
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem 1,2-sulfur migration and (aza)-Diels–Alder reaction of β-thio-α-diazoimines: rhodium catalyzed synthesis of (fused)-polyhydropyridines, and cyclohexenes†
Dongari
Yadagiri
and
Pazhamalai
Anbarasan
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: anbarasansp@iitm.ac.in
First published on 10th July 2015
Abstract
Rhodium catalyzed synthesis of substituted tetrahydropyridines was accomplished from readily accessible thio-tethered N-sulfonyl-1,2,3-triazoles. The reaction involves tandem rhodium catalyzed 1,2-sulfur migration in β-thio-α-diazoimines, generated from thio-tethered N-sulfonyl-1,2,3-triazoles, to thio-substituted 1-azadiene and subsequent self aza-Diels–Alder reaction. Interestingly, the methodology was effectively extended to the synthesis of fused tetrahydropyridines, dihydropyridines and cyclohexenes through the in situ trapping of the intermediate, 1-azadiene, with various dienophiles such as enol ether, enamine, ketene S,S-acetal, alkyne, alkene and diene. Furthermore, the direct conversion of propargyl sulfides to (fused)-tetrahydropyridines was also achieved through the successful integration of copper and rhodium catalysts in one-pot.
Introduction
1,2-Migration of atoms or groups is one of the fundamental reactions in organic synthesis. Particularly, 1,2-sulfur migration in various 1,2-functionalized thio-derivatives is an important tool for the construction of sulfur-containing building blocks and heterocycles.1 For example, rhodium catalyzed 1,2-sulfur migrations in α-thio carbenes were documented for the synthesis of α-thio-α,β-unsaturated carbonyl compounds2 and 3-alkoxycarbonyl β-lactam derivatives from β-thio-α-diazo carbonyl compounds.3 Although 1,2-sulfur migrations in α-thio metalcarbenes have been reported, they are rather limited to only β-thio-α-diazo carbonyl compounds.In the past few years, the use of readily accessible N-sulfonyl-1,2,3-triazoles4 as a source of α-diazoimines has been greatly exploited in organic synthesis as a vital reactive intermediate.5 Unlike traditional α-oxo metal carbenoids, metal carbenoids generated from α-diazoimines possess both an electrophilic carbon atom and nucleophilic nitrogen atom, which allows interesting inter/intramolecular reactions with various functional groups, like nitriles,6 alkynes,7 alkenes,8 enol ethers9/enamines,10 carbonyl compounds,11 cumulenes,12 C–H,13 X–H14 and C–X15 bonds and other carbene induced transformations to functionalized carbo(hetero)cycles16 and heteroatom-based building blocks.17 Interestingly, 1,2-hydride or alkyl migrations onto the electrophilic carbenoid carbon of α-imino metalcarbenes were also reported.18 Our contribution in this interesting field includes [2,3]-sigmatropic rearrangement with aryl allyl sulfides,15a chemo- and regioselective insertion into the para C–H bond of aniline derivatives13c and transannulation with enol ethers.9b Based on our continuous interest in the functionalization of N-sulfonyl-1,2,3-triazoles and potential 1,2-sulfur rearrangement, we envisioned rhodium catalyzed 1,2-sulfur migration in β-thio-α-diazoimines, generated from suitably substituted thio-tethered N-sulfonyl-1,2,3-triazoles, for the synthesis of thio-substituted 1-azadienes (Scheme 1).19
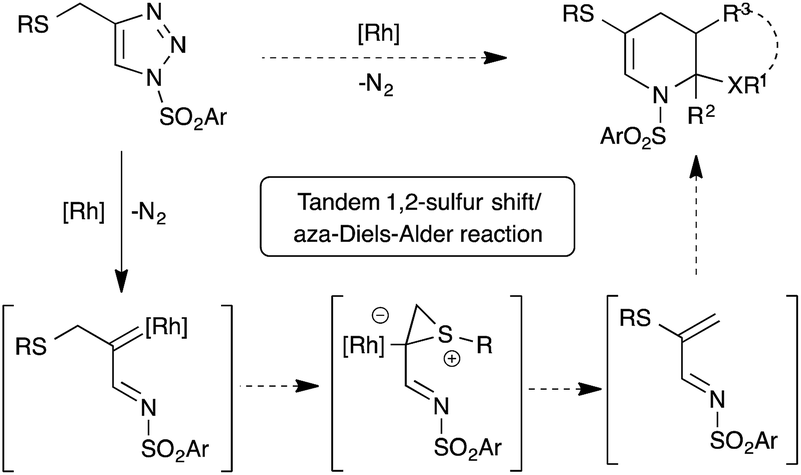 | ||
| Scheme 1 Rhodium catalyzed tandem 1,2-sulfur migration and aza-Diels–Alder reaction of β-thio-α-diazoimines derived from N-sulfonyl-1,2,3-triazoles. | ||
Thio-substituted 1-azadienes are potential building blocks for the synthesis of various polyhydropyridines20via (aza)-Diels–Alder reaction21 (Scheme 1). These substituted polyhydropyridines are the most common heterocyclic scaffolds encountered in naturally occurring bioactive compounds and pharmaceutical drugs (Fig. 1).22 In addition, they are also exploited as vital intermediates for the construction of piperidine and pyridine derivatives and organic hydride donors in biomimetic asymmetric transfer hydrogenation reactions.23 Hence, development of new strategies to access these important scaffolds is of continued interest in contemporary organic and medicinal chemistry. We herein disclose the rhodium catalyzed synthesis of substituted polyhydropyridines from readily accessible thio-tethered N-sulfonyl-1,2,3-triazoles via tandem 1,2-sulfur migration and (aza)-Diels–Alder reaction.
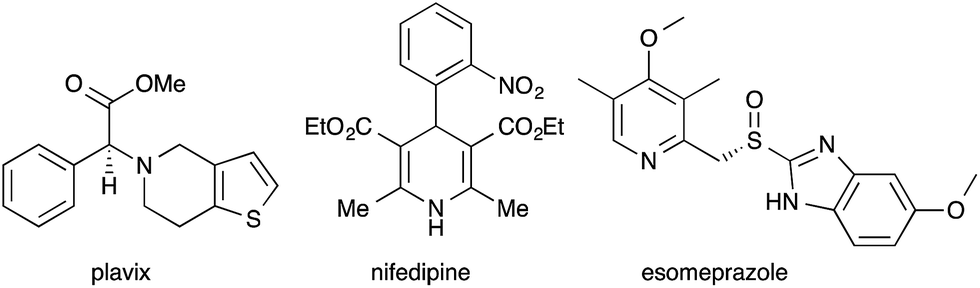 | ||
| Fig. 1 Representative examples of commercialized polyhydropyridine and pyridine derivatives. | ||
Results and discussion
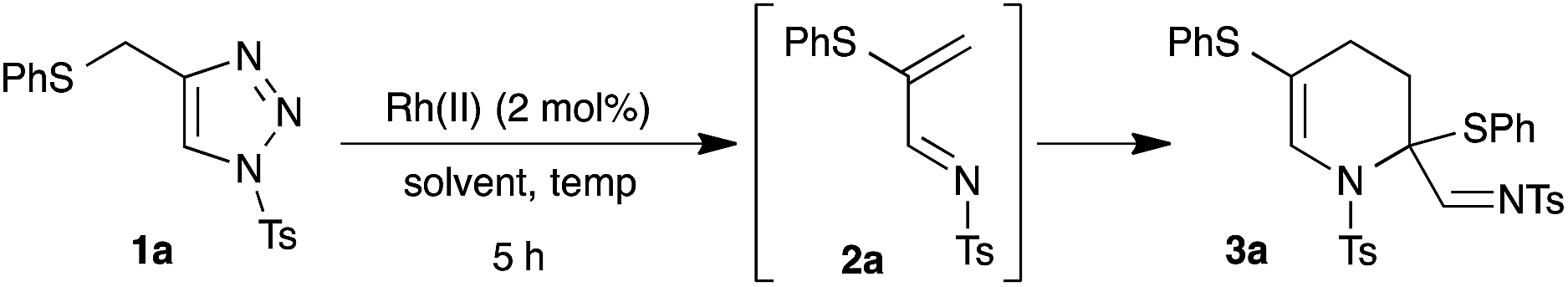
To test our hypothesis, we synthesized thio-tethered N-sulfonyl-1,2,3-triazole 1a from phenyl propargyl sulfide and tosyl azide under CuAAC conditions4a,4b and 1a was used as a suitable model substrate. The initial reaction of 1a with 2 mol% of rhodium acetate at 75 °C in CHCl3 for 5 h directly afforded the tetrahydropyridine 3a in 43% yield (Table 1, entry 1). The structure of 3a was unambiguously confirmed by X-ray analysis (Fig. 2).24 The formation of 3a can be explained through the initial rhodium catalyzed 1,2-sulfur migration in β-thio-α-diazoimines to 2a followed by the concomitant self aza-Diels–Alder reaction of 2a.|
|
||||
|---|---|---|---|---|
| Entry | Rh(II) (2 mol%) | Solvent | Temp. (°C) | Yieldb (%) |
| a Reaction conditions: triazole 1a (0.15 mmol), Rh(II) (2 mol%), solvent (1 mL), temp., 5 h. b All are isolated yield of 3a. c 3 h. | ||||
| 1 | Rh2(OAc)4 | CHCl3 | 75 | 43 |
| 2 | Rh2(OAc)4 | CHCl3 | 90 | 55 |
| 3 | Rh2(OAc)4 | Toluene | 90 | 80 |
| 4 | Rh2(OAc)4 | 1,2-DCE | 90 | 94 |
| 5 | Rh2(Oct)4 | 1,2-DCE | 90 | 85 |
| 6 | Rh2(DOSP)4 | 1,2-DCE | 90 | 93 |
| 7 | Rh2(TBSP)4 | 1,2-DCE | 90 | 94 |
| 8 | Rh2(OAc)4 | 1,2-DCE | 90 | 83c |
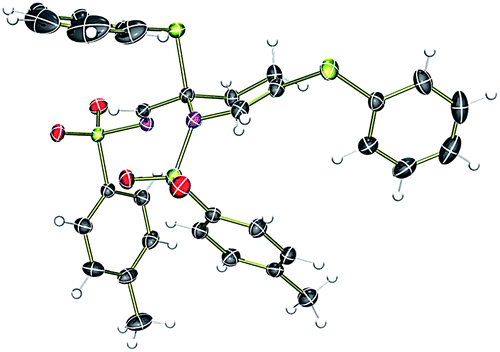 | ||
| Fig. 2 ORTEP diagram of compound 3a. The thermal ellipsoids are set at 30% probability. | ||
To improve the reaction conditions, various critical parameters were examined. Increasing the temperature to 90 °C gave 3a in 55% yield (Table 1, entry 2). A drastic increase in the yield to 80 and 94% was observed while changing the solvents to toluene and 1,2-dichloroethane (1,2-DCE), respectively, at 90 °C (Table 1, entries 3 and 4). Shortening the reaction time to 3 h led to a slight decrease in the yield (Table 1, entry 8). Similar results were observed with other rhodium based catalysts such as Rh2(Oct)4, Rh2(DOSP)4 and Rh2(TBSP)4 (Table 1, entries 5–7). Based on these studies, we chose the following reaction conditions for studying the scope and limitations of the present reaction: 2 mol% of Rh2(OAc)4 in 1,2-DCE at 90 °C.‡
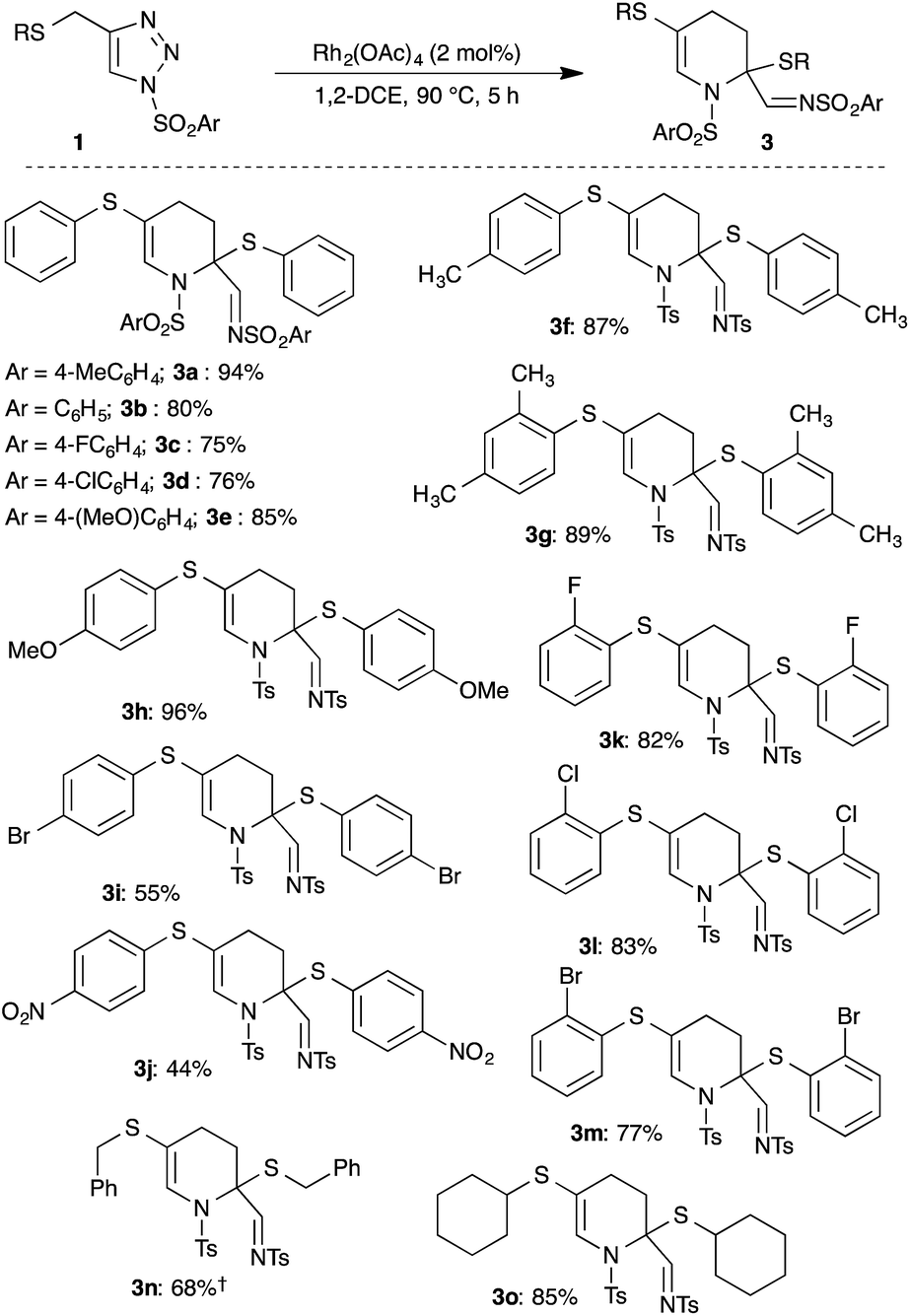
After identifying the best reaction conditions, the scope and limitations of various thio-tethered N-sulfonyl-1,2,3-triazoles 1 were examined. An initial change at the aryl moiety of the sulfonyl group did not show any influence on the outcome of the reaction and facilitated the formation of corresponding tetrahydropyridines (3a–3e) in comparable yields (Scheme 2).
 | ||
| Scheme 2 Rhodium catalyzed synthesis of tetrahydropyridines 3: scope and limitations. †10 h. | ||
Subsequently, the effect of substitution on the thio-side chain was investigated (Scheme 2). The presence of simple aryl groups like 4-methylphenyl and 2,4-dimethylphenyl were well tolerated and gave the corresponding products 3f and 3g in 87% and 89% yield, respectively. Both electron-rich and electron-deficient aryl sulfides were also tolerated under the optimized conditions, but an electron deficient aryl sulfide furnished the product 3j in low yield compared to an electron rich aryl sulfide (3h). Interestingly, synthetically useful halogen-substituted aryl-containing tetrahydropyridines 3i, 3k–3m were achieved in good to excellent yield. Also, sterically hindered aryl sulfides underwent smooth reaction to afford products 3g, 3k–3m in good yield. Furthermore, benzyl and alkyl sulfides also gave the products 3n and 3o in 68% and 85% yield, respectively.
After successful demonstration of generation of various 1-azadienes and the self aza-Diels–Alder reaction, we were interested in the in situ trapping of the formed 1-azadiene 2 with external dienophiles, such as dihydrofuran 4. Gratifyingly, the reaction of triazole 1a with 4 equivalents of dihydrofuran 4 under the optimized conditions at prolonged reaction time (10 h) afforded the bicyclic compound, tetrahydrofuran fused tetrahydropyridine 5a in 90% yield (Scheme 3).§
 | ||
Scheme 3 Rhodium catalyzed synthesis of fused-tetrahydropyridines 5: scope and limitations. †Isolated as mixture of 5g![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3g in 1:0.18 ratio. :3g in 1:0.18 ratio. | ||
Having shown the efficient synthesis of fused-tetrahydropyridines through in situ trapping of 1-azadiene, we focused our attention on the generality of the reaction. Keeping the dienophile, dihydrofuran 4 (4 equiv.) as constant, various substituted thio-tethered N-sulfonyl-1,2,3-triazoles 1 were studied under the optimized conditions (Scheme 3). Electronically and sterically different aryl substituted sulfonyl triazoles furnished the fused tetrahydropyridines 5a–5e in good to excellent yield. Similarly, various thioaryl containing tetrahydropyridines (5f–5i and 5k–5m) were also achieved in good yields from corresponding 1,2,3-triazoles. Interestingly, heteroaryl, benzoxazol-2-yl substituted sulfide-containing triazole was also tolerated under the reaction conditions to give the corresponding product 5p in 91% yield. Cyclohexyl sulfide, an alkyl sulfide substituted triazole also furnished the product 5o in 86% yield. Furthermore, synthesis of pyran-fused tetrahydropyridine 5a′ was also achieved on replacement of dihydrofuran with dihydropyran.
All the above studies prompted us to investigate various other potential dienophiles under the rhodium catalyzed tandem 1,2-sulfur migration and aza-Diels–Alder reaction with 1a to further widen the scope of the reaction. Thus, reaction of 1a and ethylvinyl ether in the presence of Rh2(OAc)4 at 90 °C for 12 h gave the tetrahydropyridine 6 in 84% yield (Scheme 4).
 | ||
| Scheme 4 Rhodium catalyzed reaction of 1a with various dienophiles. †Yield of 3a. ‡NMR yield. | ||
β-Aryl substituted enol ethers underwent smooth reaction at prolonged reaction time (12 h) to afford the expected products (7–9) in good to moderate yield, along with an isolable amount of self aza-Diels–Alder reaction product 3a. Next, reaction of α,β-disubstituted enol ether with 1a gave the corresponding tetrahydropyridine 10 in 36% yield. Interestingly, the present method also allows the synthesis of tricyclic compound 11 in 46% yield from the cyclic enol ether derived from β-tetralone. Reaction of ketene S,S-acetal25 with 1a furnished the product 12 in 69% 1H NMR yield, as an inseparable mixture along with ketene S,S-acetal. Similarly, enamine was also tolerated under the optimized conditions to afford the corresponding product 13. Consequently, neutral dienophiles such as alkynes and alkenes were also examined (Scheme 4). Reaction of 1a with phenylacetylene and p-methoxyphenylacetylene gave dihydropyridines 14 and 15, respectively, in low yield along with decomposition of starting material. Similar results were observed on reaction of 1a with styrene, which led to the formation of tetrahydropyridine 16 in 25% yield along with 61% of 3a.
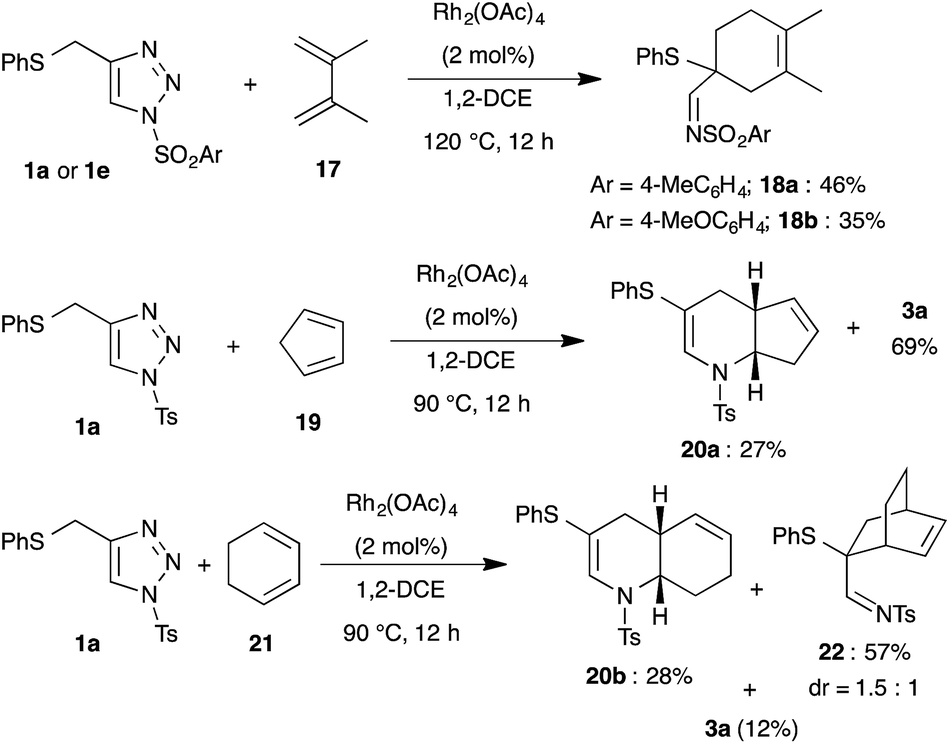
Likewise, dienes were also employed as reaction partners to examine the mode of reactivity (Scheme 5). Treatment of 1a and 2,3-dimethyl-1,3-butadiene 17 in the presence of Rh2(OAc)4 at 90 °C led to the formation of cyclohexene 18a in very low yield following normal Diels–Alder mode, wherein the formed 1-azadiene 2a acted as a dienophile instead of diene. Increasing the reaction temperature to 120 °C afforded the product 18a in 46% yield along with decomposition of the starting material. A similar result was also observed with 1,2,3-triazole 1e and 17.
 | ||
| Scheme 5 Rhodium catalyzed reaction of 1a with dienes. | ||
Interestingly, bicyclic[4.3.0]-compound 20a was isolated in moderate yield upon reaction with cyclopentadiene 19, where cyclopentadiene acted as a dienophile. Reaction of 1a with cyclohexadiene 21 furnished a mixture of bicyclic[4.4.0]-compound 20b, bridged bicyclic[2.2.2]-compound 22 and 3a in 28%, 57% and 12% yield, respectively. Thus, the isolated product showed that cyclohexadiene reacted mainly as a diene similar to 17.
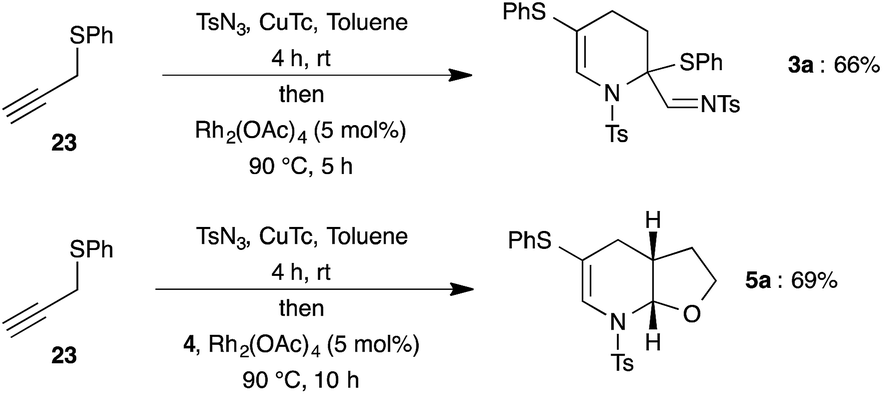
After successful demonstration of reactivity of various dienophiles and dienes with N-sulfonyl-1,2,3-triazoles, we focused our attention on the direct synthesis of (fused)-tetrahydropyridines (3a and 5a) from propargyl sulfide 23 through integration of copper catalyzed azide–alkyne cycloaddition and rhodium catalyzed tandem 1,2-sulfur migration cum aza-Diels–Alder reaction (Scheme 6). To our delight, treatment of phenyl propargyl sulfide 23 with TsN3 in the presence of CuTc for 4 h at room temperature in toluene followed by addition of Rh2(OAc)4 and subsequent elevation of temperature to 90 °C afforded the expected product 3a in 66% yield. Likewise, fused tetrahydropyridine 5a was also isolated in 69% yield from phenyl propargyl sulfide 23 on reaction with CuTc followed by addition of Rh2(OAc)4 and dihydrofuran 4, in one-pot.
 | ||
| Scheme 6 One-pot conversion of propargyl sulfide 23 to 3a and 5a. | ||
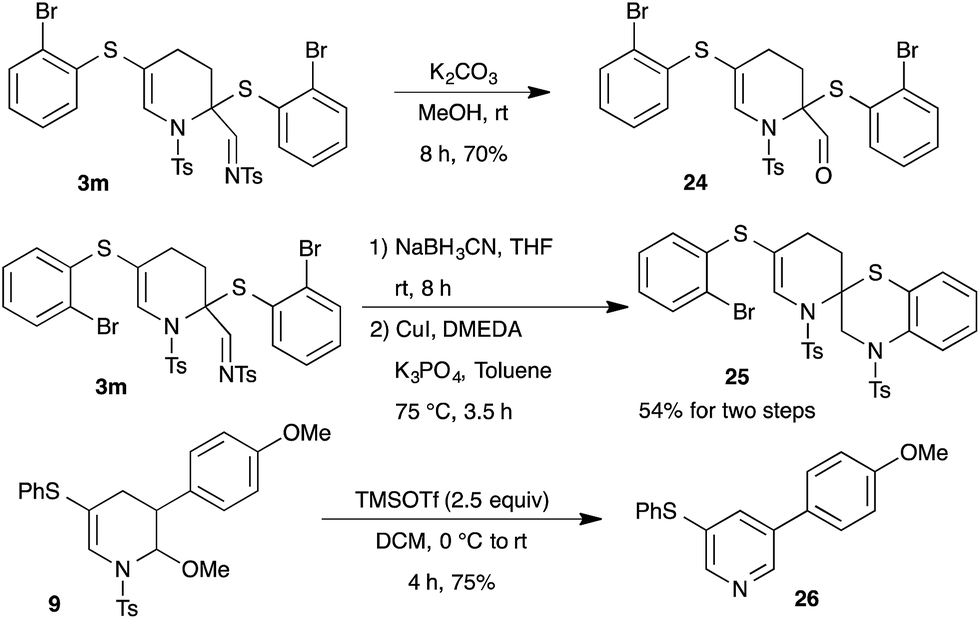
Next, the synthetic potential of the developed methodology was investigated (Scheme 7). The imine moiety in the product 3m can be readily converted to aldehyde functionality in high yield through treatment with K2CO3 in methanol. Interestingly, spiro-bicyclic compound 25 was accomplished from the tetrahydropyridine 3m through NaBH3CN reduction of imine followed by copper catalyzed intramolecular C–N bond formation (Scheme 7). Additionally, the synthesized tetrahydropyridines can also be readily transformed to pyridine derivatives. Thus, the reaction of 9 with TMSOTf furnished the aromatized product 26 in 75% yield.
 | ||
| Scheme 7 Synthetic applications. | ||
Based on the earlier report of 1,2-sulfur migration1a and denitrogenative functionalization of 1,2,3-triazoles,5a,5b we postulated the mechanism for the synthesis of tetrahydropyridines from N-sulfonyl-1,2,3-triazoles (Scheme 8). First, trapping of α-diazoimines A, generated from 1,2,3-triazole 1, with the rhodium catalyst would afford the reactive rhodium carbenoid B. Intramolecular attack of the sulfur atom onto the electrophilic carbenoid carbon would lead to the formation of thiiranium intermediate C, which upon rearrangement would furnish the 1-azadiene 2 and active rhodium species for the next catalytic cycle. Dimerization of the formed 1-azadiene 2 under thermal aza-Diels–Alder conditions furnishes the expected tetrahydropyridines 3. On the other hand, in situ trapping of 1-azadiene 2 with other dienophiles and dienes would afford the corresponding heterocycles and carbocycles.
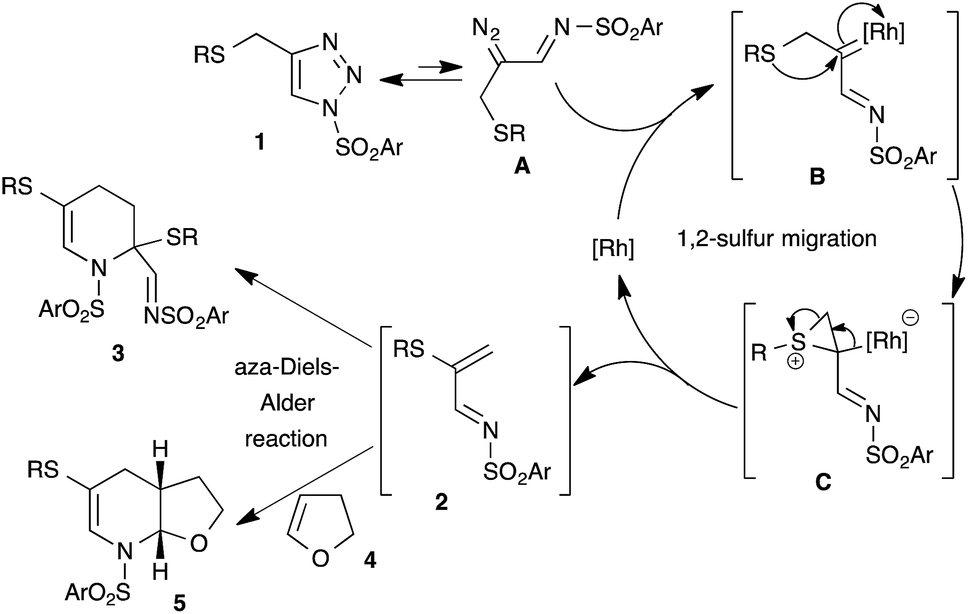 | ||
| Scheme 8 Plausible mechanism. | ||
Conclusions
In conclusion, we developed an efficient rhodium catalyzed tandem 1,2-sulfur migration and aza-Diels–Alder reaction of thio-tethered N-sulfonyl-1,2,3-triazoles. The present method allows the synthesis of various substituted tetrahydropyridines, fused tetrahydropyridines, tricyclic and (bridged)bicyclic compounds and cyclohexenes in moderate to excellent yield. Interestingly, the developed strategy was successfully integrated with copper catalyzed azide–alkyne cycloaddition for the direct conversion of propargyl sulfide to the target compounds. Furthermore, the synthetic potential of the methodology was demonstrated through the synthesis of spiro-bicyclic and substituted pyridine derivatives.Acknowledgements
We thank Department of Science and Technology (DST), New Delhi, India (Project No. SR/S1/OC-48/2012) for funding this work. DY thanks Indian Institute of Technology Madras (IITM) for a fellowship. We thank Mr. Ramkumar (IIT Madras) for single-crystal analysis support.Notes and references
- (a) A. Sromek and V. Gevorgyan, in Sulfur-Mediated Rearrangements I, ed. E. Schaumann, Springer, Berlin Heidelberg, 2007, vol. 274, ch. 57, pp. 77–124 Search PubMed; (b) A. S. Dudnik, A. W. Sromek, M. Rubina, J. T. Kim, A. V. Kel'i and V. Gevorgyan, J. Am. Chem. Soc., 2008, 130, 1440–1452 CrossRef CAS PubMed.
- F. Xu, W. Shi and J. Wang, J. Org. Chem., 2005, 70, 4191–4194 CrossRef CAS PubMed.
- L. Jiao, Q. Zhang, Y. Liang, S. Zhang and J. Xu, J. Org. Chem., 2006, 71, 815–818 CrossRef CAS PubMed.
- (a) E. J. Yoo, M. Ahlquist, S. H. Kim, I. Bae, V. V. Fokin, K. B. Sharpless and S. Chang, Angew. Chem., Int. Ed., 2007, 46, 1730–1733 CrossRef CAS PubMed; (b) J. Raushel and V. V. Fokin, Org. Lett., 2010, 12, 4952–4955 CrossRef CAS PubMed; (c) Y. Liu, X. Wang, J. Xu, Q. Zhang, Y. Zhao and Y. Hu, Tetrahedron, 2011, 67, 6294–6299 CrossRef CAS PubMed.
- (a) P. Anbarasan, D. Yadagiri and S. Rajasekar, Synthesis, 2014, 3004–3023 CrossRef CAS; (b) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162 RSC; (c) A. V. Gulevich and V. Gevorgyan, Angew. Chem., Int. Ed., 2013, 52, 1371–1373 CrossRef CAS PubMed.
- (a) T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972–14974 CrossRef CAS PubMed; (b) M. Zibinsky and V. V. Fokin, Org. Lett., 2011, 13, 4870–4872 CrossRef CAS PubMed.
- (a) T. Miura, M. Yamauchi and M. Murakami, Chem. Commun., 2009, 1470–1471 RSC; (b) B. Chattopadhyay and V. Gevorgyan, Org. Lett., 2011, 13, 3746–3749 CrossRef CAS PubMed; (c) Y. Shi and V. Gevorgyan, Org. Lett., 2013, 15, 5394–5396 CrossRef CAS PubMed.
- (a) S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034–18035 CrossRef CAS PubMed; (b) J. S. Alford and H. M. L. Davies, Org. Lett., 2012, 14, 6020–6023 CrossRef CAS PubMed; (c) J. S. Alford, J. E. Spangler and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 11712–11715 CrossRef CAS PubMed; (d) J. C. Culhane and V. V. Fokin, Org. Lett., 2011, 13, 4578–4580 CrossRef CAS PubMed.
- (a) C.-E. Kim, S. Park, D. Eom, B. Seo and P. H. Lee, Org. Lett., 2014, 16, 1900–1903 CrossRef CAS PubMed; (b) S. Rajasekar and P. Anbarasan, J. Org. Chem., 2014, 79, 8428–8434 CrossRef CAS PubMed; (c) R.-Q. Ran, J. He, S.-D. Xiu, K.-B. Wang and C.-Y. Li, Org. Lett., 2014, 16, 3704–3707 CrossRef CAS PubMed; (d) J. Feng, Y. Wang, Q. Li, R. Jiang and Y. Tang, Tetrahedron Lett., 2014, 55, 6455–6458 CrossRef CAS PubMed; (e) C.-E. Kim, Y. Park, S. Park and P. H. Lee, Adv. Synth. Catal., 2014, 357, 210–220 CrossRef PubMed.
- J. E. Spangler and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6802–6805 CrossRef CAS PubMed.
- (a) M. Zibinsky and V. V. Fokin, Angew. Chem., Int. Ed., 2013, 52, 1507–1510 CrossRef CAS PubMed; (b) T. Miura, T. Tanaka, K. Hiraga, S. G. Stewart and M. Murakami, J. Am. Chem. Soc., 2013, 135, 13652–13655 CrossRef CAS PubMed; (c) X.-Y. Tang, Y.-S. Zhang, L. He, Y. Wei and M. Shi, Chem. Commun., 2015, 51, 133–136 RSC; (d) H. Shen, J. Fu, J. Gong and Z. Yang, Org. Lett., 2014, 16, 5588–5591 CrossRef CAS PubMed; (e) Y.-S. Zhang, X.-Y. Tang and M. Shi, Chem. Commun., 2014, 50, 15971–15974 RSC.
- (a) E. E. Schultz and R. Sarpong, J. Am. Chem. Soc., 2013, 135, 4696–4699 CrossRef CAS PubMed; (b) T. Miura, K. Hiraga, T. Biyajima, T. Nakamuro and M. Murakami, Org. Lett., 2013, 15, 3298–3301 CrossRef CAS PubMed; (c) S. Chuprakov, S. W. Kwok and V. V. Fokin, J. Am. Chem. Soc., 2013, 135, 4652–4655 CrossRef CAS PubMed; (d) T. Miura, T. Nakamuro, T. Biyajima and M. Murakami, Chem. Lett., 2015, 44, 700–702 CrossRef CAS.
- (a) S. Chuprakov, J. A. Malik, M. Zibinsky and V. V. Fokin, J. Am. Chem. Soc., 2011, 133, 10352–10355 CrossRef CAS PubMed; (b) J.-M. Yang, C.-Z. Zhu, X.-Y. Tang and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 5142–5146 CAS; (c) D. Yadagiri and P. Anbarasan, Org. Lett., 2014, 16, 2510–2513 CrossRef CAS PubMed; (d) B. Rajagopal, C.-H. Chou, C.-C. Chung and P.-C. Lin, Org. Lett., 2014, 16, 3752–3755 CrossRef CAS PubMed; (e) S. Park, W.-S. Yong, S. Kim and P. H. Lee, Org. Lett., 2014, 16, 4468–4471 CrossRef CAS PubMed; (f) B. Seo, W. H. Jeon, J. Kim, S. Kim and P. H. Lee, J. Org. Chem., 2015, 80, 722–732 CrossRef CAS PubMed; (g) M.-H. Shen, Y.-P. Pan, Z.-H. Jia, X.-T. Ren, P. Zhang and H.-D. Xu, Org. Biomol. Chem., 2015, 13, 4851–4854 RSC; (h) S. Shin, Y. Park, C.-E. Kim, J.-Y. Son and P. H. Lee, J. Org. Chem., 2015, 80, 5859–5869 CrossRef CAS PubMed.
- (a) T. Miura, T. Biyajima, T. Fujii and M. Murakami, J. Am. Chem. Soc., 2012, 134, 194–196 CrossRef CAS PubMed; (b) T. Miura, T. Tanaka, T. Biyajima, A. Yada and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 3883–3886 CrossRef CAS PubMed; (c) S. Chuprakov, B. T. Worrell, N. Selander, R. K. Sit and V. V. Fokin, J. Am. Chem. Soc., 2014, 136, 195–202 CrossRef CAS PubMed; (d) H. J. Jeon, D. J. Jung, J. H. Kim, Y. Kim, J. Bouffard and S.-G. Lee, J. Org. Chem., 2014, 79, 9865–9871 CrossRef CAS PubMed; (e) D. J. Lee and E. J. Yoo, Org. Lett., 2015, 17, 1830–1833 CrossRef CAS PubMed.
- (a) D. Yadagiri and P. Anbarasan, Chem.–Eur. J., 2013, 19, 15115–15119 CrossRef CAS PubMed; (b) T. Miura, T. Tanaka, A. Yada and M. Murakami, Chem. Lett., 2013, 42, 1308–1310 CrossRef CAS; (c) A. Boyer, Org. Lett., 2014, 16, 1660–1663 CrossRef CAS PubMed; (d) F. Medina, C. Besnard and J. Lacour, Org. Lett., 2014, 16, 3232–3235 CrossRef CAS PubMed; (e) X. Ma, S. Pan, H. Wang and W. Chen, Org. Lett., 2014, 16, 4554–4557 CrossRef CAS PubMed; (f) A. Boyer, Org. Lett., 2014, 16, 5878–5881 CrossRef CAS PubMed; (g) Y. Wang, X. Lei and Y. Tang, Chem. Commun., 2015, 51, 4507–4510 RSC.
- (a) B. T. Parr, S. A. Green and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 4716–4718 CrossRef CAS PubMed; (b) H. Shang, Y. Wang, Y. Tian, J. Feng and Y. Tang, Angew. Chem., Int. Ed., 2014, 53, 5662–5666 CrossRef CAS PubMed; (c) D. J. Lee, H. S. Han, J. Shin and E. J. Yoo, J. Am. Chem. Soc., 2014, 136, 11606–11609 CrossRef CAS PubMed; (d) B. T. Parr and H. M. L. Davies, Angew. Chem., Int. Ed., 2013, 52, 10044–10047 CrossRef CAS PubMed; (e) T. Miura, Y. Funakoshi and M. Murakami, J. Am. Chem. Soc., 2014, 136, 2272–2275 CrossRef CAS PubMed; (f) E. E. Schultz, V. N. G. Lindsay and R. Sarpong, Angew. Chem., Int. Ed., 2014, 53, 9904–9908 CrossRef CAS PubMed; (g) J. S. Alford and H. M. L. Davies, J. Am. Chem. Soc., 2014, 136, 10266–10269 CrossRef CAS PubMed; (h) H.-D. Xu, K. Xu, H. Zhou, Z.-H. Jia, H. Wu, X.-L. Lu and M.-H. Shen, Synthesis, 2015, 641–646 CAS; (i) Y. Yang, M.-B. Zhou, X.-H. Ouyang, R. Pi, R.-J. Song and J.-H. Li, Angew. Chem., Int. Ed., 2015, 54, 6595–6599 CrossRef CAS PubMed; (j) T. Miura, Y. Funakoshi, Y. Fujimoto, J. Nakahashi and M. Murakami, Org. Lett., 2015, 17, 2454–2457 CrossRef CAS PubMed; (k) E. Lee, T. Ryu, E. Shin, J.-Y. Son, W. Choi and P. H. Lee, Org. Lett., 2015, 17, 2470–2473 CrossRef CAS PubMed.
- (a) N. Selander, B. T. Worrell, S. Chuprakov, S. Velaparthi and V. V. Fokin, J. Am. Chem. Soc., 2012, 134, 14670–14673 CrossRef CAS PubMed; (b) D. J. Jung, H. J. Jeon, J. H. Kim, Y. Kim and S.-G. Lee, Org. Lett., 2014, 16, 2208–2211 CrossRef CAS PubMed; (c) D. J. Lee, J. Shin and E. J. Yoo, Chem. Commun., 2014, 50, 6620 RSC; (d) T. Miura, T. Nakamuro, K. Hiraga and M. Murakami, Chem. Commun., 2014, 50, 10474–10477 RSC; (e) J. He, Z. Man, Y. Shi and C.-Y. Li, J. Org. Chem., 2015, 80, 4816–4823 CrossRef CAS PubMed.
- (a) T. Miura, Y. Funakoshi, M. Morimoto, T. Biyajima and M. Murakami, J. Am. Chem. Soc., 2012, 134, 17440–17443 CrossRef CAS PubMed; (b) N. Selander, B. T. Worrell and V. V. Fokin, Angew. Chem., Int. Ed., 2012, 51, 13054–13057 CrossRef CAS PubMed; (c) R. Liu, M. Zhang, G. Winston-McPherson and W. Tang, Chem. Commun., 2013, 49, 4376 RSC.
- Y. Jiang, X.-Y. Tang and M. Shi, Chem. Commun., 2015, 51, 2122–2125 RSC.
- J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed.
- (a) T. Mesganaw and J. A. Ellman, Org. Process Res. Dev., 2014, 18, 1097–1104 CrossRef CAS PubMed; (b) J. M. Neely and T. Rovis, Org. Chem. Front., 2014, 1, 1010–1015 RSC; (c) G. Masson, C. Lalli, M. Benohoud and G. Dagousset, Chem. Soc. Rev., 2013, 42, 902–923 RSC; (d) J.-C. M. Monbaliu, K. G. R. Masschelein and C. V. Stevens, Chem. Soc. Rev., 2011, 40, 4708 RSC.
- (a) T. D. Bailey, G. L. Goe and E. F. V. Scriven, in Chem. Heterocycl. Compd., John Wiley & Sons, Inc., 1985, pp. 1–252 Search PubMed; (b) G. R. Newkome, in Chem. Heterocycl. Compd., John Wiley & Sons, Inc., 1985, pp. 635–658 Search PubMed; (c) N. N. Mateeva, L. L. Winfield and K. K. Redda, Curr. Med. Chem., 2005, 12, 551–571 CAS.
- C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498 RSC.
- CCDC 1040728 (3a). See ESI†.
- M. Yokoyama, H. Togo and S. Kondo, Sulfur Rep., 1990, 10, 23–43 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, characterizations data, and 1H and 13C NMR spectra of isolated compounds. CCDC 1040728. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02379c |
| ‡ Synthesis of tetrahydropyridine 3a: to an oven dried 10 mL reaction tube equipped with stir bar, N-sulfonyl-1,2,3-triazole 1a (50 mg, 0.144 mmol) and Rh2(OAc)4 (1.28 mg, 0.0028 mmol, 2 mol%) were added under a nitrogen atmosphere and dissolved in 1,2-DCE (1 mL). The reaction tube was sealed and the reaction mixture was stirred at 90 °C for 5 h. After the completion of reaction, based on TLC analysis, the reaction mixture was cooled to room temperature and the solvent was removed under reduced pressure. The resultant crude was purified by column chromatography using hexane/ethyl acetate (9:1) as eluant to afford tetrahydropyridine 3a in 94% yield. Mp: 131–133 °C; FTIR (KBr): 3101, 2956, 1620, 1595, 1472, 1439, 1331, 1163, 1088, 1040, 960, 835, 812, 787, 743, 691, 664, 566, 538, 502 cm−1; 1H NMR (400 MHz, CDCl3, 24 °C): δ 8.71 (s, 1H), 7.87 (d, 2H, J = 8.24 Hz), 7.44–7.34 (m, 7H), 7.32–7.25 (m, 6H), 7.22–7.20 (m, 3H), 7.13 (s, 1H), 2.49 (s, 3H), 2.44 (s, 1H), 2.41 (s, 3H), 2.05 (dd, 1H, J = 6.09, 18.12 Hz), 1.92 (dd, 1H, J = 6.92, 14.66 Hz), 1.66–1.61 (m, 1H); 13C NMR (100 MHz, CDCl3, 24 °C): δ 168.7, 145.2, 144.9, 137.5, 135.3, 134.8, 133.1, 130.6, 130.0, 129.3, 129.3, 129.2, 129.1, 128.6, 127.7, 127.0, 126.7, 119.0, 71.8, 28.0, 25.3, 21.8, 21.7; HRMS: calcd for C32H30N2O4S4 + H: 635.1161; found: 635.1152. |
| § Synthesis of fused tetrahydropyridines 5a: to an oven dried 10 mL reaction tube equipped with stir bar, N-sulfonyl-1,2,3-triazole 1a (50 mg, 0.144 mmol) and Rh2(OAc)4 (1.28 mg, 0.0028 mmol, 2 mol%) were added under a nitrogen atmosphere and dissolved in 1,2-DCE (1 mL). Next, 2,3-dihydrofuran 4 (40 mg, 0.576 mmol, 4 equiv.) was introduced through a syringe. The reaction tube was sealed and the reaction mixture was stirred at 90 °C for 10 h. After the completion of reaction, based on TLC analysis, the reaction mixture was cooled to room temperature and the solvent was removed under reduced pressure. The resultant crude was purified by column chromatography using hexane/ethyl acetate (92:8) as eluant to furnish the fused tetrahydropyridine 5a in 90% yield. Mp: 121–123 °C; FTIR (KBr): 3052, 2941, 2889, 1640, 1598, 1475, 1436, 1404, 1351, 1240, 1106, 1027, 967, 862, 803, 746, 695, 667, 587, 549 cm−1; 1H NMR (400 MHz, CDCl3, 24 °C): δ 7.83 (d, 2H, J = 8.16 Hz), 7.33 (d, 2H, J = 8.36 Hz), 7.29–7.26 (m, 2H), 7.23–7.17 (m, 3H), 7.14–7.13 (m, 1H), 5.55 (d, 1H, J = 4.36 Hz), 3.84–3.77 (m, 1H), 2.45 (s, 3H), 2.35–2.32 (m, 1H), 2.22 (dd, 1H, J = 6.64, 17.14 Hz), 2.13–2.08 (m, 1H), 1.98–1.92 (m, 1H), 1.74–1.70 (m, 1H); 13C NMR (100 MHz, CDCl3, 24 °C): δ 144.4, 136.8, 135.5, 129.8, 129.1, 128.7, 128.6, 127.6, 126.3, 108.9, 83.9, 64.4, 35.8, 29.9, 28.4, 21.7; HRMS: calcd for C20H21NO3S2 + H: 388.1041; found: 388.1056. |
| This journal is © The Royal Society of Chemistry 2015 |