chiba, Y.-F. Wang, G. Lapointe and K. Narasaka, Org. Lett., 2008, 10, 313 CrossRef CAS PubMed; (c) Y.-F. Wang, K. K. Toh, S. Chiba and K. Narasaka, Org. Lett., 2008, 10, 5019 CrossRef CAS PubMed; (d) S. Chiba, Y.-J. Xu and Y.-F. Wang, J. Am. Chem. Soc., 2009, 131, 12886 CrossRef CAS PubMed; (e) H.-Y. Wang, D. S. Mueller, R. M. Sachwani, H. N. Londino and L. L. Anderson, Org. Lett., 2010, 12, 2290 CrossRef CAS PubMed; (f) T. J. Donohoe, N. J. Race, J. F. Bower and C. K. A. Callens, Org. Lett., 2010, 12, 4094 CrossRef CAS PubMed; (g) H.-Y. Wang, D. S. Mueller, R. M. Sachwani, R. Kapadia, H. N. Londino and L. L. Anderson, J. Org. Chem., 2011, 76, 3203 CrossRef CAS PubMed; (h) S. Shafi, M. Kedziorek and K. Grela, Synlett, 2011, 124 CAS; (i) K. Iida, T. Miura, J. Ando and S. Saito, Org. Lett., 2013, 15, 1436 CrossRef CAS PubMed.
- (a) M. R. Rivero and S. L. Buchwald, Org. Lett., 2007, 9, 973 CrossRef CAS PubMed; (b) D. Ciez, Org. Lett., 2009, 11, 4282 CrossRef CAS PubMed; (c) Q. Li, A. Fan, Z. Lu, Y. Cui, W. Lin and Y. Jia, Org. Lett., 2010, 12, 4066 CrossRef CAS PubMed.
- (a) X. Qi, X. Xu and C.-M. Park, Chem. Commun., 2012, 48, 3996 RSC; (b) D. Hong, Y. Zhu, Y. Li, X. Lin, P. Lu and Y. Wang, Org. Lett., 2011, 13, 4668 CrossRef CAS PubMed; (c) X. Xu, M. O. Ratnikov, P. Y. Zavalij and M. P. Doyle, Org. Lett., 2011, 13, 6122 CrossRef CAS PubMed; (d) E. Lourdusamy, L. Yao and C.-M. Park, Angew. Chem., Int. Ed., 2010, 49, 7963 CrossRef CAS PubMed; (e) C. Dong, G. Deng and J. Wang, J. Org. Chem., 2006, 71, 5560 CrossRef CAS PubMed; (f) M. N. Eberlin and C. Kascheres, J. Org. Chem., 1988, 53, 2084 CrossRef CAS.
- (a) S. T. Heller, T. Kiho, A. R. H. Narayan and R. Sarpong, Angew. Chem., Int. Ed., 2013, 52, 11129 CrossRef CAS PubMed; (b) H. Mcnab and L. C. Monahan, J. Chem. Soc., Perkin Trans. 1, 1988, 869 RSC; (c) T. Eicher and D. Krause, Synthesis, 1986, 899 CrossRef CAS.
- Other metal triflates also changed the regioselectivity in favor of the product 3aq. See the ESI† for details.
- (a) A. Heim, A. Terpin and W. Steglich, Angew. Chem., Int. Ed., 1997, 36, 155 CrossRef CAS PubMed; (b) F. Ishibashi, Y. Miyazaki and M. Iwao, Tetrahedron, 1997, 53, 5951 CrossRef CAS; (c) M. Banwell, B. Flynn and D. Hockless, Chem. Commun., 1997, 2259 RSC.
- (a) S. T. Handy, Y. N. Zhang and H. Bregman, J. Org. Chem., 2004, 69, 2362 CrossRef CAS PubMed; (b) D. Pla, A. Marchal, C. A. Olsen, F. Albericio and M. Alvarez, J. Org. Chem., 2005, 70, 8231 CrossRef CAS PubMed; (c) P. Ploypradith, T. Petchmanee, P. Sahakitpichan, N. D. Litvinas and S. Ruchirawat, J. Org. Chem., 2006, 71, 9440 CrossRef CAS PubMed; (d) J. T. Gupton, E. J. Banner, M. D. Sartin, M. B. Coppock, J. E. Hempel, A. Kharlamova, D. C. Fisher, B. C. Giglio, K. L. Smith, M. J. Keough, T. M. Smith, R. P. F. Kanters, R. N. Dominey and J. A. Sikorski, Tetrahedron, 2008, 64, 5246 CrossRef CAS PubMed; (e) Q. Li, J. Jiang, A. Fan, Y. Cui and Y. Jia, Org. Lett., 2011, 13, 312 CrossRef CAS PubMed; (f) K. Hasse, A. C. Willis and M. G. Banwell, Eur. J. Org. Chem., 2011, 88 CrossRef CAS PubMed; (g) D. Imbri, J. Tauber and T. Opatz, Chem.–Eur. J., 2013, 19, 15080 CrossRef CAS PubMed; (h) M. Komatsubara, T. Umeki, T. Fukuda and M. Iwao, J. Org. Chem., 2014, 79, 529 CrossRef CAS PubMed; (i) K. Ueda, K. Amaike, R. M. Maceiczyk, K. Itami and J. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 13226 CrossRef CAS PubMed.
- D. Pla, A. Marchal, C. A. Olsen, A. Francesch, C. Cuevas, F. Albericio and M. Alvarez, J. Med. Chem., 2006, 49, 3257 CrossRef CAS PubMed.
- V. A. Khlebnikov, M. S. Novikov, A. F. Khlebnikov and N. V. Rostovskii, Tetrahedron Lett., 2009, 50, 6509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1040843 and 1063222. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02322j |
| This journal is © The Royal Society of Chemistry 2015 |