
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnesium-catalyzed hydrosilylation of α,β-unsaturated esters†
Nicole L.
Lampland
a,
Aradhana
Pindwal
a,
Steven R.
Neal‡
b,
Shealyn
Schlauderaff
a,
Arkady
Ellern
a and
Aaron D.
Sadow
*a
aDepartment of Chemistry, Iowa State University, 1605 Gilman Hall, Ames, IA 50011, USA. E-mail: sadow@iastate.edu
bDepartment of Chemistry, University of Tennessee, 515 Dabney-Buehler Hall, 1420 Circle Dr., Knoxville, TN 37996, USA
First published on 26th August 2015
Abstract
ToMMgHB(C6F5)3 (1, ToM = tris(4,4-dimethyl-2-oxazolinyl)phenylborate) catalyzes the 1,4-hydrosilylation of α,β-unsaturated esters. This magnesium hydridoborate compound is synthesized by the reaction of ToMMgMe, PhSiH3, and B(C6F5)3. Unlike the transient ToMMgH formed from the reaction of ToMMgMe and PhSiH3, the borate adduct 1 persists in solution and in the solid state. Crystallographic characterization reveals tripodal coordination of the HB(C6F5)3 moiety to the six-coordinate magnesium center with a ∠Mg–H–B of 141(3)°. The pathway for formation of 1 is proposed to involve the reaction of ToMMgMe and a PhSiH3/B(C6F5)3 adduct because the other possible intermediates, ToMMgH and ToMMgMeB(C6F5)3, react to give an intractable black solid and ToMMgC6F5, respectively. Under catalytic conditions, silyl ketene acetals are isolated in high yield from the addition of hydrosilanes to α,β-unsaturated esters with 1 as the catalyst.
Introduction
Catalytic addition reactions, such as hydrosilylation1 and hydroboration2 are important synthetic tools for the reduction of unsaturated moieties. These reactions also provide carbon-element, oxygen-element, and nitrogen-element bonds (element = silicon, boron, hydrogen) that allow further elaboration of organic and inorganic substances through cross-coupling3 or oxidation.4 Transition-metal, main-group metal, and rare earth metal complexes catalyze hydrosilylation through a range of pathways including 2-electron metal-centered redox chemistry, single-electron processes, σ-bond metathesis, or hydride abstraction reactions involving Lewis acid sites. Even a single compound can be involved in catalytic additions through a number of pathways that vary depending on the substrates, reductants, conditions and/or co-catalysts. For example, B(C6F5)3 catalyzes hydrosilylation of alkenes and carbonyls by action upon silanes,5 through frustrated Lewis Pairs in the presence of a bulky base,6 or through its combination with a metal center.7The availability of many reaction pathways creates a challenge to control the selective conversion of carbonyl or olefin functional groups in substrates that contain both moieties. α,β-Unsaturated carbonyls can be particularly difficult because they may be susceptible to 1,2-addition to the carbonyl, 1,4-additions, α- or β-additions to the olefin, or polymerizations. The 1,4-addition products, silyl enol ethers or silyl ketene acetals, are valuable versatile nucleophiles in Mukaiyama aldol, Michael reactions,8 arylations,9 and haloketone or ketol formations. Since Wilkinson's and Karstedt's catalysts were shown to give selective 1,4-addition of R3SiH to α,β-unsaturated ketones,10 mainly platinum-group metals have been studied as catalysts for 1,4-hydrosilylation of α,β-unsaturated esters.11 Examples using more earth-abundant metals, such as main group or first row transition-metals, are less common and largely limited to Cu systems.12
There are only a few examples of alkene hydrosilylation catalyzed by heavy group 2 metal complexes (Ca, Sr, Ba),13 and carbonyl hydrosilylation is even less common. This is likely a result of the oxophilicity of magnesium and its heavier congeners. In fact, [{Me-NacnacDipp}CaH·THF]2 (Me-NacnacDipp = ((2,6-iPr2C6H3)NCMe)2CH) provides a rare example of a group 2 catalyzed 1,2-hydrosilylation of ketones.14 In the stoichiometric dearomatization of pyridine and quinoline derivatives utilizing [{Me-NacnacDipp}MgnBu] and PhSiH3, it was found that PhSiH3 is insufficiently reactive to provide catalytic turnover.15 To the best of our knowledge, there are no previous reports of hydrosilylation catalyzed by homogeneous magnesium complexes.
More often, esters are cleaved under hydrosilylation conditions with first-row transition-metal catalysts,16 or with main group catalysts in hydroborations.17 In a magnesium catalyzed hydroboration of esters, the α,β-unsaturated ester reacts through C–O bond cleavage while the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is unaffected.17b In that system, an important postulated intermediate, ToMMg{H(RO)Bpin} (ToM = tris(4,4-dimethyl-2-oxazolinyl)phenylborate; Bpin = boron pinacol ester), contains a boron–hydrogen bond. The [M]{H(RO)Bpin} motif contains features also associated with [M]{HB(C6F5)3} complexes,18 including oxygen or fluorine coordination to the metal center and a B–H → M interaction featuring a long M–H distance and nonlinear B–H–M angle. Recently, a {Me-NacnacDipp}MHB(C6F5)3 complex (M = Mg, Ca) was reported to catalyze the hydroboration of carbon dioxide,19 and this may suggest that hydroborates derived from B(C6F5)3 or HBpin may lead to new chemistry. Alternatively, a terminal magnesium hydride supported by a tetradentate monoanionic trimethylated tetraazacyclododecane ligand is stabilized by AliBu3, which coordinates to the amide moiety in the ancillary ligand rather than the nucleophilic hydride.20 The tris(oxazolinyl)borato magnesium catalyst precursors studied for hydroboration, namely ToMMgMe or ToMMgOR, do not mediate hydrosilylation of esters under the conditions tested, further suggesting that the boron center in ToMMg{H(RO)Bpin} provides a key feature for magnesium-catalyzed conversions of oxygenates.
C bond is unaffected.17b In that system, an important postulated intermediate, ToMMg{H(RO)Bpin} (ToM = tris(4,4-dimethyl-2-oxazolinyl)phenylborate; Bpin = boron pinacol ester), contains a boron–hydrogen bond. The [M]{H(RO)Bpin} motif contains features also associated with [M]{HB(C6F5)3} complexes,18 including oxygen or fluorine coordination to the metal center and a B–H → M interaction featuring a long M–H distance and nonlinear B–H–M angle. Recently, a {Me-NacnacDipp}MHB(C6F5)3 complex (M = Mg, Ca) was reported to catalyze the hydroboration of carbon dioxide,19 and this may suggest that hydroborates derived from B(C6F5)3 or HBpin may lead to new chemistry. Alternatively, a terminal magnesium hydride supported by a tetradentate monoanionic trimethylated tetraazacyclododecane ligand is stabilized by AliBu3, which coordinates to the amide moiety in the ancillary ligand rather than the nucleophilic hydride.20 The tris(oxazolinyl)borato magnesium catalyst precursors studied for hydroboration, namely ToMMgMe or ToMMgOR, do not mediate hydrosilylation of esters under the conditions tested, further suggesting that the boron center in ToMMg{H(RO)Bpin} provides a key feature for magnesium-catalyzed conversions of oxygenates.
The present study follows this idea to develop magnesium-catalyzed reductions of oxygenates employing organosilanes, rather than pinacolborane, as stoichiometric reductants. Here, we have incorporated the [M]HB(C6F5)3 motif into the complex ToMMgHB(C6F5)3 (1) and report its reactivity as the first magnesium-catalyzed hydrosilylation. This transformation provides silyl ketene acetals through 1,4-hydrosilylation of α,β-unsaturated esters.
Results and discussion
The monomeric magnesium methyl ToMMgMe reacts slowly with organosilanes to provide Me–Si bond-containing compounds. For example, ToMMgMe and PhSiH3 react in toluene-d8 to form PhMeSiH2 over 3 h at 100 °C (eqn (1)).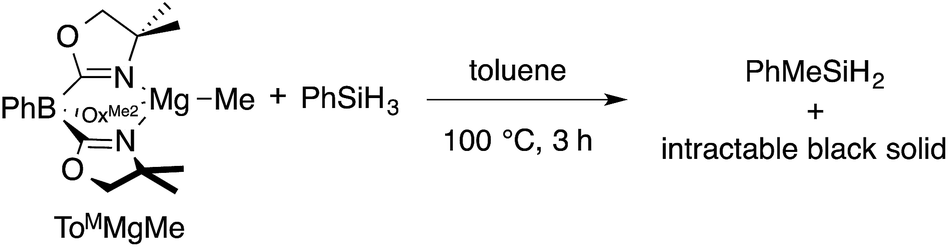 | (1) |
The presumed magnesium-containing product, ToMMgH, is rapidly converted into an intractable black solid under these conditions. This black material is also formed as a byproduct in room temperature reactions of ToMMgNHR and hydrosilanes that provide Si–N bond-containing products21 and in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reactions of ToMMgMe and HBpin that afford Me-Bpin.17b As a result, the identity of ToMMgH is assumed based on reaction stoichiometry and its apparent reactivity as a catalytic intermediate.21 In order to obtain more evidence for ToMMgH, we attempted to trap it as a Lewis acid adduct with B(C6F5)3.
:1 reactions of ToMMgMe and HBpin that afford Me-Bpin.17b As a result, the identity of ToMMgH is assumed based on reaction stoichiometry and its apparent reactivity as a catalytic intermediate.21 In order to obtain more evidence for ToMMgH, we attempted to trap it as a Lewis acid adduct with B(C6F5)3.
A mixture of ToMMgMe, PhSiH3, and B(C6F5)3 gives PhMeSiH2 and 1 (eqn (2)). Notably, this reaction occurs at room temperature over 10 min, whereas the direct interaction of ToMMgMe and PhSiH3 requires the forcing conditions noted above. The optimized preparation of 1 involves dropwise addition of ToMMgMe to a mixture of PhSiH3 and B(C6F5)3 dissolved in benzene.
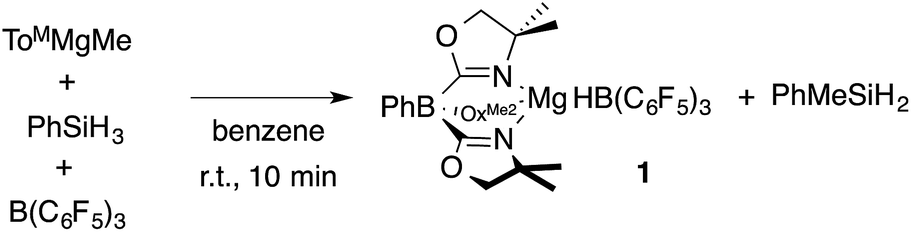 | (2) |
The 1H NMR spectrum of 1 (benzene-d6, r.t.) contained one set of oxazoline resonances, which is consistent with a pseudo-C3v-symmetric structure and tridentate coordination of ToM to the magnesium center. In addition, the hydrogen bonded to boron was observed at 2.7 ppm as a 1:1:1:1 quartet (1JBH = 69 Hz). In the 11B NMR spectrum, a singlet at −18.2 ppm was assigned to the tris(oxazolinyl)borate ligand, and a doublet at −21.1 ppm (1JHB = 69 Hz) characterized the HB(C6F5)3 group. The C6F5 are equivalent and freely rotating on the NMR timescale at room temperature, as indicated by the three resonances observed in the 19F NMR spectrum at −134.2, −156.5 and −161.4 ppm. The chemical shift of the ortho-F are similar to Cp*2ZrH{HB(C6F5)3} (Cp* = C5Me5), while the meta-F and para-F signals of 1 are downfield with respect to the zirconium hydride complex.22 The δpara−δmeta of 5 ppm23 suggests coordination of HB(C6F5)3 to the Mg center. On the basis of these data and the single-crystal X-ray diffraction study (see below), 19F NMR spectra were acquired from 298 to 180 K; however, these signals did not vary over that temperature range. A single infrared band at 1579 cm−1 assigned to the oxazoline νCN also supported the assignment of tridentate ToM-coordination. In addition, B–H bond formation was evidenced by an IR band at 2372 cm−1.
A single crystal X-ray diffraction study confirms the identity of compound 1 as ToMMgHB(C6F5)3, the tridentate coordination mode of the ToM ligand, and the tripodal Mg–HB(C6F5)3 interaction (Fig. 1). The six coordinating groups (three N from ToM, two F and one H from HB(C6F5)3) form a distorted octahedral coordination geometry. Thus, the pseudo-trans disposed N1–Mg1–H1 angle is 162.3(7) and the N2–Mg1–F10 and N3–Mg1–F11 angles are 169.28(9) and 173.33(9)°. The Mg1–H1 and B1–H1 interatomic distances are 2.06(3) Å and 1.24(3) Å, respectively. The Mg1–H1 distance is longer than in the bridging Mg–H–Mg of [{Me-NacnacDipp}Mg(μ-H)]2 (1.95(3) Å)24 and [{tBu-NacnacDipp}Mg(μ-H)]2 (1.80(5) and 1.91(5) Å; tBu-NacnacDipp = ((2,6-iPr2C6H3)NCtBu)2CH). It is also longer than in the terminal magnesium hydride {tBu-NacnacDipp}MgH(DMAP) (1.75(7) Å).25 The Mg1–H1 distance, however, is shorter than the related Mg–H distance of 2.19(3) Å in ToMMgH2Bpin.17b In [{Me-NacnacDipp}MgBH4]2, there are two types of Mg–H–B interactions, a Mg–H–B bridge (1.95(2) and 1.96(2) Å) containing shorter distances than in 1, and Mg,Mg,B-μ3-H with magnesium–hydrogen distances of 2.20(2) and 2.34(2) Å that are longer than 1.26 The B1–H1 distance of 1 is between the bridging (1.33(2) Å) and terminal (1.19(3) Å) B–H distances in diborane27 and much longer than in the terminal B–H (1.06(6) Å) of Cp*2ZrH{HB(C6F5)3}.22 Additionally, the B–H distance in 1 is similar to that of Cp*2SmHB(C6F5)3 (1.18(5) A),18b Cp*2ScHB(C6F5)3 (1.14(3) Å),18a and {Me-NacnacDipp}CaHB(C6F5)3 (1.16(2) Å).19
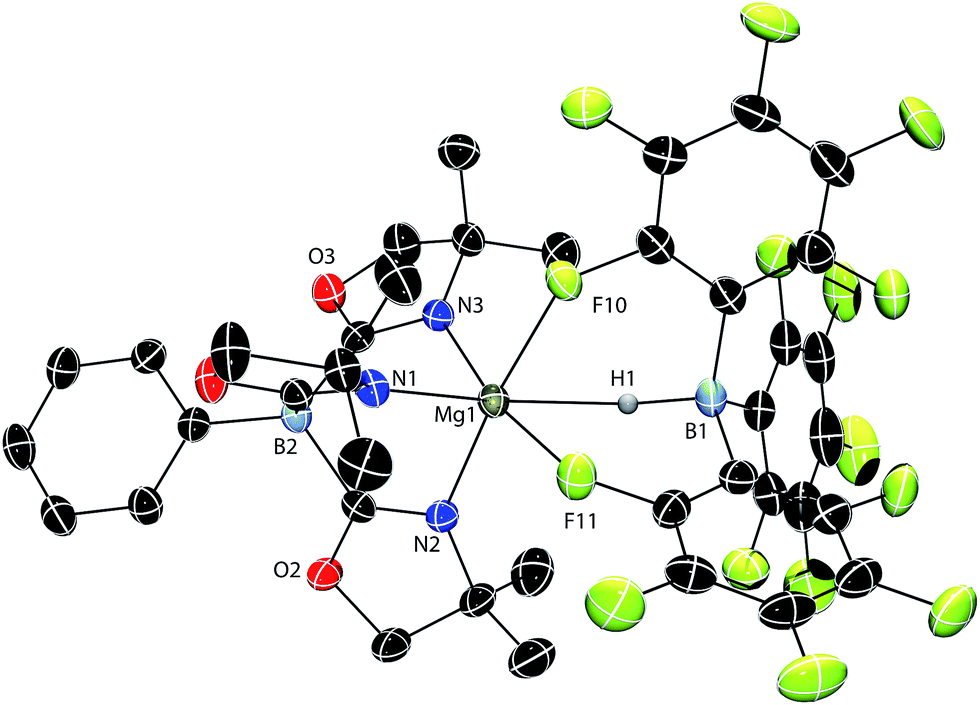 | ||
| Fig. 1 Rendered thermal ellipsoid diagram of ToMMgHB(C6F5)3 (1) plotted at 50% probability. The H1, bridging between Mg1 and B1, was located objectively in the difference Fourier map and was refined isotropically. Two molecules of toluene and the H atoms on ToM are not included in the depiction for clarity. | ||
The nonlinear ∠Mg1–H1–B1 (141(3)°) angle is likely strongly influenced by the magnesium–fluorine interactions rather than from a Mg-(η2-H–B) interaction because the Mg1–B1 distance is long (3.149(4) Å). However, the Mg–H–B angle in ToMMgH2Bpin of 93(2)° is much smaller, and as a result the Mg–B distance of 2.520(8) Å in the pinacol borane compound is shorter than in 1. The tridentate coordination mode of HB(C6F5)3 is similar in 1, MC(SiHMe2)3{HB(C6F5)3}THF2 (M = Ca, Yb),18d and {Me-NacnacDipp}CaHB(C6F5)3.19 Cp*2SmHB(C6F5)3 contains two Sm−F interactions from the aryl rings and a possible interaction between Sm and the hydride.18b Despite the size difference and the bulky tridentate oxazolinylborate ligand, Mg2+ still forms an analogous structure to these larger divalent metal cations. In contrast, Cp*2ScHB(C6F5)3 (ref. 18a) and Cp*2ZrH{HB(C6F5)3} (ref. 22) are bidentate through two M–F interactions.
Three pathways were considered for the formation of 1 (Scheme 1). The first one involves the reaction of ToMMgMe and B(C6F5)3 to give ToMMgMeB(C6F5)3 (2), followed by reaction of this species with PhSiH3 to give PhMeSiH2 and 1 (Path A). In Path B, the reaction of ToMMgMe and PhSiH3 forms ToMMgH, which is trapped by B(C6F5)3 to give 1. Alternatively, PhSiH3 and B(C6F5)3 could interact to give a transient adduct [PhH2SiHB(C6F5)3], and this intermediate reacts with ToMMgMe to give the products (Path C). Methide abstraction by B(C6F5)3 in Path A is well established,28 supporting the possible intermediate ToMMgMeB(C6F5)3. Furthermore, Cp*2ZrMe{(μ-Me)B(C6F5)3} is reported to undergo hydrogenation with H2 to give Cp*2ZrH{HB(C6F5)3},22,28 and (C5R5)2MMe{(μ-Me)B(C6F5)3} (M = Zr, Hf; C5R5 = C5H5, C5H4Me, C5Me5) and silanes react to give (C5R5)2MH{HB(C6F5)3}.29 These reactions, however, may involve methyl-hydride exchange through the conversion of [M]H{(μ-Me)B(C6F5)3} to [M]Me{(μ-H)B(C6F5)3} rather than direct hydrogenolysis of a M–Me–B bridge required for Path A. Path C is supported by proposed silane-borane adducts in B(C6F5)3-catalyzed hydrosilylations with tertiary silanes,5b and recently a tris(pentafluorophenyl)-boraindene and triethylsilane adduct was isolated and fully characterized.30
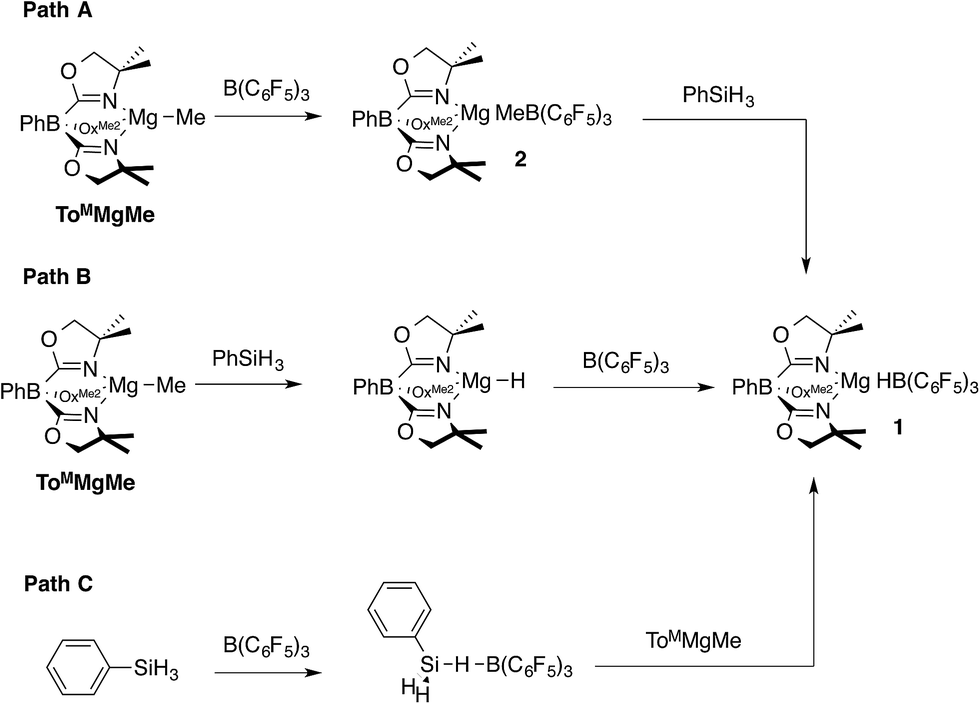 | ||
| Scheme 1 Possible pathways to ToMMgHB(C6F5)3 (1). | ||
Path B is immediately ruled out by the apparent reaction kinetics, which require forcing conditions to slowly generate ToMMgH from PhSiH3 and ToMMgMe. This reaction time and temperature contrasts the rapid formation of 1 from ToMMgMe and PhSiH3 in the presence of B(C6F5)3. To test the feasibility of Path A, the proposed intermediate, ToMMgMeB(C6F5)3 (2), was independently synthesized by addition of B(C6F5)3 dissolved in pentane to a benzene solution containing ToMMgMe (eqn (3)).
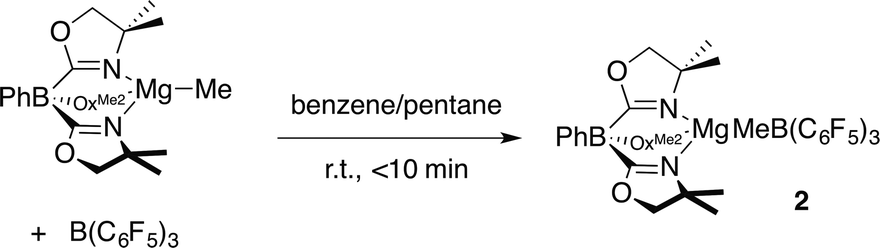 | (3) |
The product immediately precipitates giving analytically pure 2. Reactions in benzene-d6 or methylene chloride-d2 provide ToMMgMeB(C6F5)3 as a partially soluble species that may be quickly characterized by solution-phase spectroscopy. However, once solvent is removed and ToMMgMeB(C6F5)3 is isolated, it becomes insoluble in benzene and methylene chloride and only partially redissolves in THF. As in 1, 1H NMR spectra of in situ generated 2 revealed equivalent oxazoline groups. In an 1H–11B HMBC experiment, the resonance assigned to the MeB(C6F5)3 at 1.27 ppm correlated with a singlet 11B NMR signal at −15.5 ppm. However as 2 stands in benzene-d6, the signals for ToMMgMeB(C6F5)3 decrease as the new species ToMMgC6F5 (3) and BMe3 form. After 7 h, ToMMgMeB(C6F5)3 is still the major component, but it is completely consumed over 20 h. This transformation occurs more rapidly in methylene chloride-d2 (t1/2 = 1 h).
Compound 3 is most conveniently prepared and isolated by the reaction of 1 equiv. of ToMMgMe and 1 equiv. of B(C6F5)3 in benzene-d6 over 24 h, but also forms from the reaction of 0.3 equiv. of B(C6F5)3 with ToMMgMe (eqn (4)). Solid ToMMgC6F5 was purified from the BMe3 side product by washing with pentane.
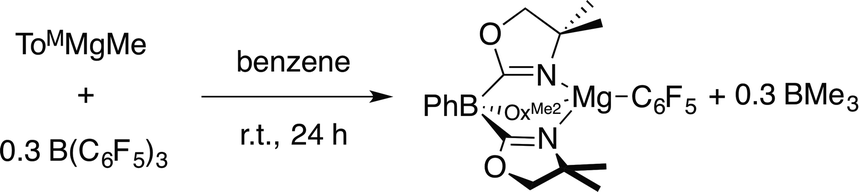 | (4) |
The 1H NMR spectrum of the crude reaction mixture contained a broad signal at 0.74 ppm assigned to BMe3 (ref. 31) and singlet resonances at 0.98 and 3.38 ppm assigned to the ToM ancillary ligand. Two peaks were observed in the 11B NMR spectrum at 86.5 and −18.3 ppm assigned to BMe3 and ToM, respectively. In addition, the tridentate coordination of the tris(oxazolinyl)borate ligand is supported by the 15N NMR chemical shift of −158 ppm and the νCN band in the infrared spectrum at 1594 cm−1. These values are similar to those of crystallographically characterized ToMMgMe (15N NMR: −157 ppm; νCN: 1592 cm−1).32 Three signals in the 19F NMR spectrum included a downfield signal at −110 ppm assigned to the ortho-fluorine. For comparison, C6F5MgBr provides three sets of 19F NMR signals, with ortho-F resonance appearing 45 ppm upfield of the para-F peak.33
The reaction of in situ generated 2 and PhSiH3 at room temperature in benzene-d6 gives only starting materials after 30 min. Over ca. 24 h, ToMMgMeB(C6F5)3 undergoes C6F5 transfer to the magnesium center, and PhSiH3 remains unconsumed. Micromolar-scale reactions in methylene chloride-d2 yield a mixture of ToMMgC6F5, BMe3, B(C6F5)3, and PhSiH3 after 2 h. On the basis of these observations, 2 is not an intermediate in the formation of the magnesium hydridoborate 1, and Path A is ruled out. Therefore, the currently preferred pathway for the formation of 1 involves methide abstraction by a transient borane–silane adduct (Scheme 1, Path C). In fact, the aryl group transfer from boron to magnesium may be a decomposition pathway for 1 in catalytic reactions (see below).
α,β-Unsaturated esters and silanes react through selective 1,4-hydrosilylation in the presence of catalytic amounts of ToMMgHB(C6F5)3 (1). For instance, the reaction of methyl methacrylate, Ph2SiH2, and 1 mol% 1 gives complete conversion of methyl methacrylate after 30 min in benzene-d6, as determined by 1H NMR spectroscopy (eqn (5)).
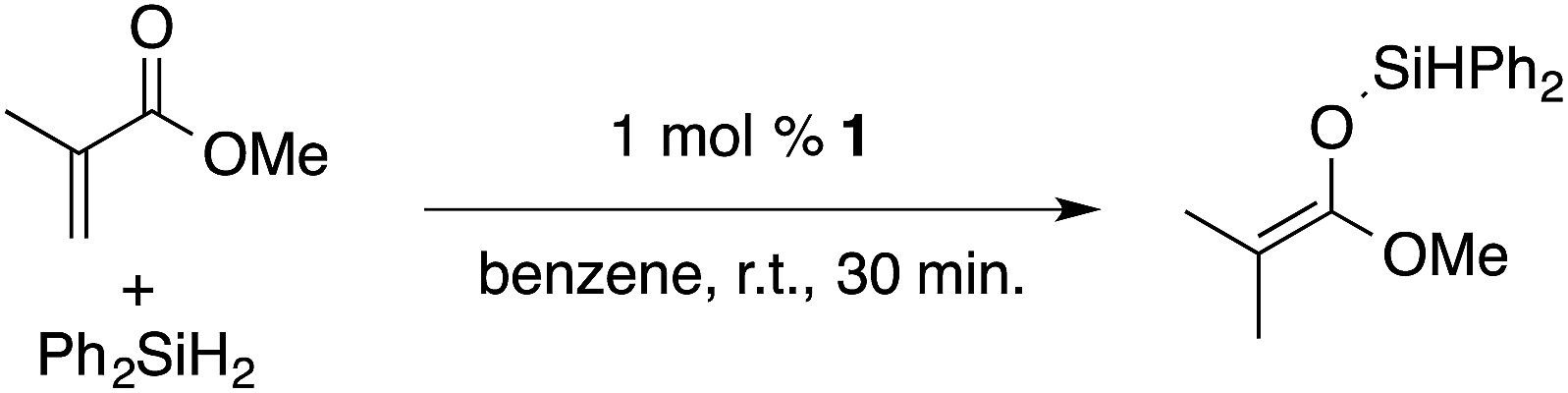 | (5) |
A 1H NMR spectrum of the isolated silyl ketene acetal product contained inequivalent methyl signals at 1.64 and 1.69 ppm, and singlets at 3.29 (3H) and 5.84 ppm (1H) assigned to the OMe and SiH groups. Olefinic signals, however, are not present in the product's 1H NMR spectrum. The 13C{1H} NMR spectrum contained a resonance at 150.93 ppm assigned to the acetal carbon. In an 1H–29Si HMBC experiment, a 29Si NMR signal at −14.5 ppm correlated to the SiH, inequivalent methyl signals, and phenyl resonances.
A range of silyl ketene acetals are prepared using 1 as the hydrosilylation catalyst (Table 1). Although transformations proceed with the low catalyst loadings of Table 1, scaled up reactions were performed with 20 mol% 1 to increase the rate of conversion. Secondary and tertiary silanes effectively hydrosilylate methyl methacrylate, and the products are isolated in good yield. In addition, the cyclic α,β-unsaturated ester 5,6-dihydro-2H-pyran-2-one react with PhMeSiH2 or BnMe2SiH in the presence of 1.
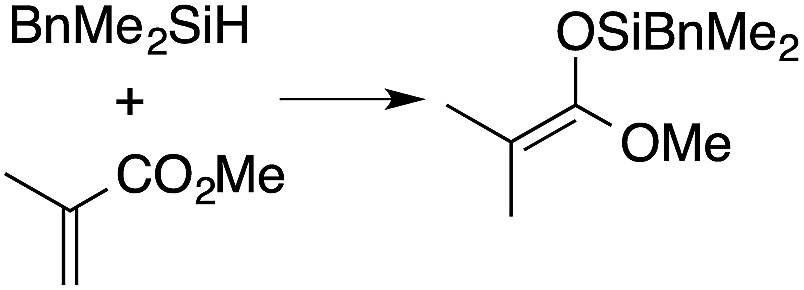
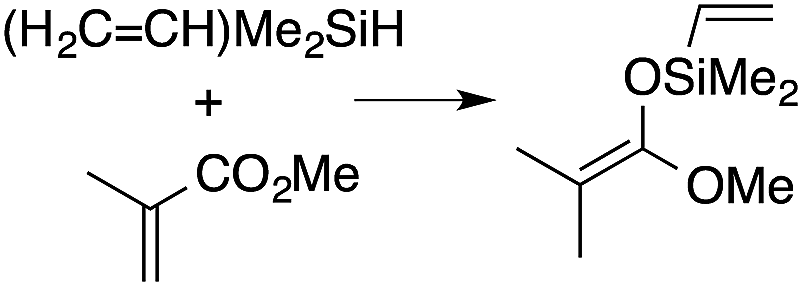
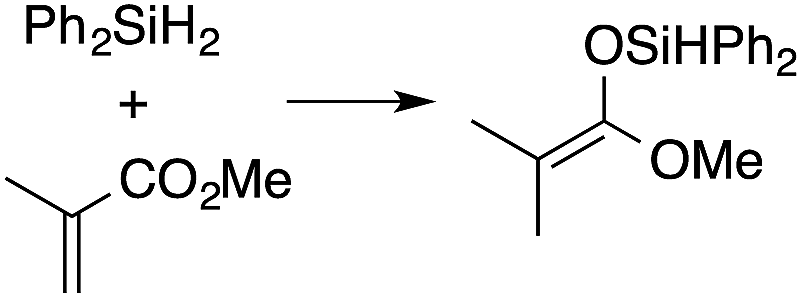
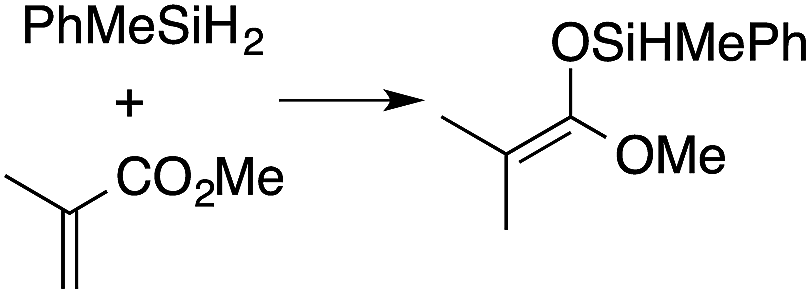
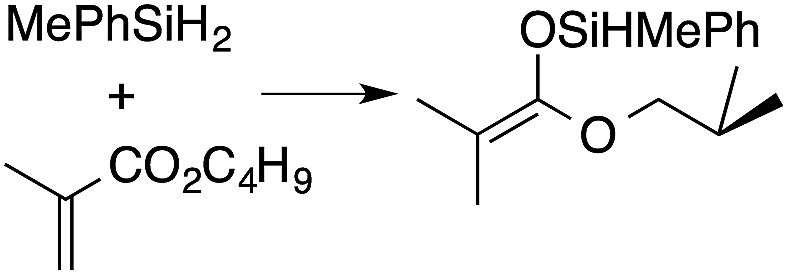
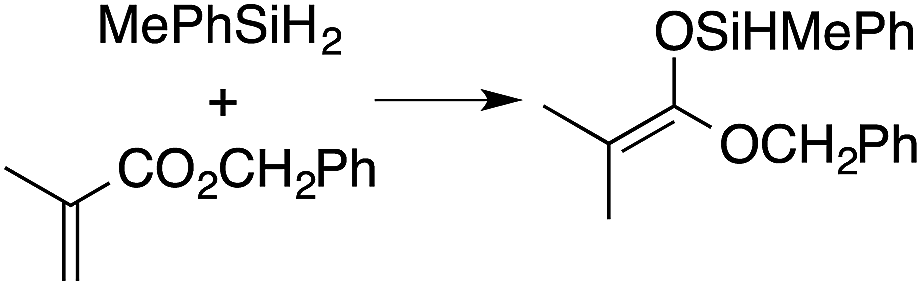
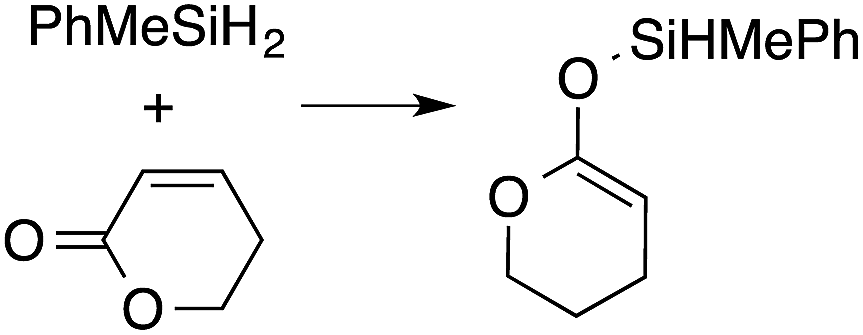
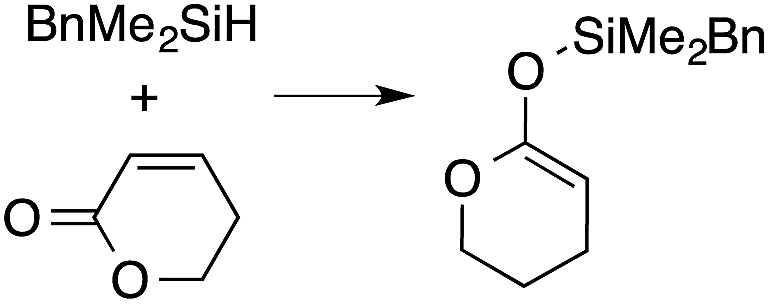
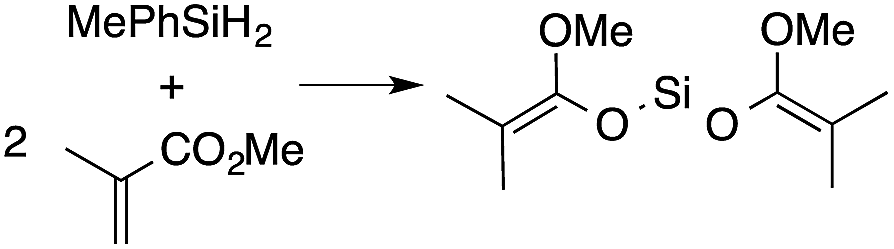
A number of experiments further test the key features of the catalyst structure and the reaction pathway. First, a series of ToMMgX compounds (X = Me, C6F5, MeB(C6F5)3, B(C6F5)4) were investigated as catalysts for hydrosilylation of methyl methacrylate. A catalytic amount of ToMMgMe reacts instantaneously with methyl methacrylate and PhMeSiH2 in benzene-d6 to give insoluble materials likely resulting from polymerization. Even though some of the silane is consumed in this reaction, neutral ToMMgMe is not a viable hydrosilylation catalyst. Moreover, this further demonstrates that the silicon–oxygen bond formation is unlikely to involve σ-bond metathesis of silanes and a magnesium alkoxide.
In addition, 1H NMR spectra of catalytic mixtures of methyl methacrylate, PhMeSiH2 and 10 mol% ToMMgMeB(C6F5)3 show only resonances assigned to methyl methacrylate and PhMeSiH2, and signals associated with the hydrosilylation product were not detected. ToMMgMeB(C6F5)3 is converted to ToMMgC6F5 under these conditions, and independent experiments show that ToMMgC6F5 is also not catalytically active. Hydridoborate-free magnesium compounds were tested next. The reaction of ToMMgMe and [Ph3C][B(C6F5)4] in benzene-d6 at room temperature gives [ToMMg][B(C6F5)4] as a precipitate after 15 min. However, this complex is not an ester hydrosilylation catalyst, and PhMeSiH2 and methyl methacrylate are unchanged after 2 d at 80 °C in the presence of 10 mol% [ToMMg][B(C6F5)4].
Alternatively, B(C6F5)3 is known as a hydrosilylation catalyst that mediates 1,2-addition of tertiary silanes to esters.5b Free B(C6F5)3 might be present in the reaction mixture as a result of its dissociation from 1, so its catalytic mode of action in mixtures of silanes and α,β-unsaturated esters was probed. However upon treatment with 10 mol% B(C6F5)3, BnMe2SiH or (H2CCH)Me2SiH and methacrylates provide mixtures containing the 1,4-addition product contaminated with at least 2 other species (see ESI† for spectra). The reactions of PhMeSiH2 and methyl methacylate, as catalyzed by 1 or 1 mol% B(C6F5)3, give inequivalent products. The product from the strong Lewis acid catalyst, in this case, does not contain an SiH, but is instead the double addition product PhMeSi{OC(OMe)CMe2}2 formed as part of a mixture. The B(C6F5)3 catalyzed reaction of PhMeSiH2 and benzyl methacylate gives a complicated mixture. Interestingly, lower B(C6F5)3 loadings generally result in increased amounts of the side products with respect to silyl ketene acetal. These data indicate that the hydrosilylation of the methacrylates is not catalyzed by B(C6F5)3 when 1 is used as the catalyst. The B(C6F5)3-catalyzed reaction of Et3SiH and methyl methacrylate, however, gives the silyl ketene acetal quantitatively, as does the same conversion catalyzed by 1. Thus, B(C6F5)3-catalyzed hydrosilylations are more sensitive to the substitution of the organosilane than conversions catalyzed by 1.
Next, the interaction of 1 and organosilane was probed by 1H and 11B NMR spectroscopy. In the 1H NMR spectrum, the intensity of methyl and methylene signals associated with the oxazoline ligand in 1 diminish by ca. 70% upon addition of 10 equiv. of BnMe2SiH, and new, albeit small, oxazoline methyl and methylene signals were observed. The new oxazoline signals are not sufficiently abundant to account for all of the previous ToM signals. Moreover, the quartet at 2.7 ppm for HB(C6F5)3 was not visible after addition of excess organosilane, although a number of broad signals appeared in that region. The SiH of BnMe2SiH appeared as a sharp multiplet and was apparently unchanged in the presence of 1. The broad doublet at −21 ppm in the 11B NMR spectrum of 1 decreased in intensity, and a new signal at −24 ppm appeared. The new upfield 11B NMR signal appeared in the region typical of HB(C6F5)3, but H–B coupling was not resolved in the broad signal. At low temperature (190 K), the 11B NMR signal at −24 was not detected, and the doublet at −21 is the major HB(C6F5)3 resonance. As the temperature increased to 260 K, the broad signal at −24 ppm appeared while the doublet at −21 diminished. At the same time, the 11B NMR signal at −18 ppm for ToM was sharp at 190 K, broad at 260 K, and again sharpened at 280 K. These data suggest that BnMe2SiH and ToMMgHB(C6F5)3 interact to disrupt the hydridoborate coordination to magnesium resulting in a dynamic system, but the HB(C6F5)3 moiety remains intact. Moreover, 11B NMR spectra acquired during catalytic conversions reveal signals at −18 and −24 ppm assigned to the boron centers in ToM and HB(C6F5)3. These two 11B NMR signals were also observed after complete conversion of methyl methacrylate via hydrosilylation. 1H NMR spectra of the catalytic reaction mixture, however, do not contain signals associated with 1. These data suggest that a fluxional derivative of 1 is involved in the catalytic conversion.
Under pseudo-first order conditions (using toluene-d8 as solvent) with excess methyl methacrylate, the half-life for the disappearance of Ph2SiH2 is ∼3 min at 64 °C, and over several minutes the silane is completely consumed. However, a methacrylate polymerization side-reaction interferes with kinetic measurements under these conditions. In the presence of excess Ph2SiH2 with respect to the methacrylate, zero-order, first-order, and second-order kinetic plots of methyl methacrylate concentration vs. time are non-linear, and complete conversion of the methacrylate is not obtained. The decrease in catalytic rate is even more prominent in methylene-chloride-d2 than in benzene-d6. In benzene-d6, the addition of methyl methacrylate and PhMeSiH2 is catalyzed by 10 mol% 1 in fewer than 10 min, while equivalent reaction conditions in methylene chloride-d2 give only 50% conversion after 24 h. Furthermore, the only ToM-containing 1H NMR resonances observed in the catalytic reaction mixture (in methylene chloride-d2) were those assigned to ToMMgC6F5. On the basis of faster conversion of ToMMgMeB(C6F5)3 to ToMMgC6F5 in methylene chloride than in benzene, the lack of activity of ToMMgC6F5 as a hydrosilylation catalyst, and the lower catalytic activity in methylene chloride than in benzene, we suggest that catalyst deactivation occurs through C6F5 migration from boron to magnesium.
Conclusions
The catalytic results above represent an unusual example of a magnesium-catalyzed hydrosilylation of CO containing compounds. This catalytic transformation is particularly noteworthy in the context of the oxophilic magnesium center, and the general challenge of catalytic turnover under such reducing conditions. While a kinetically-characterized catalytic mechanism is not accessible in the current system, plausible intermediates can be considered, and some may be ruled out, on the basis of the observed reactivity of ToMMgMe, ToMMgHB(C6F5)3 (1), ToMMgMeB(C6F5)3 (2), and ToMMgC6F5 (3). The catalytic intermediates might involve the coordination of the ester oxygen to the magnesium center, a boron–carbon bond-containing species, a silane adduct of a cationic magnesium center, and/or an enolate of magnesium or boron. As one possibility, a magnesium enolate and a borane–silane adduct might interact to give Si–O bond formation and regenerate 1, following the proposed pathway for the formation of 1 from PhSiH3, ToMMgMe, and B(C6F5)3. The catalysis requires [HB(C6F5)3]−, and no catalysis is observed with [B(C6F5)4]− or with neutral magnesium alkyls ToMMgMe or ToMMgC6F5, providing additional support for the bifunctional role of 1 in this hydrosilylation, as proposed in frustrated Lewis pair chemistry.34 Moreover, ToMMgMeB(C6F5)3 is not a viable hydrosilylation precatalyst, in contrast to ToMMgHB(C6F5)3. This result further supports the postulate that the hydridoborate is key to accessing the active magnesium species.
A catalyst deactivation pathway is suggested to involve the transfer of C6F5 from boron to magnesium to give ToMMgC6F5. ToMMgC6F5 is shown to be catalytically inert and to form more rapidly in methylene chloride than in benzene; the trend of faster catalyst deactivation in methylene chloride than in benzene parallels the faster formation of ToMMgC6F5 in the former solvent. These observations are taken as evidence in support of C6F5 transfer as a pathway to catalyst deactivation. This catalyst deactivation pathway is somewhat unexpected, given that magnesium alkyls are much more potent nucleophiles and bases than magnesium alkoxides. That is, in the presence of oxygen-containing substrates, a magnesium catalyst is deactivated by magnesium–carbon bond formation rather than magnesium–oxygen bond formation. This, and the catalytic hydrosilylation of oxygenates employing a highly oxophilic metal center, further indicates that the combination of a strong Lewis acid with early metal centers can access new reaction pathways through cooperation between the metal center and non-innocent counterion.
Experimental
ToMMgHB(C6F5)3 (1)
A solution of ToMMgMe (0.134 g, 0.32 mmol) dissolved in benzene was added in a dropwise fashion into a benzene solution containing PhSiH3 (0.069 g, 0.64 mmol) and B(C6F5)3 (0.162 g, 0.32 mmol). A white precipitate formed as the reaction mixture stirred for 30 min. The precipitate settled after centrifugation, and the supernatant was decanted. The white solid was washed with pentane (3 × 5 mL) and dried under vacuum, providing analytically pure ToMMgHB(C6F5)3 (0.286 g, 0.31 mmol, 97.6%). Once isolated, ToMMgHB(C6F5)3 is soluble in benzene or toluene, and X-ray quality single crystals were grown from a concentrated toluene solution at −30 °C. 1H NMR (600 MHz, benzene-d6): δ 0.82 (s, 18H, CNCMe2CH2O), 2.72 (br q, 1JBH = 69 Hz, 1H, MgHB(C6F5)3), 3.30 (s, 6H, CNCMe2CH2O), 7.38 (m, 3JHH = 7.2 Hz, 1H, para-C6H5), 7.56 (m, 3JHH = 7.6 Hz, 2H, meta-C6H5), 8.25 (d, 3JHH = 7.2 Hz, 2H, ortho-C6H5). 13C{1H} NMR (150 MHz, THF-d8): δ 27.35 (CNCMe2CH2O), 66.13 (CNCMe2CH2O), 79.28 (CNCMe2CH2O), 130.24 (para-C6H5), 133.26 (meta-C6H5), 134.79 (C6F5), 135.74 (C6F5), 136.81 (C6F5), 137.49 (ortho-C6H5), 138.41 (C6F5), 142 (br, ipso-C6H5), 147.78 (C6F5), 149.35 (C6F5), 191 (CNCMe2CH2O). 11B NMR (192 MHz, benzene-d6): δ −18.2 (ToM), −21.1 (d, 1JHB = 69 Hz, MgHB(C6F5)3). 19F NMR (544 MHz, benzene-d6): δ −134.2 (ortho-C6F5), −156.5 (para-C6F5), −161.4 (meta-C6F5). 15N NMR (60 MHz, benzene-d6): δ −162. IR (KBr, cm−1): ν 2976 (s), 2937 (s), 2372 (w br, BH), 1642 (m), 1579 (s), 1511 (s), 1459 (s br), 1373 (m), 1271 (m), 1199 (m), 1180 (m), 1161 (m), 1087 (s), 965 (s br), 843 (w), 804 (w), 735 (w), 705 (w). Anal. calcd for C39H30B2F15MgN3O3: C, 50.94; H, 3.29; N, 4.57. Found C, 51.38; H, 3.41; N, 4.31. Mp: 166–167 °C.Crystallography
![[P with combining macron]](https://www.rsc.org/images/entities/i_char_0050_0304.gif) 1, Z = 2, 15346 reflections measured, 9197 unique (Rint = 0.0303). The final R1(F2) and wR2(F2) for I > 2σ(I) were 0.0492 and 0.161.
1, Z = 2, 15346 reflections measured, 9197 unique (Rint = 0.0303). The final R1(F2) and wR2(F2) for I > 2σ(I) were 0.0492 and 0.161.
Representative catalytic hydrosilylation
CMe2), 1.69 (s, 3H, CCMe2), 3.29 (s, 3H, OMe), 5.84 (s, 1H, SiH), 7.17 (m, 6H, C6H5), 7.74 (m, 4H, C6H5). 13C{1H} NMR (150 MHz, benzene-d6): δ 16.79 (CCMe2), 17.42 (CCMe2), 57.92 (OMe), 91.74 (CCMe2), 130.47 (C6H5), 131.12 (C6H5), 134.16 (ipso-C6H5), 135.49 (C6H5), 136.39 (C6H5), 150.93 (CCMe2). 29Si (119 MHz, benzene-d6) δ −14.5 (d, 1JSiH = 201 Hz). IR (KBr, cm−1): ν 3094 (m), 2931 (s), 2158 (s), 1716 (s), 1661 (w), 1598 (m), 1566 (w), 1548 (w), 1528 (w), 1437 (s), 1263 (m), 1169 (br s), 1029 (m), 949 (m), 858 (s), 738 (s), 701 (s), 671 (w). Anal. calcd for C17H20O2Si: C, 71.79; H, 7.09. Found C, 71.61; H, 7.32.
Acknowledgements
The authors gratefully thank the National Science Foundation (CHE-0955635) for financial support. Dr Sarah Cady and Dr Shu Xu are thanked for valuable NMR assistance.References
- B. Marciniec, Hydrosilylation : a comprehensive review on recent advances, Springer, Berlin, 2009 Search PubMed.
- C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS.
- (a) J.-P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS PubMed; (b) S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2008, 41, 1486–1499 CrossRef CAS PubMed; (c) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (d) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed.
- (a) I. Fleming, R. Henning and H. Plaut, J. Chem. Soc., Chem. Commun., 1984, 29–31 RSC; (b) G. R. Jones and Y. Landais, Tetrahedron, 1996, 52, 7599–7662 CrossRef CAS; (c) K. Tamao, T. Kakui, M. Akita, T. Iwahara, R. Kanatani, J. Yoshida and M. Kumada, Tetrahedron, 1983, 39, 983–990 CrossRef CAS.
- (a) D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed; (b) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS; (c) W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Inorg. Chem., 2011, 50, 12252–12262 CrossRef CAS PubMed; (d) M. Rubin, T. Schwier and V. Gevorgyan, J. Org. Chem., 2002, 67, 1936–1940 CrossRef CAS PubMed.
- A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS PubMed.
- J. Koller and R. G. Bergman, Organometallics, 2011, 31, 2530–2533 CrossRef.
- (a) I. Kuwajima and E. Nakamura, Acc. Chem. Res., 1985, 18, 181–187 CrossRef CAS; (b) R. Mahrwald, Chem. Rev., 1999, 99, 1095–1120 CrossRef CAS PubMed; (c) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS PubMed.
- F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082–1146 CrossRef CAS PubMed.
- (a) I. Ojima and T. Kogure, Organometallics, 1982, 1, 1390–1399 CrossRef CAS; (b) I. Ojima, M. Nihonyanagi, T. Kogure, M. Kumagai, S. Horiuchi, K. Nakatsugawa and Y. Nagai, J. Organomet. Chem., 1975, 94, 449–461 CrossRef CAS; (c) I. Ojima, M. Nihonyanagi and Y. Nagai, J. Chem. Soc., Chem. Commun., 1972, 938 RSC; (d) C. R. Johnson and R. K. Raheja, J. Org. Chem., 1994, 59, 2287–2288 CrossRef CAS.
- A. Revis and T. K. Hilty, J. Org. Chem., 1990, 55, 2972–2973 CrossRef CAS.
- (a) S. Díez-González and S. P. Nolan, Acc. Chem. Res., 2008, 41, 349–358 CrossRef PubMed; (b) S. Huang, K. R. Voigtritter, J. B. Unger and B. H. Lipshutz, Synlett, 2010, 2041–2044 CAS; (c) V. Jurkauskas, J. P. Sadighi and S. L. Buchwald, Org. Lett., 2003, 5, 2417–2420 CrossRef CAS PubMed; (d) H. Kim and J. Yun, Adv. Synth. Catal., 2010, 352, 1881–1885 CrossRef CAS PubMed.
- (a) F. Buch and S. Harder, Z. Naturforsch., 2008, 63b, 169–177 Search PubMed; (b) F. Buch, J. Brettar and S. Harder, Angew. Chem., Int. Ed., 2006, 45, 2741–2745 CrossRef CAS PubMed; (c) V. Leich, T. P. Spaniol, L. Maron and J. Okuda, Chem. Commun., 2014, 50, 2311–2314 RSC; (d) A. G. M. Barrett, M. R. Crimmin, M. S. Hill and P. A. Procopiou, Philos. Trans. R. Soc., A, 2010, 466, 927–963 CrossRef CAS PubMed.
- J. Spielmann and S. Harder, Eur. J. Inorg. Chem., 2008, 1480–1486 CrossRef CAS PubMed.
- M. S. Hill, G. Kociok-Kohn, D. J. MacDougall, M. F. Mahon and C. Weetman, Dalton Trans., 2011, 40, 12500–12509 RSC.
- (a) S. Das, K. Möller, K. Junge and M. Beller, Chem.–Eur. J., 2011, 17, 7414–7417 CrossRef CAS PubMed; (b) K. Junge, B. Wendt, S. Zhou and M. Beller, Eur. J. Org. Chem., 2013, 2061–2065 CrossRef CAS PubMed; (c) S. Díez-González, N. M. Scott and S. P. Nolan, Organometallics, 2006, 25, 2355–2358 CrossRef; (d) A. J. Ruddy, C. M. Kelly, S. M. Crawford, C. A. Wheaton, O. L. Sydora, B. L. Small, M. Stradiotto and L. Turculet, Organometallics, 2013, 32, 5581–5588 CrossRef CAS.
- (a) M. Arrowsmith, M. S. Hill, T. Hadlington, G. Kociok-Köhn and C. Weetman, Organometallics, 2011, 30, 5556–5559 CrossRef CAS; (b) D. Mukherjee, A. Ellern and A. D. Sadow, Chem. Sci., 2014, 5, 959–964 RSC.
- (a) A. Berkefeld, W. E. Piers, M. Parvez, L. Castro, L. Maron and O. Eisenstein, J. Am. Chem. Soc., 2012, 134, 10843–10851 CrossRef CAS PubMed; (b) W. J. Evans, K. J. Forrestal, M. A. Ansari and J. W. Ziller, J. Am. Chem. Soc., 1998, 120, 2180–2181 CrossRef CAS; (c) K. Yan, G. Schoendorff, B. M. Upton, A. Ellern, T. L. Windus and A. D. Sadow, Organometallics, 2013, 32, 1300–1316 CrossRef CAS; (d) K. Yan, B. M. Upton, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2009, 131, 15110–15111 CrossRef CAS PubMed.
- M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. Kociok-Kohn, D. J. Liptrot, M. F. Mahon and C. Weetman, Chem. Sci., 2014, 5, 2826–2830 RSC.
- S. Schnitzler, T. P. Spaniol, L. Maron and J. Okuda, Chem.–Eur. J., 2015, 21, 11330–11334 CrossRef CAS PubMed.
- J. F. Dunne, S. R. Neal, J. Engelkemier, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2011, 133, 16782–16785 CrossRef CAS PubMed.
- X. Yang, C. L. Stern and T. J. Marks, Angew. Chem., Int. Ed. Engl., 1992, 31, 1375–1377 CrossRef PubMed.
- A. D. Horton and J. de With, Chem. Commun., 1996, 1375–1376 RSC.
- S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079–9083 CrossRef CAS PubMed.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem.–Eur. J., 2010, 16, 938–955 CrossRef CAS PubMed.
- J. Spielmann, M. Bolte and S. Harder, Chem. Commun., 2009, 6934–6936 RSC.
- K. Hedberg and V. Schomaker, J. Am. Chem. Soc., 1951, 73, 1482–1487 CrossRef CAS.
- X. Yang, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10015–10031 CrossRef CAS.
- (a) A. D. Sadow and T. D. Tilley, Organometallics, 2003, 22, 3577–3585 CrossRef CAS; (b) F. Wu and R. F. Jordan, Organometallics, 2005, 24, 2688–2697 CrossRef CAS.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- (a) C. Krempner, M. Köckerling, H. Reinke and K. Weichert, Inorg. Chem., 2006, 45, 3203–3211 CrossRef CAS PubMed; (b) D. A. Walker, T. J. Woodman, D. L. Hughes and M. Bochmann, Organometallics, 2001, 20, 3772–3776 CrossRef CAS.
- J. F. Dunne, D. B. Fulton, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2010, 132, 17680–17683 CrossRef CAS PubMed.
- D. F. Evans and M. S. Khan, Chem. Commun., 1966, 67–68 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental, synthesis and characterization of magnesium compounds and catalysis products. CCDC 1411027. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02435h |
| ‡ Current address: Department of Chemistry, University of Tennessee, 515 Dabney-Buehler Hall, 1420 Circle Dr., Knoxville, TN 37996, USA. |
| This journal is © The Royal Society of Chemistry 2015 |