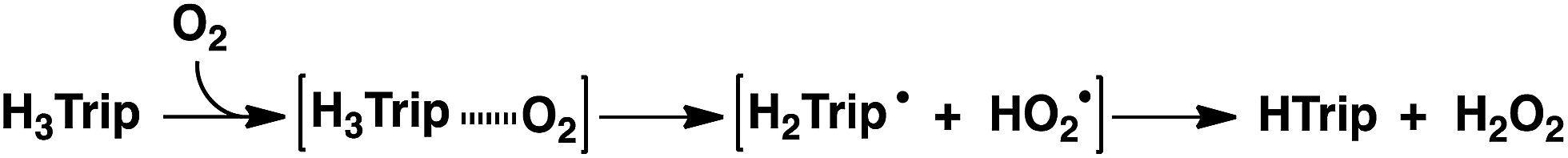DOI:
10.1039/C5SC02465J
(Edge Article)
Chem. Sci., 2015,
6, 6496-6504
Catalytic two-electron reduction of dioxygen catalysed by metal-free [14]triphyrin(2.1.1)†
Received
8th July 2015
, Accepted 2nd August 2015
First published on 3rd August 2015
Abstract
The catalytic two-electron reduction of dioxygen (O2) by octamethylferrocene (Me8Fc) occurs with a metal-free triphyrin (HTrip) in the presence of perchloric acid (HClO4) in benzonitrile (PhCN) at 298 K to yield Me8Fc+ and H2O2. Detailed kinetic analysis has revealed that the catalytic two-electron reduction of O2 by Me8Fc with HTrip proceeds via proton-coupled electron transfer from Me8Fc to HTrip to produce H3Trip˙+, followed by a second electron transfer from Me8Fc to H3Trip˙+ to produce H3Trip, which is oxidized by O2via formation of the H3Trip/O2 complex to yield H2O2. The rate-determining step in the catalytic cycle is hydrogen atom transfer from H3Trip to O2 in the H3Trip/O2 complex to produce the radical pair (H3Trip˙+ HO2˙) as an intermediate, which was detected as a triplet EPR signal with fine-structure by the EPR measurements at low temperature. The distance between the two unpaired electrons in the radical pair was determined to be 4.9 Å from the zero-field splitting constant (D).
Introduction
Utilization of natural energy to produce chemical energy consisting of earth-abundant elements is an essential technology for building a society based on the sustainable use of materials. Hydrogen peroxide (H2O2) produced by two-electron reduction of O2 is a versatile and environmentally benign oxidant, which is widely used on a large industrial scale.1,2 Furthermore, H2O2 has been proposed as a sustainable energy carrier that can be used in fuel cells, where direct and efficient conversion of chemical to electrical energy is required.3–5 However, the anthraquinone process, currently used to produce H2O2 in industry, requires potentially explosive hydrogen and a noble metal catalyst.6 Extensive efforts have so far been devoted to provide an alternative way to produce H2O2 photochemically or thermally without the use of noble metal catalysts.7–13 In many cases, redox-active transition metal-based complexes such as cobalt,14–23 iron,24–27 and copper complexes,28–31 have been employed as O2 reduction catalysts, because triplet O2 is inactive towards organic compounds due to spin restriction in the absence of an appropriate catalyst.32
Recently, nitrogen-doped carbon materials have attracted increasing attention as an efficient metal-free catalyst for the catalytic reduction of O2.33–35 However, the catalytic mechanism has yet to be well understood, because few spectroscopic studies to detect reaction intermediates in a catalytic cycle have been performed on heterogeneous systems. In homogeneous systems, reduced flavin analogues involved in flavoenzymes have so far been known to play a pivotal role in the catalytic reduction of O2, which is a key step of biological oxidation.36,37 In particular, the deprotonated states of reduced flavin analogues, which are thermodynamically more able to reduce O2via an electron-transfer process, are considered to be a reactive intermediate in the reduction of O2.38
On the other hand, Girault and coworkers recently reported that the free base porphyrin has the ability to catalyse the two-electron reduction of O2 using one-electron reductants such as ferrocene at liquid–liquid interfaces.39 In such systems, although the catalytic mechanism of metal-free organocatalysts has yet to be clarified, the oxidation state of the organocatalyst is thought to remain the same during the catalytic reduction of O2. Thus, no electron-transfer reduction of organic catalysts has been reported in relation to the catalytic reduction of O2.
In this context, Nocera and coworkers recently reported the stabilization of the peroxide dianion within the cavity of a hexacarboxamide cryptand,40 where strong hydrogen bond donors are arranged to completely surround the peroxide dianion with a partial positive charge. This result provides support for the proposal that metal-free organocatalysts, which have multiple hydrogen bonding moieties, can efficiently catalyse O2 reduction.
We report herein the catalytic two-electron reduction of O2 by an one-electron reductant, octamethylferrocene (Me8Fc), with metal-free [14]triphyrin(2.1.1) (denoted as HTrip in Chart 1)41 in the presence of HClO4 in benzonitrile (PhCN) at 298 K. The catalytic mechanism for the O2 reduction by Me8Fc is clarified on the basis of a detailed kinetic study. Proton-coupled electron-transfer reduction of HTrip by Me8Fc results in the formation of the reduced state of HTrip, and this resulting reduced HTrip is oxidized by O2 to reproduce HTrip, indicating that HTrip acts as a metal-free catalyst for the reduction of O2 by Me8Fc in the presence of HClO4 in PhCN. This discovery of a reactive intermediate in the catalytic O2 reduction with a molecular organic catalyst provides valuable insight into the development of an efficient metal-free catalyst for the reduction of O2.
 |
| | Chart 1 Structure of HTrip. | |
Results and discussion
Protonation of HTrip with HClO4
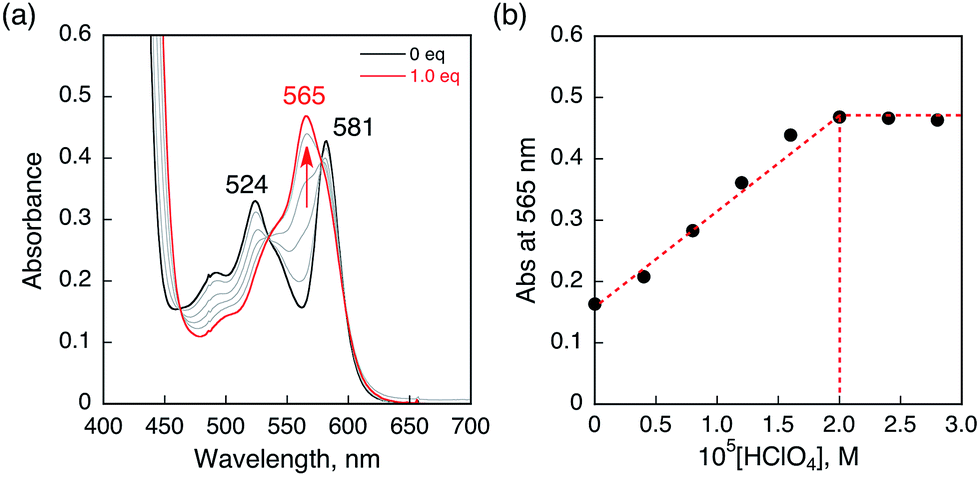
HTrip was protonated by addition of perchloric acid (HClO4) to an air-saturated benzonitrile (PhCN) solution of HTrip. The characteristic absorption bands for HTrip at 524 and 581 nm decreased in intensity, with an increase in the absorption band at 565 nm, exhibiting clean isosbestic points, as shown in Fig. 1a. As can be seen in Fig. 1b, the absorbance change at 565 nm is saturated in the presence of 1 equiv. of HClO4. Thus, HTrip is protonated to afford H2Trip+, as given by eqn (1).The pKa value of H2Trip+ in PhCN was estimated to be 15.6 from the titration of HTrip with trifluoroacetic acid (TFA), as shown in Fig. S1 in the ESI.† The pKa value of H2Trip+ is slightly larger than that of free base porphyrin analogues.42 There is no further protonation due to strong repulsion between NH protons in the small macrocyclic ligand, as reported previously.41
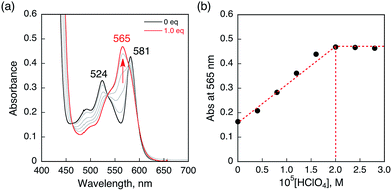 |
| | Fig. 1 (a) Absorption spectral changes of HTrip (2.0 × 10−5 M) upon the addition of HClO4 in air-saturated PhCN at 298 K. (b) Absorbance change profile at 565 nm. | |
Electrochemical measurements of HTrip in the presence of HClO4
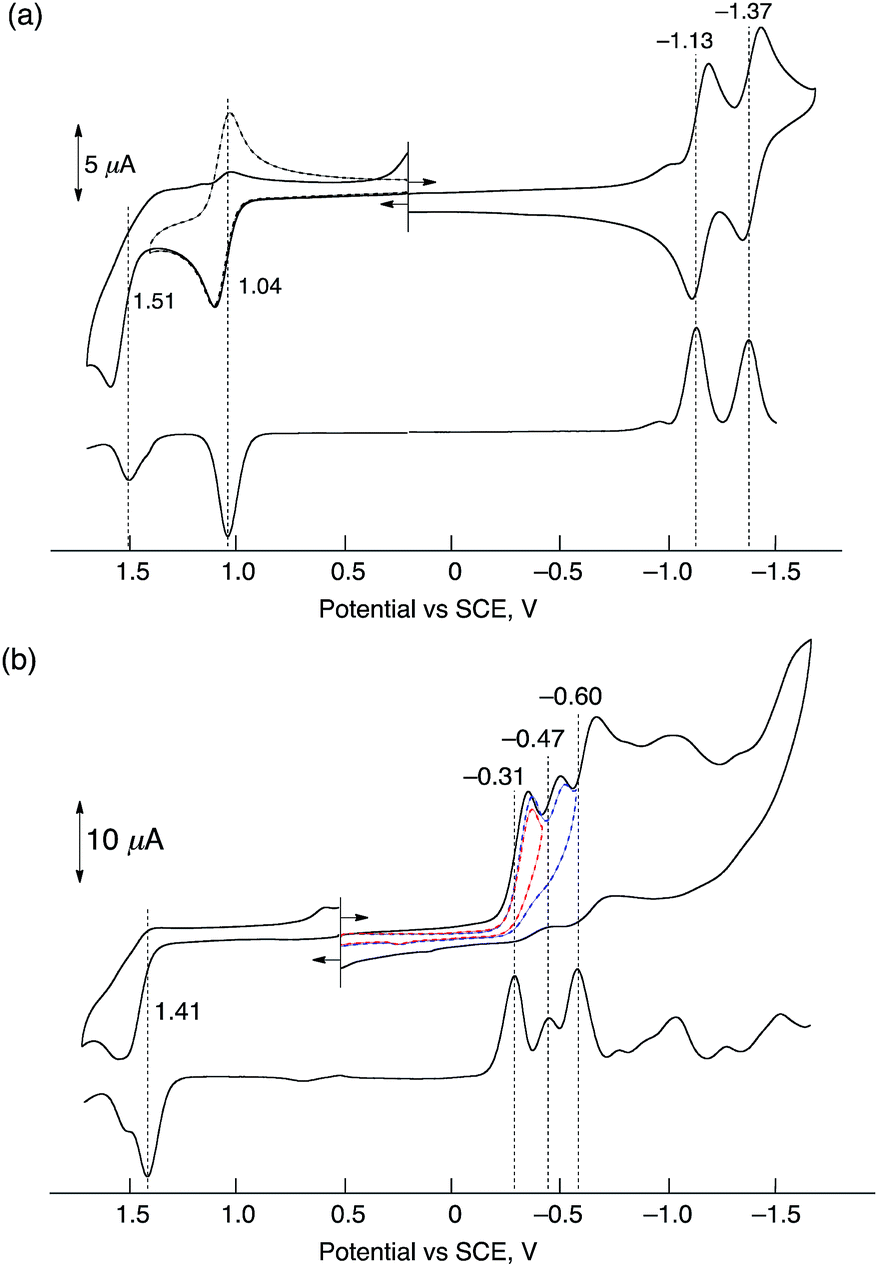
Electrochemical measurements of HTrip were performed in deaerated PhCN containing 0.10 M TBAPF6, as shown in Fig. 2. A cyclic voltammogram of HTrip exhibits reversible reduction waves at E1/2 = −1.13 and −1.37 V (vs. SCE), which correspond to the first and second one-electron reduction of HTrip. The first one-electron oxidation occurs at E1/2 = 1.04 V, which is followed by an irreversible oxidation (Fig. 2a). The formation of HTrip˙− was detected by UV-vis absorption spectra in the electrochemical reduction of HTrip at a controlled potential of −1.25 V vs. SCE in the thin-layer cell, as shown in Fig. S2 in the ESI.† By addition of HClO4, the first reduction potential of HTrip was positively shifted from E1/2 = −1.13 V to −0.31 V (vs. SCE) because of the protonation of HTrip, but the reduction became irreversible (Fig. 2b). In such a case, proton-coupled electron transfer from an electron donor with the one-electron oxidation potential, which is less negative than −0.31 V, to HTrip may be thermodynamically feasible (vide infra).
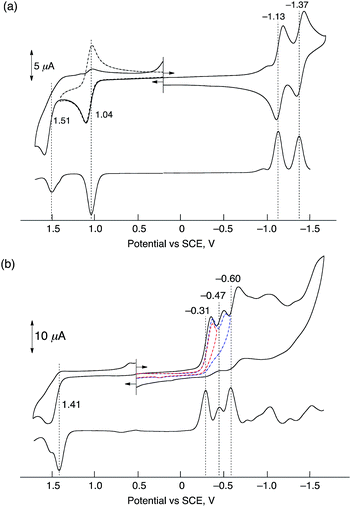 |
| | Fig. 2 Cyclic voltammograms (upper) and differential pulse voltammograms (lower) of deaerated PhCN solutions of HTrip (1.0 × 10−3 M) recorded in the presence of TBAPF6 (0.10 M) (a) without HClO4 and (b) with HClO4 (1.0 × 10−2 M); sweep rate: 100 mV s−1 for CV and 4 mV s−1 for DPV. | |
Electron-transfer reduction of HTrip in the presence of HClO4
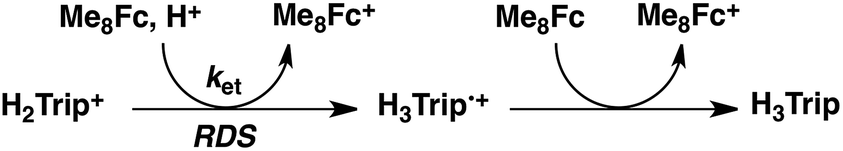
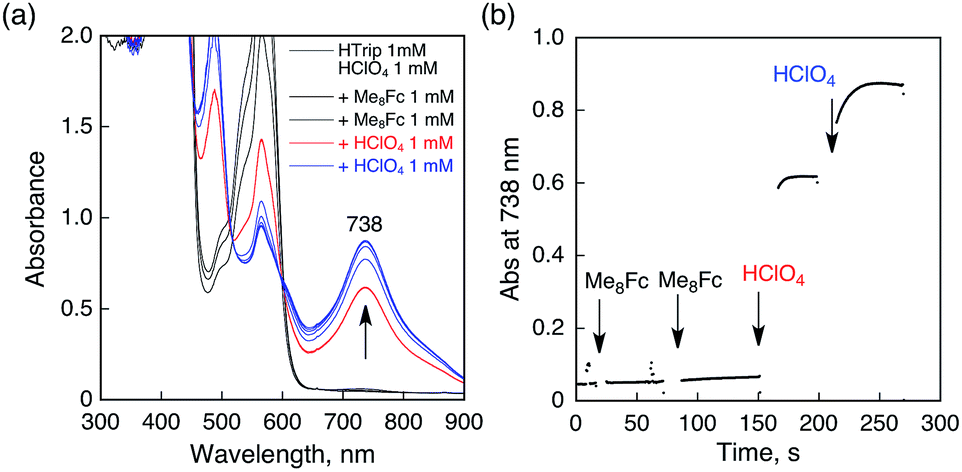
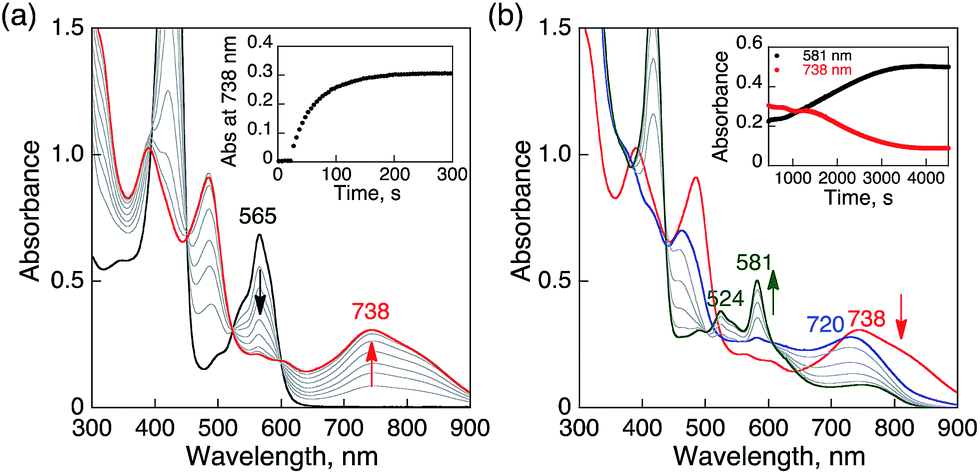
No electron transfer from Me8Fc to HTrip occurred in the absence of HClO4 in PhCN at 298 K, as indicated by the more negative E1/2 value of HTrip (−1.13 V vs. SCE) as compared with that of Me8Fc (−0.04 V vs. SCE).8 However, the addition of more than two equiv. of HClO4 to a deaerated PhCN solution of Me8Fc and HTrip resulted in the appearance of an absorption band at 738 nm due to H3Trip with clean isosbestic points, as shown in Fig. 3. It should be noted that no electron transfer from Me8Fc to H2Trip+ occurred in the presence of one equiv. of HClO4, as shown in Fig. 3b. These results indicate that uphill electron transfer from Me8Fc to H2Trip+ is coupled with protonation of H2Trip˙ to produce H3Trip˙+, followed by fast electron transfer from Me8Fc to H3Trip˙+ to yield H3Trip. Thus, the second protonation in fact occurs by coupling with reduction of H2Trip+ (i.e. H3Trip˙+ is accessible but not H3Trip2+). The stoichiometry of the overall reaction is given in Scheme 1.
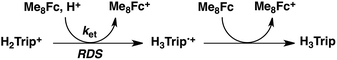 |
| | Scheme 1 | |
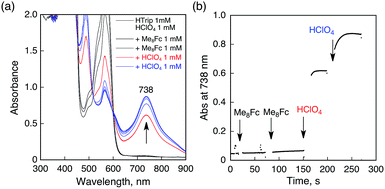 |
| | Fig. 3 (a) Absorption spectral changes upon addition of Me8Fc (1.0 × 10−3 and 2.0 × 10−3 M) to a deaerated PhCN solution of H2Trip+ (1.0 × 10−3 M) in the presence of HClO4 (1.0 × 10−3 M) at 298 K in a quartz cuvette (light path length = 1 mm) (black); absorption spectral change upon addition of HClO4 (1.0 × 10−3 M) to the solution indicated by the black line (red); absorption spectral change upon addition of HClO4 (1.0 × 10−3 M) to the solution indicated by the red line (blue). (b) Absorption change at 738 nm upon addition of various concentrations of Me8Fc and HClO4. | |
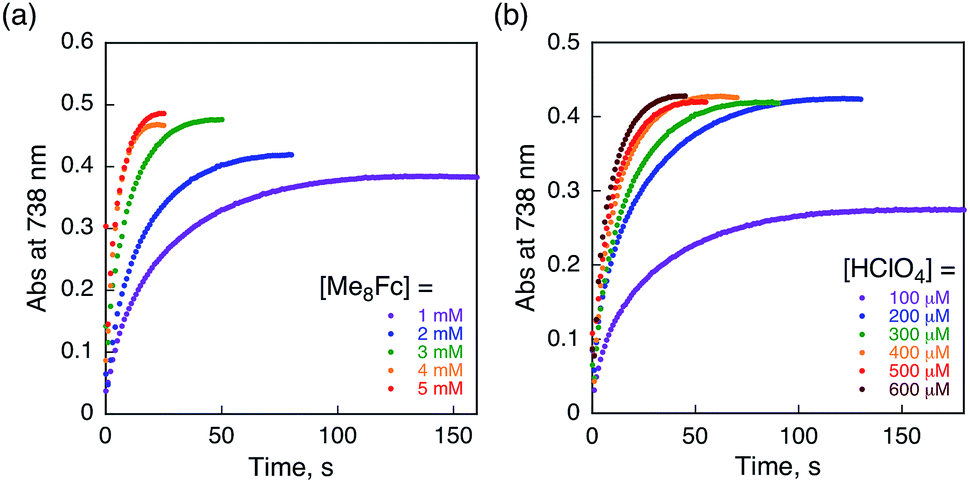
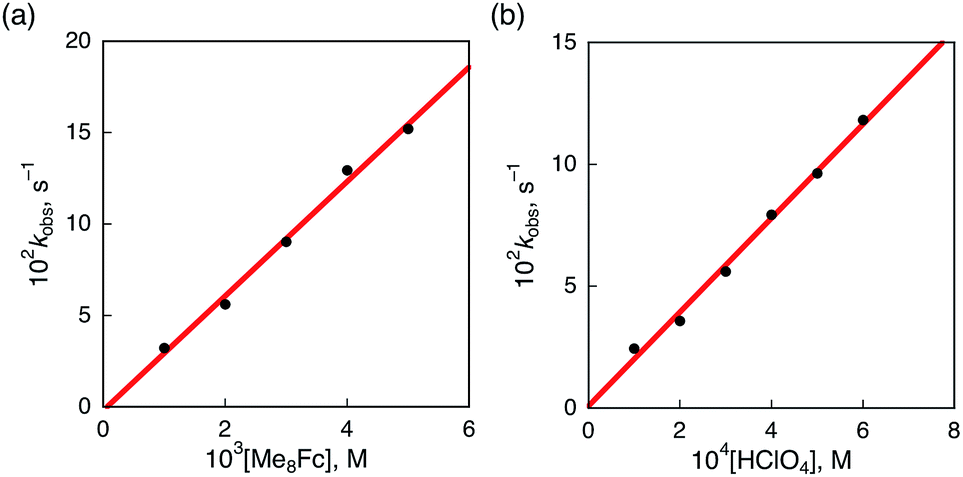
The rate of proton-coupled electron-transfer reduction of H2Trip+ (ket) to form H3Trip˙+ was determined from the dependence of the observed rate constant (kobs) on concentrations of Me8Fc and HClO4, as shown in Fig. 4. The kobs value was determined from the increase in absorbance at 738 nm due to H3Trip, which obeyed first-order kinetics (Fig. S3 in the ESI†). The kobs value increased linearly with increasing concentrations of Me8Fc and HClO4, as shown in Fig. 5. Thus, the rate of formation of H3Trip is given by eqn (2).
| | | d[H3Trip]/dt = ket[H2Trip+][HClO4][Me8Fc] | (2) |
The
ket value is determined from the slope of the linear plot of
kobsvs. [Me
8Fc] and [HClO
4] to be (9.8 ± 0.2) × 10
4 M
−2 s
−1. The
ket value of the proton-coupled electron-transfer reduction of H
2Trip
+ by Me
10Fc was also determined from the slope of the linear plot of
kobsvs. [Me
10Fc] and [HClO
4] to be (3.1 ± 0.3) × 10
5 M
−2 s
−1 (Fig. S4–S6 in the ESI
†). The
ket value for Me
10Fc is larger than that for Me
8Fc because Me
10Fc (
Eox = −0.08 V
vs. SCE) is a stronger electron donor than Me
8Fc (−0.04 V
vs. SCE).
28
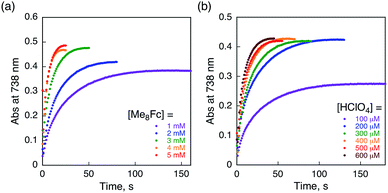 |
| | Fig. 4 Time profiles of absorbance at 738 nm due to H3Trip in the reduction of H2Trip+ (2.5 × 10−5 M) (a) by various concentrations of Me8Fc in the presence of HClO4 (3.0 × 10−4 M) and (b) by Me8Fc (2.0 × 10−3 M) in the presence of various concentrations of HClO4 in deaerated PhCN at 298 K. | |
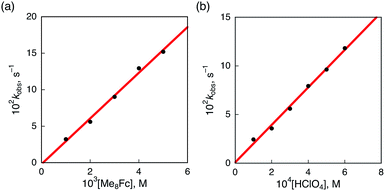 |
| | Fig. 5 (a) Plot of kobsvs. [Me8Fc] for the reduction of H2Trip+ (2.5 × 10−5 M) by various concentrations of Me8Fc in the presence of HClO4 (3.0 × 10−4 M) in PhCN at 298 K. (b) Plot of kobsvs. [HClO4] for the reduction of H2Trip+ (2.5 × 10−5 M) by Me8Fc (2.0 × 10−3 M) in the presence of various concentrations of HClO4 in deaerated PhCN at 298 K. | |
The formation of H3Trip was also confirmed by the electrochemical reduction of H2Trip+ monitored by the UV-vis spectral change at an applied potential of −0.30 V vs. SCE in the thin-layer cell, as shown in Fig. S7 (in the ESI†). The product obtained after the electrochemical reduction of H2Trip+ at −0.30 V displayed the characteristic absorption band at 738 nm. The same absorption band was seen in the chemical reduction of H2Trip+ by Me8Fc in the presence of HClO4 (Fig. 2).
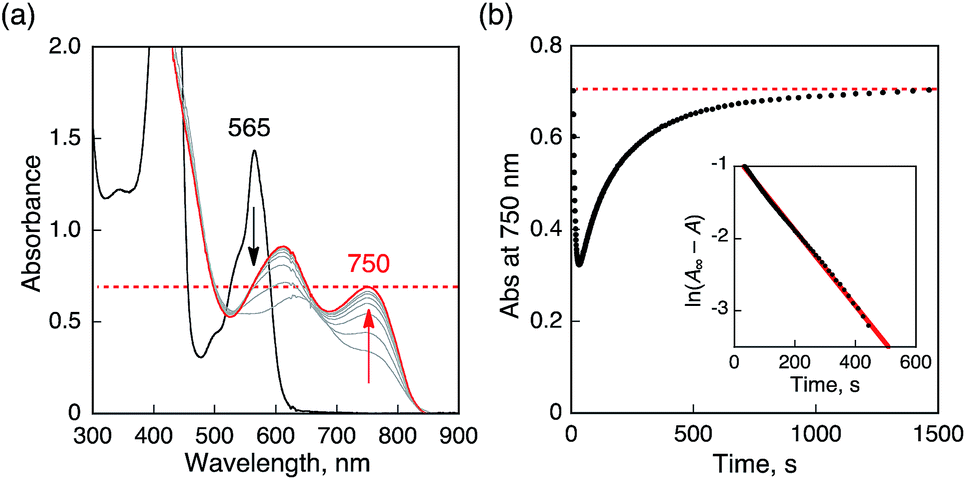
When O2 was introduced to a deaerated PhCN solution of H3Trip produced by the proton-coupled electron transfer from Me8Fc to HTrip in the presence of HClO4, the absorption band at 738 nm due to H3Trip was immediately changed to a new absorption band at 720 nm, which can be attributed to the formation of the O2 complex, as shown in Scheme 2 (vide infra). Subsequently, this spectrum decreased gradually, accompanied by the regeneration of HTrip as shown in Fig. 6. This indicates that H3Trip was readily oxidized by O2 to produce HTrip and H2O2 (Scheme 2).
 |
| | Scheme 2 | |
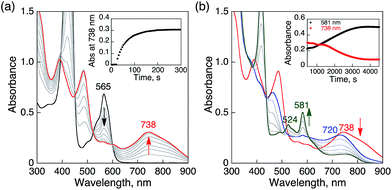 |
| | Fig. 6 (a) Absorption spectral changes produced by electron transfer from Me8Fc (1.0 × 10−4 M) to HTrip (2.5 × 10−5 M) in the presence of HClO4 (1.0 × 10−4 M) in deaerated PhCN at 298 K. (b) Absorption spectral changes upon introducing O2 to a deaerated PhCN solution of (a). The red and green lines show the spectrum of H3Trip before and after introducing O2 by O2 gas bubbling, respectively. The blue line shows the spectrum due to precursor complex. Insets show absorption time profiles. | |
Catalytic two-electron reduction of O2 by Me8Fc with HTrip in the presence of HClO4
The proton-coupled electron-transfer reduction of HTrip by Me8Fc (Scheme 1) and the oxidation of the resulting reduced HTrip (H3Trip) by O2 (Scheme 2) indicate that HTrip acts as a metal-free catalyst for the reduction of O2 by Me8Fc in the presence of HClO4 in PhCN. Indeed, the addition of Me8Fc to air-saturated PhCN at 298 K containing a catalytic amount of HTrip and a large excess of HClO4 resulted in the efficient oxidation of Me8Fc by O2 to yield Me8Fc+, as shown in Fig. 7a.
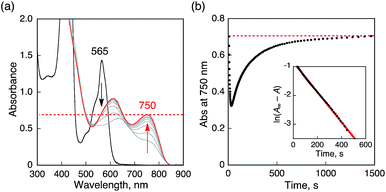 |
| | Fig. 7 (a) Absorption spectral changes in the two-electron reduction of O2 (9.4 × 10−4 M) by Me8Fc (1.0 × 10−2 M) with HTrip (5.0 × 10−5 M) in the presence of HClO4 (1.0 × 10−2 M) in PhCN at 298 K. The black and red lines show the spectra before and after addition of Me8Fc, respectively. The dotted line is the absorbance at 750 nm due to 1.9 × 10−3 M of Me8Fc+. (b) Time profile of absorbance at 750 nm due to Me8Fc+. Inset shows first-order plot. | |
The formation of Me8Fc+ was monitored by a rise in absorbance at 750 nm due to Me8Fc+ (Fig. 7b). When an excess amount of Me8Fc relative to O2 (i.e., [O2] limiting conditions) was employed, the concentration of produced Me8Fc+ (1.9 × 10−3 M) was twice that of O2 (9.4 × 10−4 M). In addition, the stoichiometric production of H2O2 was confirmed by iodometric titration, as shown in Fig. S8 (in the ESI†). In contrast, when an excess amount of O2 relative to Me8Fc (i.e., [Me8Fc] limiting conditions) was employed, the concentration of produced H2O2 (1.0 × 10−3 M) was half that of Me8Fc (2.0 × 10−3 M), where the amount of H2O2 was determined by the reaction with [(TMC)FeII](OTf)2 (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) to produce the corresponding Fe(IV)-oxo complex ([TMC]FeIV(O))2+, as shown in Fig. S9 (in the ESI†).43 Thus, the stoichiometry of the catalytic reduction of O2 by Me8Fc has been firmly established, as given in eqn (3).
| |  | (3) |
The rate of formation of Me8Fc+ in the catalytic reduction of O2 with excess Me8Fc and HClO4 in Fig. 7b obeys first-order kinetics. It should be noted that the oxidation of Me8Fc by O2 hardly occurred in the absence of HTrip under the present experimental conditions, as shown in Fig. S10 (in the ESI†). When Me8Fc was replaced by weaker one-electron reductants such as ferrocene (Fc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Eox = 0.37 V vs. SCE) and dimethylferrocene (Me2Fc:Eox = 0.26 V vs. SCE), no changes in the absorption band of H2Trip+ at 565 nm were observed, as shown in Fig. S11 (in the ESI†). When Me8Fc was replaced by a stronger one-electron reductant, i.e., decamethylferrocene (Me10Fc:Eox = −0.10 V vs. SCE), greatly enhanced oxidation of Me10Fc occurred with the decrease in absorbance at 565 nm due to H2Trip+ (Fig. S12a in the ESI†). In the case of Me10Fc, however, the oxidation of Me10Fc by O2 occurred without HTrip in the presence of HClO4 in PhCN (Fig. S12c in the ESI†). These results indicate that the reduction of H2Trip+ to produce H3Trip is essential in the catalytic reduction of O2 to produce H2O2.
:Eox = 0.37 V vs. SCE) and dimethylferrocene (Me2Fc:Eox = 0.26 V vs. SCE), no changes in the absorption band of H2Trip+ at 565 nm were observed, as shown in Fig. S11 (in the ESI†). When Me8Fc was replaced by a stronger one-electron reductant, i.e., decamethylferrocene (Me10Fc:Eox = −0.10 V vs. SCE), greatly enhanced oxidation of Me10Fc occurred with the decrease in absorbance at 565 nm due to H2Trip+ (Fig. S12a in the ESI†). In the case of Me10Fc, however, the oxidation of Me10Fc by O2 occurred without HTrip in the presence of HClO4 in PhCN (Fig. S12c in the ESI†). These results indicate that the reduction of H2Trip+ to produce H3Trip is essential in the catalytic reduction of O2 to produce H2O2.
When a metal complex of HTrip, η5-cyclopentadienyliron(II) [14]triphyrin(2.1.1) (CpFeIITrip),41c was employed as an O2 reduction catalyst instead of HTrip for comparison, however, the addition of HClO4 to an air-saturated PhCN solution of CpFeIITrip resulted in a spectral change, as shown in Fig. S13 (in the ESI†). The characteristic absorption bands of CpFeIITrip at 545 nm and 608 nm disappeared upon the addition of HClO4 with the appearance of new absorption bands at 565 nm, which can be attributed to those of H2Trip+. This indicates that CpFeIITrip was easily demetallated and protonated to afford H2Trip+ in the presence of HClO4, as shown in Fig. S13 (in the ESI†).
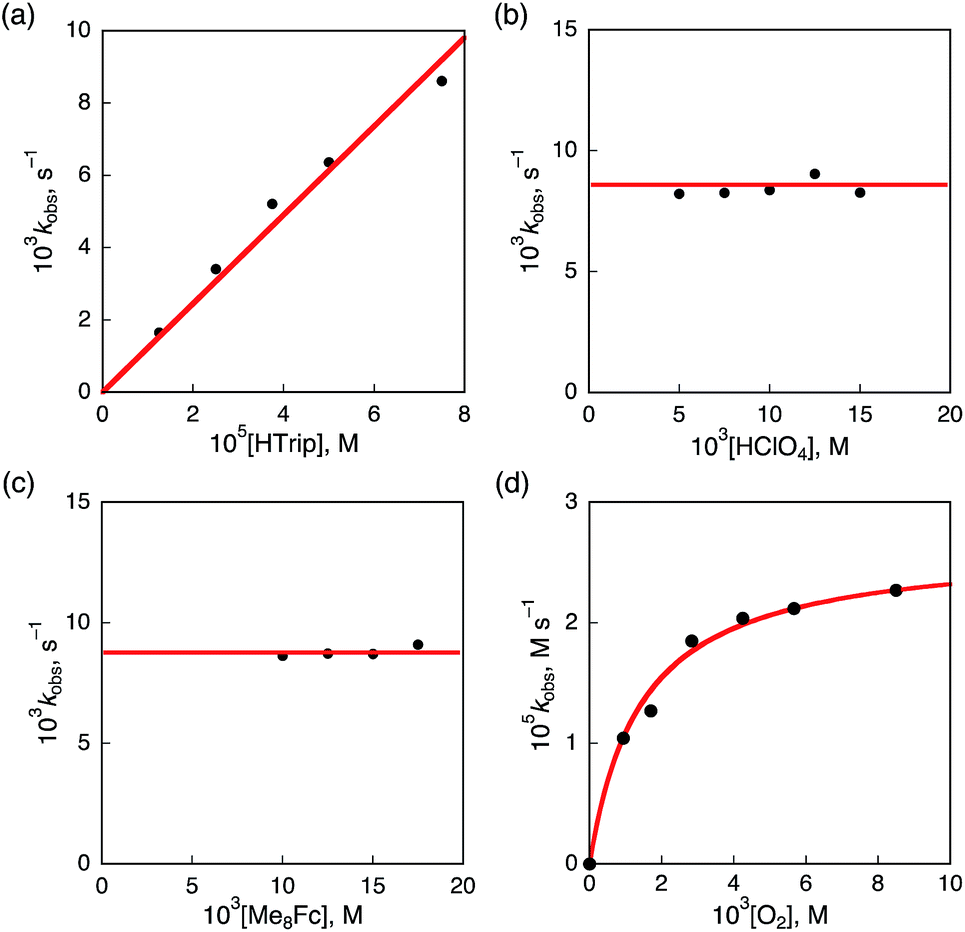
Kinetics and mechanism of the catalytic two-electron reduction of O2 by Me8Fc with HTrip
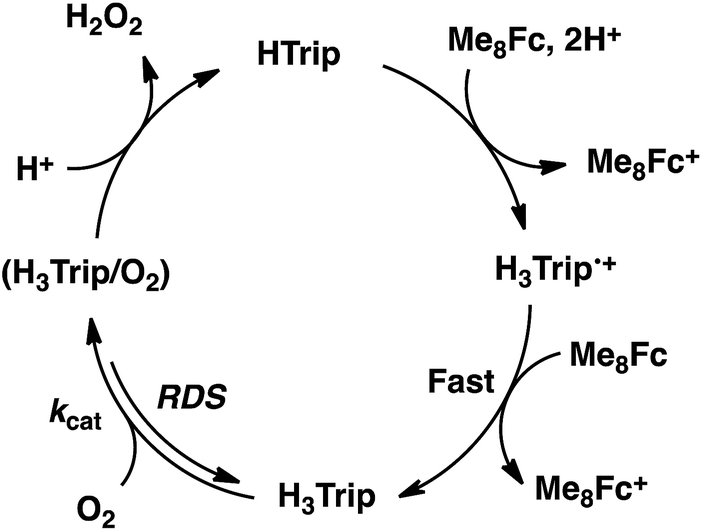
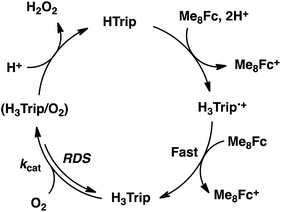
The dependence of the first-order rate constant for the formation of Me8Fc+ on the concentrations of HTrip, HClO4, Me8Fc, and O2 was examined, as shown in Fig. S14 (in the ESI†), where the first-order rate constants were determined from the initial slopes of the first-order plots in order to avoid further complication due to the deactivation of the catalyst during the reactions, as shown in Fig. S15 (in the ESI†). The observed first-order rate constant (kobs) was proportional to the concentration of HTrip, whereas the kobs value remained constant irrespective of the concentration of HClO4 or Me8Fc (Fig. 8). Although no degradation of HTrip occurred under the present acidic conditions (Fig. S16 in the ESI†), the turnover number (TON) based on HTrip was determined to be more than 40 when the lower concentration of HTrip (1.3 × 10−5 M) was employed, as shown in Fig. S14a (in the ESI†). Because the catalytic rate depends only on the concentrations of HTrip and O2, the rate-determining step in the catalytic cycle must be the reaction of H3Trip with O2 in Scheme 3. The dependence of the initial rate of formation of Me8Fc+ on the concentration of O2 shows saturation behaviour at large concentrations of O2 (Fig. 8d). Such saturation behaviour is consistent with the formation of the O2 complex (H3Trip/O2) in the oxidation of H3Trip with O2 (Fig. 6b and Scheme 3). The overall catalytic cycle is shown in Scheme 3, where proton-coupled electron transfer from Me8Fc to HTrip is followed by a second electron transfer from Me8Fc to H3Trip˙+ to produce H3Trip, which is slowly oxidized by O2via the H3Trip/O2 complex as the rate-determining step. Because the direct reaction between H3Trip and O2 in the H3Trip/O2 complex is spin-forbidden, the reaction may proceed via hydrogen atom transfer from H3Trip to O2 in the H3Trip/O2 complex to produce the (H2Trip˙/HO2˙) intermediate, followed by a rapid second hydrogen transfer from H2Trip˙ to HO2˙ to yield H2O2, accompanied by regeneration of HTrip (Scheme 3). According to Scheme 3, the rate of formation of Me8Fc+ is given by eqn (4),| | | d[Me8Fc+]/dt = kcat[H3Trip/O2], | (4) |
where kcat is the rate constant of the hydrogen atom transfer from H3Trip to O2 in the H3Trip/O2 complex. Because the concentration of the H3Trip/O2 complex is given by eqn (5)| | | [H3Trip/O2] = K[HTrip][O2]/(1 + K[O2]), | (5) |
using the formation constant (K), the initial concentration of HTrip, which is converted to H3Trip in the catalytic reaction, and the concentration of O2, eqn (4) is rewritten as eqn (6).| | | d[Me8Fc+]/dt = kcatK[HTrip][O2]/(1 + K[O2]) | (6) |
This kinetic equation agrees with the experimental observations in Fig. 8. The kcat and K values were determined from the dependence of the catalytic rate on the concentration of O2 (Fig. 8d) to be 0.5 s−1 and 8.4 × 102 M−1, respectively.
 |
| | Scheme 3 | |
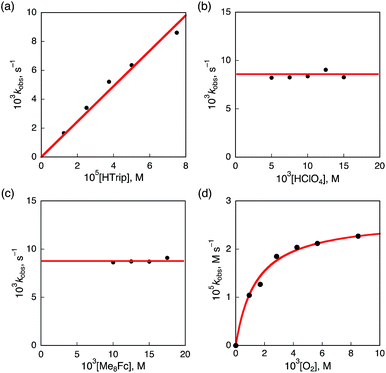 |
| | Fig. 8 Plots of (a) kobsvs. [HTrip] for the two-electron reduction of O2 (9.4 × 10−4 M) by Me2Fc (1.0 × 10−2 M) with various concentrations of HTrip in the presence of HClO4 (1.0 × 10−2 M) in PhCN; (b) kobsvs. [HClO4] for the two-electron reduction of O2 (9.4 × 10−4 M) by Me8Fc (1.0 × 10−2 M) with HTrip (5.0 × 10−5 M) in PhCN at 298 K; (c) kobsvs. [Me8Fc] for the two-electron reduction of O2 (9.4 × 10−4 M) by various concentrations of Me8Fc with HTrip (5.0 × 10−5 M) in the presence of HClO4 (1.0 × 10−2 M) in PhCN at 298 K; and (d) kobsvs. [O2] for the two-electron reduction of O2 by Me8Fc (1.0 × 10−2 M) with HTrip (5.0 × 10−5 M) in the presence of HClO4 (1.0 × 10−2 M) in PhCN at 298 K. | |
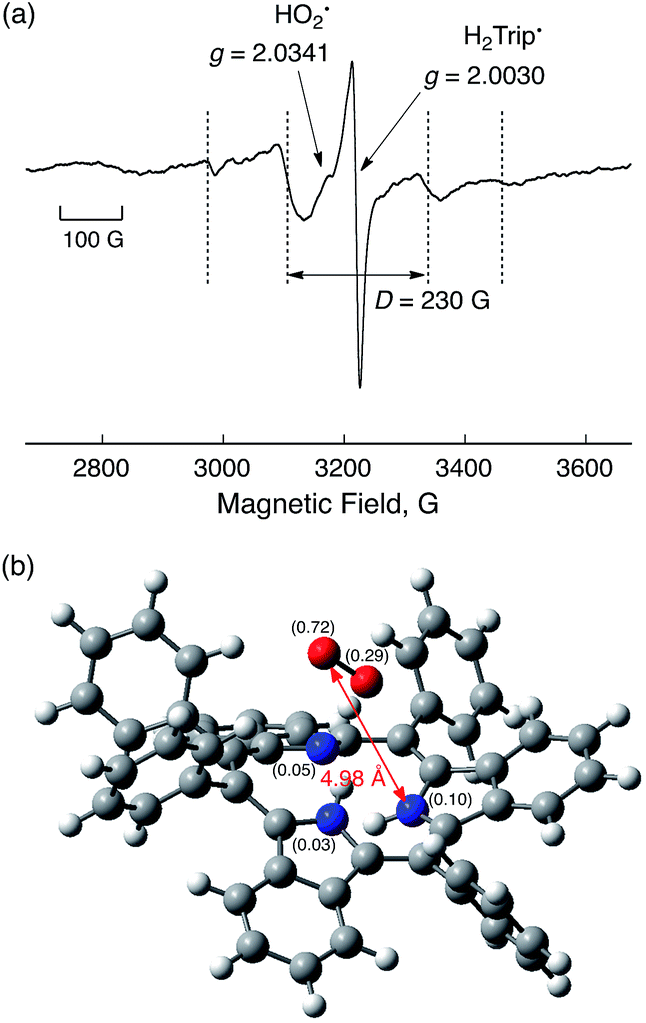
Although the radical pair (H2Trip˙/HO2˙) in Scheme 3 cannot be detected during the catalytic reaction, the formation of the radical pair (H2Trip˙/HO2˙) was successfully detected by EPR measurements using 1-benzyl-1,4-dihydronicotinamide dimer [(BNA)2]44 as an electron donor to produce H3Trip under photoirradiation at low temperature. The observed EPR spectrum in aerated PhCN in the presence of HClO4 at low temperature is shown in Fig. 9. A triplet fine structure EPR signal was observed as well as the typical anisotropic signals for HO2˙ with the g|| value of 2.0341, and isotropic signals for H2Trip˙ at 2.0030.45,46 From the zero-field splitting value (D = 230 G), the distance (r) between two unpaired electrons was determined using the relation D = 27800/r347 to be 4.9 Å. This distance is consistent with the estimated distance between O2 and H3Trip in the H3Trip/O2 complex by DFT calculations (Fig. 9b).
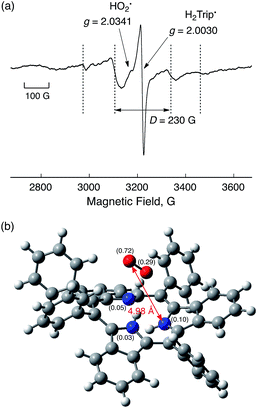 |
| | Fig. 9 EPR spectrum observed after the reduction of HTrip (1.0 × 10−3 M) by (BNA)2 (2.0 × 10−3 M) in the presence of HClO4 (1.0 × 10−3 M) in aerated PhCN under photoirradiation using a high-pressure Hg lamp (1000 W) measured at 80 K. Experimental conditions: Microwave frequency 9.0 GHz, microwave power 1.0 mW, modulation frequency 100 kHz, and modulation width 10 G. (b) Optimized structure of H3Trip/O2 calculated by DFT with calculated spin-density values given in parentheses at the UB3LYP/6-31G(d) level of theory. | |
Conclusion
Metal-free triphyrin acts as an efficient catalyst for the two-electron reduction of O2 by Me8Fc to produce H2O2 in the presence of HClO4 in PhCN at 298 K. The rate-determining step (RDS) in the catalytic cycle has been found to be hydrogen atom transfer from H3Tip to O2 in the H3Trip/O2 complex to produce the radical pair (H3Trip˙+/HO2˙), which was detected as a triplet species by EPR at 80 K. The distance between the two unpaired electrons (4.9 Å) determined from the zero-field splitting constant (D) agrees with the distance in the H3Trip/O2 complex calculated by DFT. The present study provides valuable insight into the catalytic mechanism of the two-electron reduction of O2 with an organic catalyst, and may lead to the development of more efficient metal-free organic catalysts for the selective two-electron reduction of O2 to produce H2O2.
Experimental section
General procedure
Chemicals were purchased from commercial sources and used without further purification, unless otherwise noted. Perchloric acid (HClO4, 70%), trifluoroacetic acid (TFA), ferrocene (Fc), and 1,1-dimethylferrocene (Me2Fc) were purchased from Wako Pure Chemical Industries Ltd. Octamethylferrocene (Me8Fc) and decamethylferrocene (Me10Fc) were received from Sigma Aldrich. Fc, Me2Fc, Me8Fc, and Me10Fc were purified by sublimation or recrystallization from ethanol. Benzonitrile (PhCN) used for spectroscopic and electrochemical measurements was distilled over phosphorus pentoxide prior to use.48 [14]Triphyrin(2.1.1) [HTrip] was synthesized according to the reported procedure.41 Fe(II)(TMC)(OTf)2 (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane; OTf = CF3SO3) was prepared according to a literature method.43 Tetra-n-butylammonium hexafluorophosphate (TBAPF6) was twice recrystallized from ethanol and dried in vacuo prior to use. 1H NMR spectra (300 MHz) were recorded on a JEOL AL-300 spectrometer at room temperature and chemical shifts (ppm) were determined relative to tetramethylsilane (TMS). UV-vis absorption spectroscopy was carried out on a Hewlett Packard 8453 diode array spectrophotometer at room temperature using a quartz cell (light path length = 1 cm).
Spectroscopic measurements
The amount of hydrogen peroxide (H2O2) produced was determined by titration with iodide ion: a dilute CH3CN solution (2.0 mL) of the product mixture (50 μL) was treated with an excess amount of NaI, and the amount of I3− formed was determined from the absorption spectrum (λmax = 361 nm, ε = 2.8 × 104 M−1 cm−1).49 The formation of H2O2 in the catalytic O2 reduction with HTrip was again confirmed by the reaction between H2O2 and Fe(II)(TMC)(OTf)2 to afford the corresponding Fe(IV)-oxo species. The amount of the Fe(IV)-oxo species produced was determined from the absorption spectrum (λmax = 820 nm, ε = 400 M−1 cm−1).43
The turnover numbers (TON = the number of moles of H2O2 formed per mole of HTrip in the catalytic two-electron reduction of O2) were determined from the concentration of produced Me8Fc+ under catalytic conditions, where stoichiometric production of H2O2 was confirmed by iodometric titration.
Kinetic measurements
Rate constants of oxidation of ferrocene derivatives by O2 in the presence of a catalytic amount of HTrip and an excess amount of HClO4 in PhCN at 298 K were determined by monitoring the appearance of an absorption band due to the corresponding ferrocenium ions (Fc+, λmax = 620 nm, εmax = 330 M−1 cm−1; Me2Fc+, λmax = 650 nm, εmax = 290 M−1 cm−1; Me8Fc+, λmax = 750 nm, εmax = 410 M−1 cm−1; Me10Fc+, λmax = 780 nm, εmax = 450 M−1 cm−1).14 At the wavelengths monitored, spectral overlap was observed with H3Trip (λ = 738 nm (ε = 1.6 × 103 M−1 cm−1)), H3Trip/O2 (λ = 720 nm (ε = 1.2 × 103 M−1 cm−1)). The concentration of O2 in an air-saturated PhCN solution was determined to be 1.7 × 10−3 M as reported previously.50 The concentrations of ferrocene derivatives employed for the catalytic reduction of O2 were much larger than that of O2, as O2 is the rate-limiting reagent in the reaction solution. The PhCN solutions containing various concentrations of O2 for the kinetic measurements were prepared by N2/O2 mixed gas bubbling using a KOFLOC GASBLENDER GB-3C. Typically, a PhCN stock solution of a ferrocene derivative was added using a microsyringe to a PhCN solution containing HTrip and HClO4 in a quartz cuvette (light path length = 1 cm).
Electrochemical measurements
Cyclic voltammetry (CV) measurements were performed on an ALS 630B electrochemical analyser and voltammograms were measured in deaerated PhCN containing 0.10 M TBAPF6 as a supporting electrolyte at room temperature. A conventional three-electrode cell was used with a glassy carbon working electrode (surface area of 0.3 mm2) and a platinum wire as the counter electrode. The glassy carbon working electrode (BAS) was routinely polished with BAS polishing alumina suspension and rinsed with acetone before use. The potentials were measured with respect to the Ag/AgNO3 (1.0 × 10−2 M) reference electrode. All potentials (vs. Ag/AgNO3) were converted to values vs. SCE by adding 0.29 V.51 Redox potentials were determined using the relation E1/2 = (Epa + Epc)/2.
Spectroelectrochemical measurements
UV-visible spectroelectrochemical experiments were performed with a home-built thin-layer cell (1 mm) that had a light transparent platinum net working electrode. Potentials were applied and monitored with an ALS 730D electrochemical analyser.
EPR measurements
EPR spectra were measured on a JEOL X-band EPR spectrometer (JES-ME-LX) using a quartz EPR tube containing a deaerated frozen sample solution at 80 K. The internal diameter of the EPR tube is 4.0 mm, which is small enough to fill the EPR cavity but large enough to obtain good signal-to-noise ratios during the EPR measurements at low temperatures (at 80 K). EPR spectrum of HTrip˙− produced by the electrochemical reduction of HTrip was measured using a home-built three-electrode quartz EPR tube. Potentials were applied and monitored with an ALS 730D electrochemical analyser. EPR spectra were measured under nonsaturating microwave power conditions. The amplitude of modulation was chosen to optimize the resolution and the signal-to-noise (S/N) ratio of the observed spectra. The g values were calibrated with a Mn2+ marker.
Theoretical calculations
Density functional theory (DFT) calculations were performed on a 32CPU workstation (PQS, Quantum Cube QS8-2400C-064). Geometry optimisations were carried out using the B3LYP/6-31G(d) level of theory52 for HTrip˙−, H2Trip+, H3Trip2+, H3Trip˙+, and [H3Trip/O2]. All calculations were performed using Gaussian 09, revision A.02.53 Graphical outputs of the computational results were generated with the GaussView software program (ver. 3.09) developed by Semichem, Inc.54
Acknowledgements
This work was supported by grants from the Advanced Low Carbon Technology Research and Development (ALCA) and SENTAN programs from Japan Science Technology Agency (JST) to S. F., the Japan Society for the Promotion of Science (JSPS: Grants 26620154 and 26288037 to K. O., 25288092 and 26105004 to H. Y. and 25-727 to K. M.), and the Green Photonics Project in NAIST sponsored by the MEXT (Japan).
Notes and references
-
(a) S. Abrantes, E. Amaral, A. P. Costa, A. A. Shatalov and A. P. Duarte, Ind. Crops Prod., 2007, 25, 288–293 CrossRef CAS PubMed;
(b) S. H. Zeronian and M. K. Inglesby, Cellulose, 1995, 2, 265–272 CrossRef CAS.
- L. Li, S. Lee, H. L. Lee and H. J. Youn, BioResources, 2011, 6, 721–736 CAS.
-
(a) S. Yamazaki, Z. Siroma, H. Senoh, T. Ioroi, N. Fujiwara and K. Yasuda, J. Power Sources, 2008, 178, 20–25 CrossRef CAS PubMed;
(b) R. S. Disselkamp, Energy Fuels, 2008, 22, 2771–2774 CrossRef CAS;
(c) R. S. Disselkamp, Int. J. Hydrogen Energy, 2010, 35, 1049–1053 CrossRef CAS PubMed.
- S. Fukuzumi, Y. Yamada and K. D. Karlin, Electrochim. Acta, 2012, 82, 493–511 CrossRef CAS PubMed.
-
(a) Y. Yamada and S. Fukuzumi, Aust. J. Chem., 2014, 67, 354–364 CrossRef;
(b) Y. Yamada, M. Yoneda and S. Fukuzumi, Inorg. Chem., 2014, 53, 1272–1274 CrossRef CAS PubMed;
(c) Y. Yamada, M. Yoneda and S. Fukuzumi, Chem.–Eur. J., 2013, 19, 11733–11741 CrossRef CAS PubMed;
(d) Y. Yamada, S. Yoshida, T. Honda and S. Fukuzumi, Energy Environ. Sci., 2011, 4, 2822–2825 RSC;
(e) Y. Yamada, Y. Fukunishi, S. Yamazaki and S. Fukuzumi, Chem. Commun., 2010, 46, 7334–7336 RSC.
- E. Santacesaria, M. D. Serio, R. Velotti and U. Leone, Ind. Eng. Chem. Res., 1994, 33, 277–281 CrossRef CAS.
- M. Fukushima, K. Tatsumi, S. Tanaka and H. Nakamura, Environ. Sci. Technol., 1998, 32, 3948–3953 CrossRef CAS.
- S. Liu, K. Mase, C. Bougher, S. D. Hicks, M. M. Abu-Omar and S. Fukuzumi, Inorg. Chem., 2014, 53, 7780–7788 CrossRef CAS PubMed.
- A. J. Hoffman, E. R. Carraway and M. R. Hoffmann, Environ. Sci. Technol., 1994, 28, 776–785 CrossRef CAS PubMed.
-
(a) V. Maurino, C. Minero, G. Mariella and E. Pelizzetti, Chem. Commun., 2005, 2627–2629 RSC;
(b) M. Teranishi, S. Naya and H. Tada, J. Am. Chem. Soc., 2010, 132, 7850–7851 CrossRef CAS PubMed;
(c) D. Tsukamoto, A. Shiro, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, ACS Catal., 2012, 2, 599–603 CrossRef CAS.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756–3764 CAS.
-
(a) Y. Yamada, A. Nomura, K. Ohkubo, T. Suenobu and S. Fukuzumi, Chem. Commun., 2013, 49, 5132–5134 RSC;
(b) Y. Yamada, A. Nomura, T. Miyahigashi and S. Fukuzumi, Chem. Commun., 2012, 48, 8329–8331 RSC.
-
(a) S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561–574 RSC;
(b) S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059–6071 RSC;
(c) K. Ohkubo and S. Fukuzumi, Bull. Chem. Soc. Jpn., 2009, 82, 303–315 CrossRef CAS;
(d) H. Kotani, K. Ohkubo and S. Fukuzumi, Appl. Catal., B, 2008, 77, 317–324 CrossRef CAS PubMed;
(e) K. Ohkubo, R. Iwata, K. Mizushima, K. Souma, Y. Yamamoto, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601–603 RSC.
-
(a) K. Mase, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 2800–2808 CrossRef CAS PubMed;
(b) T. Honda, T. Kojima and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 4196–4206 CrossRef CAS PubMed;
(c) S. Fukuzumi, Chem. Lett., 2008, 37, 808–813 CrossRef CAS;
(d) S. Fukuzumi, K. Okamoto, C. P. Gros and R. Guilard, J. Am. Chem. Soc., 2004, 126, 10441–10449 CrossRef CAS PubMed;
(e) S. Fukuzumi, K. Okamoto, Y. Tokuda, C. P. Gros and R. Guilard, J. Am. Chem. Soc., 2004, 126, 17059–17066 CrossRef CAS PubMed.
-
(a) S. Fukuzumi, S. Mochizuki and T. Tanaka, Inorg. Chem., 1989, 28, 2459–2465 CrossRef CAS;
(b) S. Fukuzumi, S. Mochizuki and T. Tanaka, Inorg. Chem., 1990, 29, 653–659 CrossRef CAS;
(c) S. Fukuzumi, S. Mochizuki and T. Tanaka, J. Chem. Soc., Chem. Commun., 1989, 391–392 RSC.
-
(a) R. McGuire Jr, D. K. Dogutan, T. S. Teets, J. Suntivich, Y. Shao-Horn and D. G. Nocera, Chem. Sci., 2010, 1, 411–414 RSC;
(b) D. K. Dogutan, S. A. Stoian, R. McGuire Jr, M. Schwalbe, T. S. Teets and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 131–140 CrossRef CAS PubMed;
(c) T. S. Teets, T. R. Cook, B. D. McCarthy and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 8114–8117 CrossRef CAS PubMed.
-
(a) F. C. Anson, C. Shi and B. Steiger, Acc. Chem. Res., 1997, 30, 437–449 CrossRef CAS;
(b) C. Shi, B. Steiger, M. Yuasa and F. C. Anson, Inorg. Chem., 1997, 36, 4294–4295 CrossRef CAS;
(c) C. Shi and F. C. Anson, Inorg. Chem., 1998, 37, 1037–1043 CrossRef CAS;
(d) Z. Liu and F. C. Anson, Inorg. Chem., 2000, 39, 274 CrossRef CAS.
-
(a) A. Schechter, M. Stanevsky, A. Mahammed and Z. Gross, Inorg. Chem., 2012, 51, 22–24 CrossRef CAS PubMed;
(b) J. Masa, K. Ozoemena, W. Schuhmann and J. H. Zagal, J. Porphyrins Phthalocyanines, 2012, 16, 761–784 CrossRef CAS.
-
(a) A. J. Olaya, D. Schaming, P.-F. Brevet, H. Nagatani, T. Zimmermann, J. Vanicek, H.-J. Xu, C. P. Gros, J.-M. Barbe and H. H. Girault, J. Am. Chem. Soc., 2012, 134, 498–506 CrossRef CAS PubMed;
(b) P. Peljo, L. Murtomäki, T. Kallio, H.-J. Xu, M. Meyer, C. P. Gros, J.-M. Barbe, H. H. Girault, K. Laasonen and K. Kontturi, J. Am. Chem. Soc., 2012, 134, 5974–5984 CrossRef CAS PubMed;
(c) B. Su, I. Hatay, A. Trojánek, Z. Samec, T. Khoury, C. P. Gros, J.-M. Barbe, A. Daina, P.-A. Carrupt and H. H. Girault, J. Am. Chem. Soc., 2010, 132, 2655–2662 CrossRef CAS PubMed.
-
(a) S. Fukuzumi, S. Mandal, K. Mase, K. Ohkubo, H. Park, J. Benet-Buchholz, W. Nam and A. Llobet, J. Am. Chem. Soc., 2012, 134, 9906–9909 CrossRef CAS PubMed;
(b) T. Wada, H. Maki, T. Imamoto, H. Yuki and Y. Miyazato, Chem. Commun., 2013, 49, 4394–4396 RSC.
-
(a) K. M. Kadish, L. Fremond, J. Shen, P. Chen, K. Ohkubo, S. Fukuzumi, M. El Ojaimi, C. P. Gros, J.-M. Barbe and R. Guilard, Inorg. Chem., 2009, 48, 2571–2582 CrossRef CAS PubMed;
(b) P. Chen, H. Lau, B. Habermeyer, C. P. Gros, J.-M. Barbe and K. M. Kadish, J. Porphyrins Phthalocyanines, 2011, 15, 467–479 CrossRef CAS;
(c) K. M. Kadish, L. Fremond, Z. Ou, J. Shao, C. Shi, F. C. Anson, F. Burdet, C. P. Gros, J.-M. Barbe and R. Guilard, J. Am. Chem. Soc., 2005, 127, 5625–5631 CrossRef CAS PubMed.
-
(a) C. J. Chang, Y. Deng, D. G. Nocera, C. Shi, F. C. Anson and C. K. Chang, Chem. Commun., 2000, 1355–1356 RSC;
(b) C. J. Chang, Z. H. Loh, C. Shi, F. C. Anson and D. G. Nocera, J. Am. Chem. Soc., 2004, 126, 10013–10020 CrossRef CAS PubMed.
-
(a) E. Askarizadeh, S. B. Yaghoob, D. M. Boghaei, A. M. Slawin and J. B. Love, Chem. Commun., 2010, 46, 710–712 RSC;
(b) M. Volpe, H. Hartnett, J. W. Leeland, K. Wills, M. Ogunshun, B. J. Duncombe, C. Wilson, A. J. Blake, J. McMaster and J. B. Love, Inorg. Chem., 2009, 48, 5195–5207 CrossRef CAS PubMed.
-
(a) R. A. Decréau, J. P. Collman and A. Hosseini, Chem. Soc. Rev., 2010, 39, 1291–1301 RSC;
(b) J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS PubMed.
-
(a) J. Rosenthal and D. G. Nocera, Acc. Chem. Res., 2007, 40, 543–553 CrossRef CAS PubMed;
(b) M. Schwalbe, D. K. Dogutan, S. A. Stoian, T. S. Teets and D. G. Nocera, Inorg. Chem., 2011, 50, 1368–1377 CrossRef CAS PubMed;
(c) C. J. Chang, L. L. Chng and D. G. Nocera, J. Am. Chem. Soc., 2003, 125, 1866–1876 CrossRef CAS PubMed.
-
(a) Z. Halime, H. Kotani, Y. Li, S. Fukuzumi and K. D. Karlin, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13990–13994 CrossRef CAS PubMed;
(b) E. E. Chufan, S. C. Puiu and K. D. Karlin, Acc. Chem. Res., 2007, 40, 563–572 CrossRef CAS PubMed;
(c) E. Kim, E. E. Chufan, K. Kamaraj and K. D. Karlin, Chem. Rev., 2004, 104, 1077–1133 CrossRef CAS PubMed.
-
(a) C. T. Carver, B. D. Matson and J. M. Mayer, J. Am. Chem. Soc., 2012, 134, 5444–5447 CrossRef CAS PubMed;
(b) B. D. Matson, C. T. Carver, A. von Ruden, J. Y. Yang, S. Raugei and J. M. Mayer, Chem. Commun., 2012, 48, 11100–11102 RSC;
(c) J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
-
(a) S. Kakuda, R. L. Peterson, K. Ohkubo, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 6513–6522 CrossRef CAS PubMed;
(b) D. Das, Y.-M. Lee, K. Ohkubo, W. Nam, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 2825–2834 CrossRef CAS PubMed;
(c) D. Das, Y.-M. Lee, K. Ohkubo, W. Nam, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 4018–4026 CrossRef CAS PubMed;
(d) S. Fukuzumi, L. Tahsini, Y.-M. Lee, K. Ohkubo, W. Nam and K. D. Karlin, J. Am. Chem. Soc., 2012, 134, 7025–7035 CrossRef CAS PubMed.
-
(a) S. Fukuzumi and K. D. Karlin, Coord. Chem. Rev., 2013, 257, 187–195 CrossRef CAS PubMed;
(b) L. Tahsini, H. Kotani, Y.-M. Lee, J. Cho, W. Nam, K. D. Karlin and S. Fukuzumi, Chem.–Eur. J., 2012, 18, 1084–1093 CrossRef CAS PubMed;
(c) S. Fukuzumi, H. Kotani, H. R. Lucas, K. Doi, T. Suenobu, R. L. Peterson and K. D. Karlin, J. Am. Chem. Soc., 2010, 132, 6874–6875 CrossRef CAS PubMed.
-
(a) J. Zhang and F. C. Anson, J. Electroanal. Chem., 1993, 348, 81–97 CrossRef CAS;
(b) Y. Lei and F. C. Anson, Inorg. Chem., 1994, 33, 5003–5009 CrossRef CAS;
(c) Y. C. Weng, F.-R. F. Fan and A. J. Bard, J. Am. Chem. Soc., 2005, 127, 17576–17577 CrossRef CAS PubMed.
-
(a) M. S. Thorum, J. Yadav and A. A. Gewirth, Angew. Chem., Int. Ed., 2009, 48, 165–167 CrossRef CAS PubMed;
(b) C. C. L. McCrory, A. Devadoss, X. Ottenwaelder, R. D. Lowe, T. D. P. Stack and C. E. D. Chidsey, J. Am. Chem. Soc., 2011, 133, 3696–3699 CrossRef CAS PubMed;
(c) M. A. Thorseth, C. E. Tornow, E. C. M. Tse and A. A. Gewirth, Coord. Chem. Rev., 2013, 257, 130–139 CrossRef CAS PubMed;
(d) M. A. Thorseth, C. S. Letko, T. B. Rauchfuss and A. A. Gewirth, Inorg. Chem., 2011, 50, 6158–6161 CrossRef CAS PubMed.
- C. Kemal, T. W. Chan and T. C. Bruice, J. Am. Chem. Soc., 1977, 99, 7272–7286 CrossRef CAS.
-
(a) K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed;
(b) S. Wang, D. Yu and L. Dai, J. Am. Chem. Soc., 2011, 133, 5182–5185 CrossRef CAS PubMed.
-
(a) Q. Li, B. W. Noffke, Y. Wang, B. Menezes, D. G. Peters, K. Raghavachari and L.-S. Li, J. Am. Chem. Soc., 2014, 136, 3358–3361 CrossRef CAS PubMed;
(b) Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
- L. Wang, A. Ambrosi and M. Pumera, Angew. Chem., Int. Ed., 2013, 52, 13818–13821 CrossRef CAS PubMed.
-
(a) S. Shibata, T. Suenobu and S. Fukuzumi, Angew. Chem., Int. Ed., 2013, 52, 12327–12331 CrossRef CAS PubMed;
(b) S. Fukuzumi, K. Yasui, T. Suenobu, K. Ohkubo, M. Fujitsuka and O. Ito, J. Phys. Chem. A, 2001, 105, 10501–10510 CrossRef CAS;
(c) S. Fukuzumi, S. Kuroda, T. Goto, K. Ishikawa and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1989, 1047–1053 RSC.
-
(a) S. Ghisla and V. Massey, Eur. J. Biochem., 1989, 181, 1–17 CrossRef CAS PubMed;
(b) T. C. Bruice, Acc. Chem. Res., 1980, 13, 256–262 CrossRef CAS;
(c) V. Massey, J. Biol. Chem., 1994, 269, 22459–22462 CAS.
- S. Fukuzumi and T. Okamoto, J. Chem. Soc., Chem. Commun., 1994, 521–522 RSC.
-
(a) I. Hatay, B. Su, M. A. Méndez, C. Corminboeuf, T. Khoury, C. P. Gros, M. Bourdillon, M. Meyer, J.-M. Barbe, M. Ersoz, S. Záliš, Z. Samec and H. H. Girault, J. Am. Chem. Soc., 2010, 132, 13733–13741 CrossRef CAS PubMed;
(b) A. Trojánek, J. Langmaier and Z. Samec, Electrochim. Acta, 2012, 82, 457–462 CrossRef PubMed;
(c) A. Trojánek, J. Langmaier, B. Su, H. H. Girault and Z. Samec, Electrochem. Commun., 2009, 11, 1940–1943 CrossRef PubMed;
(d) S. Wu and B. Su, Chem.–Eur. J., 2012, 18, 3169–3173 CrossRef CAS PubMed;
(e) X. Liu, S. Wu and B. Su, J. Electroanal. Chem., 2013, 709, 26 CrossRef CAS PubMed.
- N. Lopez, D. J. Graham, R. McGuire Jr, G. E. Alliger, Y. Shao-Horn, C. C. Cummins and D. G. Nocera, Science, 2012, 335, 450–453 CrossRef CAS PubMed.
-
(a) Z.-L. Xue, J. Mack, H. Lu, L. Zhang, X.-Z. You, D. Kuzuhara, M. Stillman, H. Yamada, S. Yamauchi, N. Kobayashi and Z. Shen, Chem.–Eur. J., 2011, 17, 4396–4407 CrossRef CAS PubMed;
(b) Z.-L. Xue, Z. Shen, J. Mack, D. Kuzuhara, H. Yamada, T. Okujima, N. Ono, X.-Z. You and N. Kobayashi, J. Am. Chem. Soc., 2008, 130, 16478–16479 CrossRef CAS PubMed;
(c) Z.-L. Xue, D. Kuzuhara, S. Ikeda, Y. Sakakibara, K. Ohkubo, N. Aratani, T. Okujima, H. Uno, S. Fukuzumi and H. Yamada, Angew. Chem., Int. Ed., 2013, 52, 7306–7309 CrossRef CAS PubMed.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
-
(a) J.-U. Rohde, J.-H. In, M. H. Lim, W. W. Brennessel, M. R. Bukowski, A. Stubna, E. Münck, W. Nam and L. Que Jr, Science, 2003, 299, 1037–1039 CrossRef CAS PubMed;
(b) E. S. Yang, M.-S. Chan and A. C. Wahl, J. Phys. Chem., 1975, 79, 2049–2051 CrossRef CAS.
- S. Fukuzumi, T. Suenobu, M. Patz, T. Hirasaka, S. Itoh, M. Fujitsuka and O. Ito, J. Am. Chem. Soc., 1998, 120, 8060–8068 CrossRef CAS.
-
(a) K. Suga, K. Ohkubo and S. Fukuzumi, J. Phys. Chem. A, 2005, 109, 10168–10175 CrossRef CAS PubMed;
(b) H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 15999–16006 CrossRef CAS PubMed;
(c) S. Fukuzumi and K. Ohkubo, Chem.–Eur. J., 2000, 6, 4532–4535 CrossRef CAS.
-
(a) R. N. Bagchi, A. M. Bond, F. Scholz and R. Stösser, J. Am. Chem. Soc., 1989, 111, 8270–8271 CrossRef CAS;
(b) M. Bersohn and J. R. Thomas, J. Am. Chem. Soc., 1964, 86, 959–959 CrossRef CAS;
(c)
J. A. Howard, in Peroxyl radicals, ed. Z. B. Alfassi, Wiley, Chichester, 1997, p. 283 Search PubMed.
-
(a) J. S. Park, E. Karnas, K. Ohkubo, P. Chen, K. M. Kadish, S. Fukuzumi, C. W. Bielawski, T. W. Hudnall, V. M. Lynch and J. L. Sessler, Science, 2010, 329, 1324–1326 CrossRef CAS PubMed;
(b) S. Fukuzumi, K. Ohkubo, Y. Kawashima, D. S. Kim, J. S. Park, A. Jana, V. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2011, 133, 15938–15941 CrossRef CAS PubMed.
-
W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 7th edn, 2013 Search PubMed.
- S. Fukuzumi, S. Kuroda and T. Tanaka, J. Am. Chem. Soc., 1985, 107, 3020–3027 CrossRef CAS.
-
(a) S. Fukuzumi, H. Imahori, H. Yamada, M. E. El-Khouly, M. Fujitsuka, O. Ito and D. M. Guldi, J. Am. Chem. Soc., 2001, 123, 2571–2575 CrossRef CAS PubMed;
(b) S. Fukuzumi, M. Ishikawa and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1989, 1037–1045 RSC.
-
C. K. Mann and K. K. Barnes, in Electrochemical Reactions in Non-Aqueous Systems, Mercel Dekker, New York, 1970 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
-
R. Dennington II, T. Keith, J. Millam, K. Eppinnett, W. L. Hovell and R. Gilliland, Gaussview, Semichem, Inc., Shawnee Mission, KS, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic, kinetic and DFT data. See DOI: 10.1039/c5sc02465j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a