
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolation of +2 rare earth metal ions with three anionic carbocyclic rings: bimetallic bis(cyclopentadienyl) reduced arene complexes of La2+ and Ce2+ are four electron reductants†
Christopher M.
Kotyk
a,
Megan E.
Fieser
a,
Chad T.
Palumbo
a,
Joseph W.
Ziller
a,
Lucy E.
Darago
b,
Jeffrey R.
Long
*b,
Filipp
Furche
*a and
William J.
Evans
*a
aDepartment of Chemistry, University of California, Irvine, California 92697, USA. E-mail: wevans@uci.edu; filipp.furche@uci.edu; Fax: +1-949-824-2210; Tel: +1-949-824-5174
bDepartment of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: jrlong@berkeley.edu
First published on 21st September 2015
Abstract
A new option for stabilizing unusual Ln2+ ions has been identified in the reaction of Cp′3Ln, 1-Ln (Ln = La, Ce; Cp′ = C5H4SiMe3), with potassium graphite (KC8) in benzene in the presence of 2.2.2-cryptand. This generates [K(2.2.2-cryptand)]2[(Cp′2Ln)2(μ-η6:η6-C6H6)], 2-Ln, complexes that contain La and Ce in the formal +2 oxidation state. These complexes expand the range of coordination environments known for these ions beyond the previously established examples, (Cp′′3Ln)1− and (Cp′3Ln)1− (Cp′′ = C5H3(SiMe3)2-1,3), and generalize the viability of using three anionic carbocyclic rings to stabilize highly reactive Ln2+ ions. In 2-Ln, a non-planar bridging (C6H6)2− ligand shared between two metals takes the place of a cyclopentadienyl ligand in (Cp′3Ln)1−. The intensely colored (ε = ∼8000 M−1 cm−1) 2-Ln complexes react as four electron reductants with two equiv. of naphthalene to produce two equiv. of the reduced naphthalenide complex, [K(2.2.2-cryptand)][Cp′2Ln(η4-C10H8)].
One of the most fundamental aspects of any element is the number of oxidation states accessible to it. Since this defines the range of chemistry possible with the element, the limits of oxidation states for each element have been heavily probed for decades and are well established. Surprisingly, in the last few years, a new oxidation state has been discovered for nine elements in the rare earth series.1–4
The discovery of the nine new Ln2+ ions required,1–4 in each case, a coordination environment composed of three cyclopentadienyl rings, specifically (Cp′′3)3− or (Cp′3)3− (Cp′′ = C5H3(SiMe3)2-1,3; Cp′ = C5H4SiMe3), Scheme 1. In this tris(cyclopentadienyl) ligand field, it was found that reduction of a 4fn Ln3+ ion added an electron, not to the 4f valence orbitals to make a 4fn+1 Ln2+ ion, but to an orbital with a higher principal quantum number, a 5dz2 orbital, to give Ln2+ ions best described by 4fn5d1 electron configurations. This was rationalized by the fact that the dz2 orbital is the lowest energy d orbital in a tris(cyclopentadienyl) coordination environment complex.1–11
 | ||
| Scheme 1 Reduction of Cp′′3Ln (R = SiMe3; Ln = La) and Cp′3Ln (R = H; Ln = Y, La, Ce, Pr, Nd, Sm, Gd, Tb, Dy, Ho, Er, Tm, Lu) to form Ln2+ complexes.1–4 | ||
It was of interest to determine if these Ln2+ ions could be isolated in other coordination environments to examine the general accessibility of these new oxidation states since Ln2+ ions have proven to be broadly useful reductants as in the use of Sm2+ in organic chemistry.12,13 Crystallographic evidence that Ln2+ complexes could be made with an anionic reduced benzene ligand was previously suggested by Lappert et al.14,15 as shown in Scheme 2.
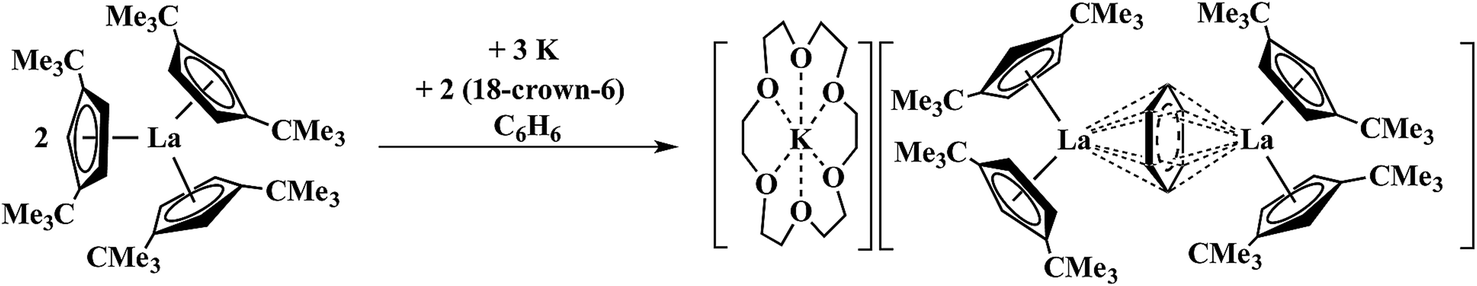 | ||
| Scheme 2 Reaction of Cptt3La Cptt = C5H3(CMe3)2]-1,3 with 1.5 equiv. of K in benzene to form a (C6H6)1− complex.14 | ||
Reduction of Cptt3La (Cptt = C5H3(CMe3)2-1,3) with 1.5 equiv. of potassium in the presence of benzene gave a compound that was postulated to be a La2+ complex of a (C6H6)1− bridging ligand rather than a Ln3+ complex of (C6H6)3−. With the silyl analogs, Cp′′3Ln (Ln = La, Ce), in toluene, similar reactions to form (C6H5CH3)1− complexes were reported.15 The known difficulty in assigning oxidation states in bridging arene systems16–25 complicated the assignments until unambiguous examples of La2+ and Ce2+ were found viaScheme 1.1
In a small variation of Scheme 2, reactions involving excess K instead of 1.5 equiv. per Ln led to the formation of Ln3+ products, rather than Ln2+ complexes and reduction of benzene to (C6H6)2− rather than (C6H6)1− in the compounds, [K(18-crown-6)][(C6H6)LnCp′′2] (Ln = La, Ce, Pr, Nd), Scheme 3.26,27 In a further variation with (C5H4SiMe2CMe3)3Ln precursors in toluene, Ln3+ hydride products were found.12 All of these reactions were postulated to involve Ln2+ intermediates.
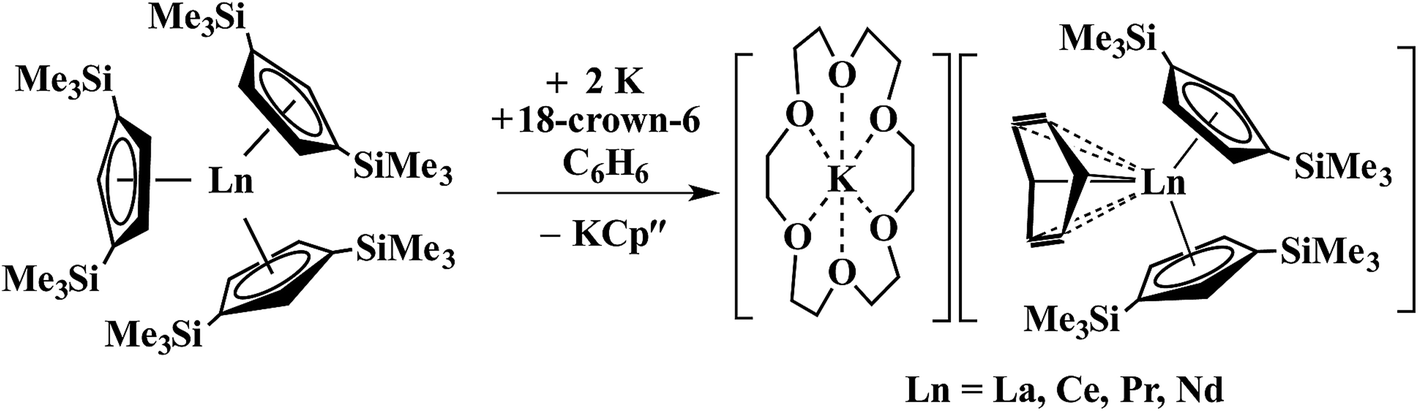 | ||
| Scheme 3 Reduction of benzene to (C6H6)2− by Cp′′3Ln/K.26,27 | ||
Further variability in this arene rare earth reduction chemistry was subsequently observed with Cp′ ligands in reactions of crystallographically characterized Ln2+ complexes, [K(2.2.2-cryptand)][Cp′3Ln], 3-Ln (Ln = Y, La, Ce, Dy).3,4 These compounds reduce naphthalene (−2.50 V vs. SCE19) and biphenyl (−2.69 V vs. SCE19), but were not observed to reduce benzene (−3.43 V vs. SCE19). These reactions differed from Schemes 2 and 3 not only in the Cp′ ligand, but also in that they started with bona fide Ln2+ precursors and not a combination of a Ln3+ precursor and potassium that could form a Ln2+ intermediate.
In light of these results, it was of interest to see how the Cp′ complexes would behave in reactions analogous to Schemes 2 and 3. This ligand has given yet another variation on benzene reduction and has led to a new series of rare earth complexes in the formal +2 oxidation state. These results show the generality of using three anionic carbocyclic ligands to stabilize Ln2+ and provide a new type of four-electron reductant.
Reactions of solutions of Cp′3Ln, 1-Ln (Ln = La, Ce), in benzene with 2 equiv. of potassium–graphite (KC8) in the presence of 2.2.2-cryptand produce thick black precipitates. After stirring at room temperature for 4 h, extraction with THF followed by centrifugation to remove graphite produces deep purple solutions from which deep purple crystals of [K(2.2.2-cryptand)]2[(Cp′2Ln)2(μ-η6:η6-C6H6)], 2-Ln, were isolated. Single crystals were grown from THF and identified by X-ray diffraction, although only 2-La gave good metrical data, Fig. 1. The byproduct [K(2.2.2-cryptand)][Cp′] was identified by 1H NMR spectroscopy.
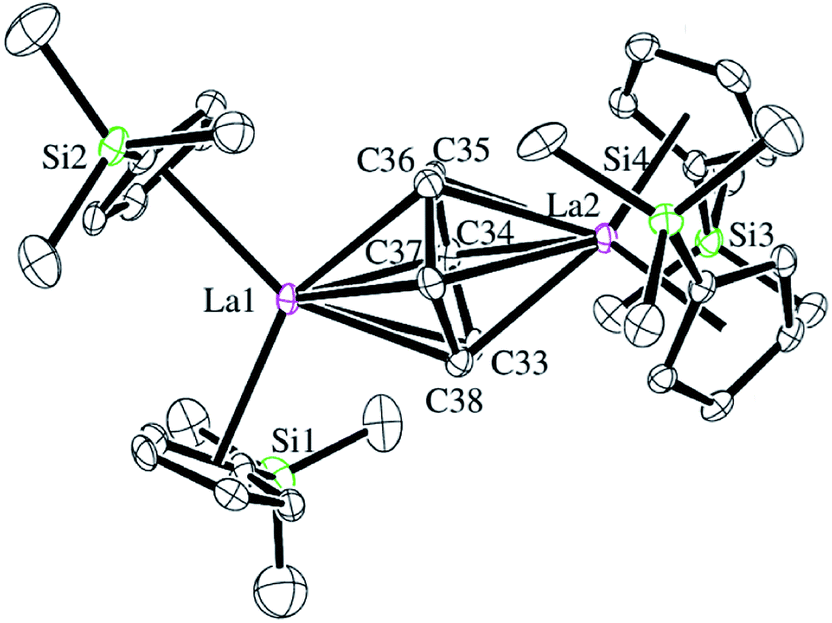 | ||
| Fig. 1 Molecular structure of the anion of [K(2.2.2-cryptand)]2[(Cp′2La)2(μ-η6:η6-C6H6)], 2-La. Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms and co-crystallized benzene and THF were omitted for clarity. | ||
The reaction formally involves the addition of four equiv. of KC8 to two equiv. of Cp′3Ln with loss of one (Cp′)1− anion per metal complex as [K(2.2.2-cryptand)][Cp′] and formation of a reduced benzene ligand bridging two bis(cyclopentadienyl) metal units, Scheme 4. The [(Cp′2Ln)2(μ-η6:η6-C6H6)]2− product could be described by two extreme forms as either two Ln2+ ions and a (C6H6)2− dianion or two Ln3+ ions and a (C6H6)4− tetraanion.
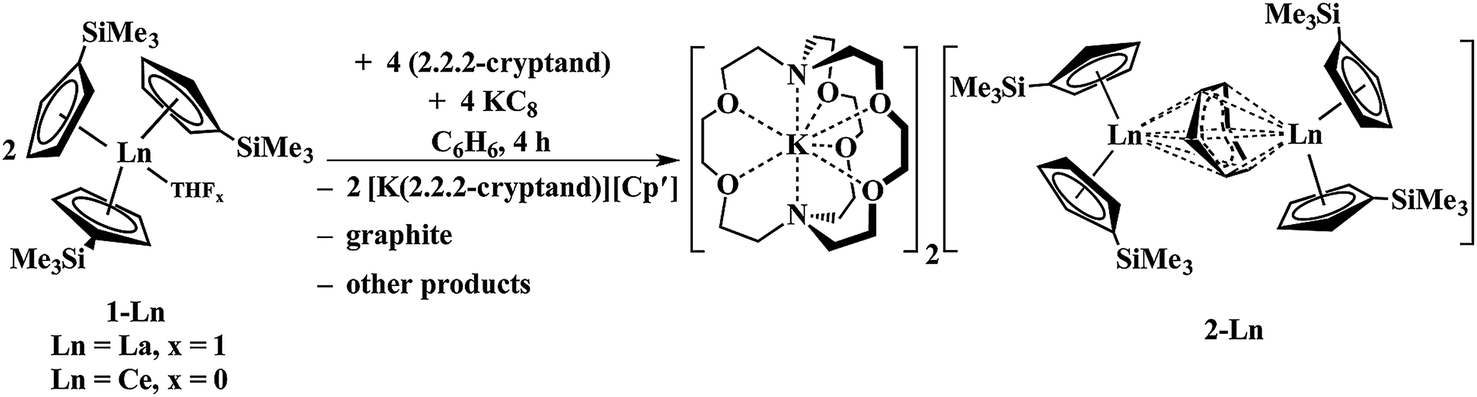 | ||
| Scheme 4 Reduction of benzene by Cp′3Ln/KC8 to form [K(2.2.2-cryptand)]2[(Cp′2La)2(μ-η6:η6-C6H6)], 2-Ln (Ln = La, Ce). | ||
1H NMR analysis of 2-La shows multiple resonances at chemical shifts typical of aromatic Cp′ protons (5.5 to 6.0 ppm), known 2.2.2-cryptand resonances, and peaks in the 0.0 to 0.5 ppm range typical of trimethylsilyl protons. A peak at 2.03 ppm in the 1H NMR spectrum was assigned to the reduced arene since it was missing in the analogous reaction using C6D6. The product of the reaction described in Scheme 4 with C6D6 gives a product with a resonance at 2.03 ppm in the 2H NMR spectrum.
The compound 2-La exhibits diamagnetic behavior, with an extremely small (∼10−2 emu K mol−1) magnetic susceptibility across the temperature range 2–300 K. The gradual increase in χMT with increasing temperature is likely due to a very small population of a triplet excited state or field-induced mixing with a triplet excited state. While the diamagnetism of 2-La may at first seem indicative of a La3+/(C6H6)4− electron configuration, we instead propose, in agreement with the crystallographic analysis, UV-vis spectroscopy, and electronic structure calculations, that extremely strong magnetic exchange coupling between the two La2+ (d1) centers and the (C6H6)2− diradical generates a well-isolated singlet ground state for 2-La. Indeed, DFT calculations reveal the energy splitting between the ground state and first excited triplet state to be 10500 cm−1, well over an order of magnitude higher than the thermal energy at 300 K. The magnetic data could also be reasonably well-simulated using the Hamiltonian Ĥ = −2JLa–C6H6 (ŜLa1 × ŜC6H6+ ŜLa2 × ŜC6H6), where JLa–C6H6 represents the coupling between the S = 1/2 La2+ centers and the S = 1 (C6H6)2− bridging unit, along with a temperature-independent paramagnetism contribution, χTIP. The best simulation was achieved using the values JLa–C6H6 = −497 cm−1 and χTIP = 0.000057 emu mol−1 (Fig. 5). Thus, the diamagnetism observed across the measured temperature range is also consistent with an assignment of La2+/(C6H6)2− together with a pairwise exchange constant of JLa–C6H6 < |−500 cm−1|.
The variable-temperature magnetic susceptibility of 2-Ce alone also does not enable a definitive assignment of the oxidation states present. At 300 K under an applied magnetic field of 0.1 T, 2-Ce exhibits a χMT product of 1.78 emu K mol−1, which then drops to 1.36 emu K mol−1 under an increased applied magnetic field of 1 T. The field dependence of 2-Ce is due to temperature-independent paramagnetism, which has been previously observed for both Ce2+ and Ce3+ compounds.28,29 The expected χMT value at 300 K for a Ce3+/(C6H6)4− configuration is 1.60 emu K mol−1. The situation for a Ce2+-based electronic configuration is much more complicated, as even for a simple mononuclear Ce2+ complex the room temperature χMT value falls in between those expected for “uncoupled” and “coupled” 4f15d1 configurations.29 The LS coupling schemes for 4fn5d1 configurations detailed in ref. 29 can be used, in tandem with the assumption that at 300 K the S = 1 (C6H6)2− moiety is not magnetically coupled to the Ce2+ ions, to predict room temperature χMT values for 2-Ce. The calculated χMT products at 300 K for 2-Ce assuming “uncoupled” and “coupled” 4f15d1 configurations plus an isolated S = 1 center are 3.36 emu K mol−1 and 1.66 emu K mol−1, respectively. Clearly the former value does not match the χMT data observed for 2-Ce. However, the latter value for the “coupled” scheme is quite close to the experimental χMT product for 2-Ce. If magnetic coupling between the Ce2+ spins and the (C6H6)2− diradical is strong, as proposed above for 2-La, the expected χMT product at 300 K for 2-Ce will be even lower than 1.66 emu K mol−1, and thus closer to the observed value of 1.36 emu K mol−1 at 1 T. If extremely strong d–π* magnetic coupling is present in 2-Ce, the resulting magnetic behavior could even appear much like that of a standard dinuclear Ce3+ (4f1) molecule. Therefore, discerning between the possible electronic configurations for 2-Ce, Ce3+/(C6H6)4− or Ce2+/(C6H6)2−, is not possible from the magnetic data alone.
The structural parameters for [K(2.2.2-cryptand)]2[(Cp′2La)2(μ-η6:η6-C6H6)], 2-La, are summarized in Table 1. The 1.446(6)–1.459(6) Å C–C bonds of the C6 unit in the solid-state structure of 2-La are longer than the bond lengths in free benzene, which has an average C–C bond length of 1.397(9) Å.30 This is consistent with reduction of C6H6. The C6H6 moiety is not planar and has a dihedral angle of 11° between the planes defined by C34–C37 and C33, C34, C37, C38. This is more consistent with (C6H6)2− than (C6H6)4−, since (C6H6)4− is reported to be planar.19 Interestingly, the 2.690 Å La–(Cp′ ring centroid) distances are significantly longer than those of either the La3+ complex, Cp′3La, 2.559 Å,31 or the La2+ complex, [K(2.2.2-cryptand)][Cp′3La], 2-La, 2.586 Å.4 Traditionally, bond distances in 4fn+1 Ln2+ complexes are 0.1–0.2 Å larger than those of 4fn Ln3+ complexes, but bond distances for 4fn5d1 complexes of the recently discovered Ln2+ ion complexes are only 0.02–0.03 Å longer.4 A referee has noted that the La1–C34 and La–C37 distances, the shortest for La1–C(C6H6), correlate with the longest La2–C(C6H6) distances, La2–C34 and La2–C37. Similarly, the shortest La2–C(C6H6) distances, La2–C33 and La2–C36, correlate with the longest La1–C(C6H6) distances, La1–C33 and La1–C36.
| 2-La | |||||
|---|---|---|---|---|---|
| a Cnt1, Cnt2, Cnt3, and Cnt4 are the centroids of the Cp′ groups. b Cnt5 is the centroid of the C6 unit labeled C33–C38. c Pln1 and Pln2 are the planes of (C33–C36) and (C33, C36, C37, C38), respectively. | |||||
| La1–Cnt1a | 2.692 | La1–C33 | 2.770(4) | Cnt1–La1–Cnt2a | 111.5 |
| La1–Cnt2a | 2.681 | La1–C34 | 2.639(4) | Cnt1–La1–Cnt5a,b | 125.1 |
| La2–Cnt3a | 2.687 | La1–C35 | 2.694(5) | Cnt2–La1–Cnt5a,b | 123.4 |
| La2–Cnt4a | 2.709 | La1–C36 | 2.777(5) | Cnt3–La2–Cnt4a | 112.6 |
| La1–Cnt5b | 2.278 | La1–C37 | 2.641(5) | Cnt3–La2–Cnt5a | 123.0 |
| La2–Cnt5b | 2.273 | La1–C38 | 2.680(4) | Cnt4–La2–Cnt5a,b | 124.5 |
| C33–C34 | 1.457(6) | La2–C33 | 2.637(5) | Pln1–Pln2c | 11.0 |
| C34–C35 | 1.448(7) | La2–C34 | 2.776(5) | ||
| C35–C36 | 1.446(6) | La2–C35 | 2.683(4) | ||
| C36–C37 | 1.459(6) | La2–C36 | 2.635(5) | ||
| C37–C38 | 1.456(6) | La2–C37 | 2.766(4) | ||
| C38–C33 | 1.454(6) | La2–C38 | 2.676(4) | ||
The UV-vis spectra for 2-Ln are shown in Fig. 2 and compared to the Ln2+ complexes, [K(2.2.2-cryptand)][Cp′3Ln], 3-Ln.3,4 The spectra of 2-Ln are similar to those of 3-Ln, but the extinction coefficients are much higher: ∼8000 M−1 cm−1 in the high-energy visible region. This is particularly unusual since the 1000–2000 M−1 cm−1 extinction coefficients for 3-Ln are already much higher than those of analogous complexes of traditional 4fn Ln2+ complexes of 3-Ln which are all lower than 900 M−1 cm−1.4
 | ||
| Fig. 2 Experimental UV-vis spectra of [K(2.2.2-cryptand)]2[(Cp′2Ln)2(μ-η6:η6-C6H6)], 2-Ln (dark), and [K(2.2.2-cryptand)][Cp′3Ln], 3-Ln (light), in THF (1 mM) at 298 K (Ln = La, solid; Ln = Ce, dotted). | ||
DFT calculations were used to examine the energies of the possible electron configurations of 2-La. All calculations were performed using the Turbomole quantum chemistry software32 and the TPSSh functional (further details are provided in the ESI†).33 Calculations on [(Cp′2Ln)2(μ-η6:η6-C6H6)]2− found an energy minimum corresponding to a diamagnetic singlet ground state that had metrical parameters that matched the crystal data better than any other electronic configuration. The calculations also showed long La–(Cp′ ring centroid) distances, similar to those found experimentally. However, the La–C(C6H6) bond lengths in the calculated minimum were 0.05 Å longer than those in the crystal structure. Single-point energy calculations indicate the triplet and quintet states are 30 kcal mol−1 (10500 cm−1) and 62 kcal mol−1 (21800 cm−1) higher than the singlet ground state, respectively. When a geometry optimization of the quintet state was performed, the optimized structure had only three carbon atoms of the bridging C6H6 ligand coordinated to one metal and the other three carbon atoms coordinated to the other metal. This large difference from the experimental structure indicated that this quintet state is not a viable description of 2-La.
The two highest occupied molecular orbitals (HOMO and HOMO−1) of the singlet state show significant mixing between the metal orbitals and the π* orbitals of the C6H6 ring, Fig. 3. Mulliken population analyses (MPA)34 of the HOMO suggest that 61% of the orbital is localized on π* orbitals of the C6H6 ring with 39% involved with the two metals centers. For HOMO−1, the orbital is 64% on the ring and 36% on the two metals. This is less than that for the HOMO of the Ln2+ ions in (Cp′3Ln)1− (from calculations with identical computational methods), which is often >70% metal-based, depending on the lanthanide, and is primarily a dz2 orbital.
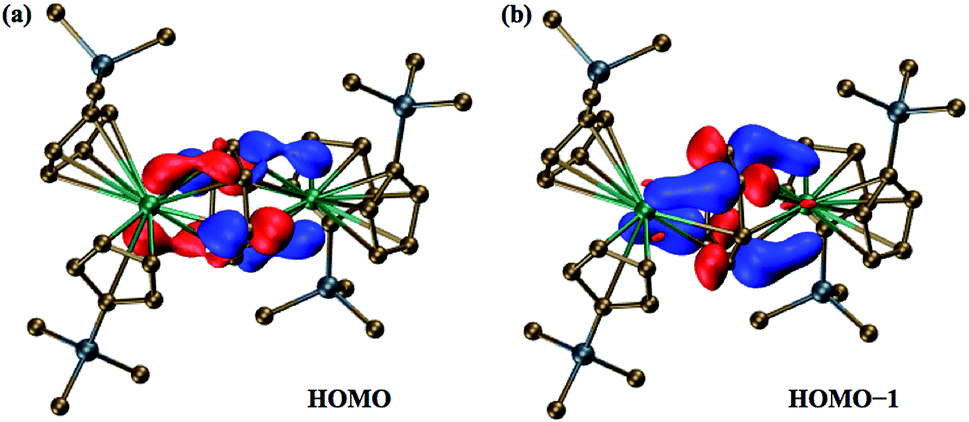 | ||
| Fig. 3 Molecular orbital plots of (a) the 183a orbital (HOMO) and (b) the 182a orbital (HOMO−1) of the dianion in 2-La, using a contour value of 0.05. | ||
Natural population analysis (NPA)35 of the dianion in 2-La suggests that each La center has approximately 1.4 5d electrons, Table 2, which is more than the 1.2 electrons and 0.9 electrons found in calculations on (Cp′3La)1− and Cp′3La, 1-La, respectively.4 Since there is more electron density on each La center in 2-La than even that for the La2+ complex, [K(2.2.2-cryptand)][Cp′3La], 3-La,4 these calculations indicate that the description involving two Ln2+ ions and a (C6H6)2− dianion is the most accurate of the two extreme structures considered above. The mixed arene/d orbital character of the HOMO and HOMO−1 orbitals suggests that it is not a rigid requirement for isolation of Ln2+ complexes of metals such as lanthanum and cerium to have ligand fields that provide a low lying dz2 orbital.
| Metal center | (HOMO–1) MPA | (HOMO) MPA | NPA total density (5d orbital) | ||
|---|---|---|---|---|---|
| % metal | % d | % metal | % d | ||
| La1 | 18 | 13 | 20 | 18 | 1.4 |
| La2 | 18 | 13 | 19 | 18 | 1.4 |
Time dependent DFT (TDDFT)36 calculations were performed to simulate the UV-vis spectrum of 2-La. The simulated spectrum of the singlet state gave the best match to the experimental spectrum as shown in Fig. 4. The predicted UV-vis spectrum contains three broad absorptions, one of which matches the low energy absorption in the experimental spectrum, while the combination of the other two fit under the experimentally-determined high-energy absorption. The excitations between 300 and 1000 nm are comprised of excitations from the HOMO and HOMO−1 orbitals shown in Fig. 3. The lowest energy transitions (>440 nm) involve transitions to primarily metal-based orbitals with s and/or d character, while the highest energy transitions (<440 nm) involve transitions to primarily ligand-based orbitals.
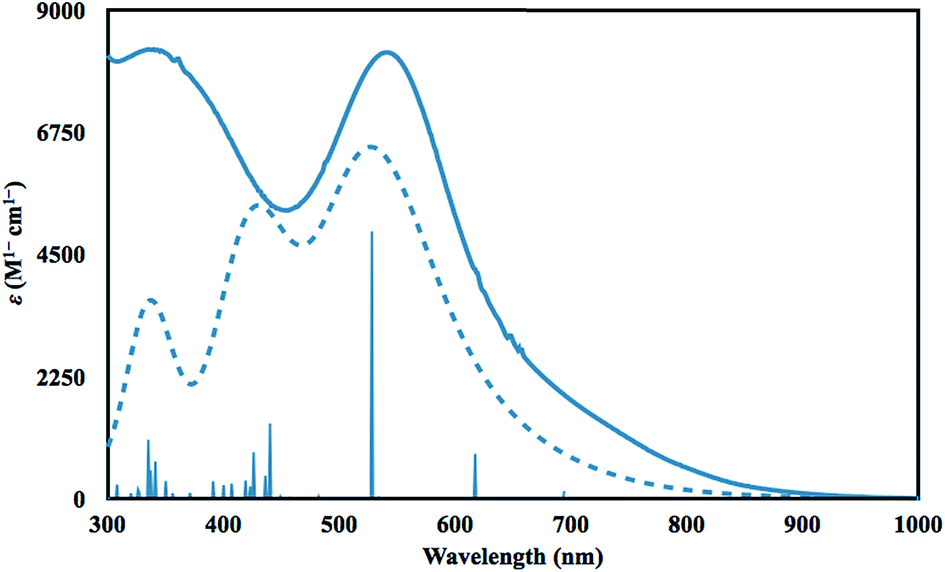 | ||
| Fig. 4 Experimental (solid) and calculated (dotted) UV-vis spectra of 2-La in THF at 298 K, with pertinent theoretical excitations shown as vertical lines and theoretical extinction coefficients scaled down by a factor of 4.7. | ||
 | ||
| Fig. 5 Variable-temperature magnetic susceptibility data collected for 2-La under Hdc = 1 T (green). Simulation of the data using the model described in the text is represented by a solid black line. Variable-temperature magnetic susceptibility data collected for 2-Ce under Hdc = 0.1 T (orange) and 1 T (blue). | ||
The reductive reactivity of 2-Ln was probed by examining the reaction with naphthalene. Two equiv. of naphthalene are reduced by four electrons to produce 2 equiv. of [K(2.2.2-cryptand)][Cp′2Ln(η4-C10H8)], 4-Ln (Ln = La, Ce), Scheme 5.37 The four electron reduction is consistent with the presence of two Ln2+ ions and a (C6H6)2− dianion, but does not provide definitive evidence on this because a (C6H6)4− anion would also be a 4-electron reductant. This reaction does provide a clean route to 4-Ln, which are originally made from 3-Ln in a reaction that has an inseparable byproduct, [K(2.2.2-cryptand)][Cp′4Ln] (Ln = Y, La) that required 4-La to be separated a la Pasteur.37
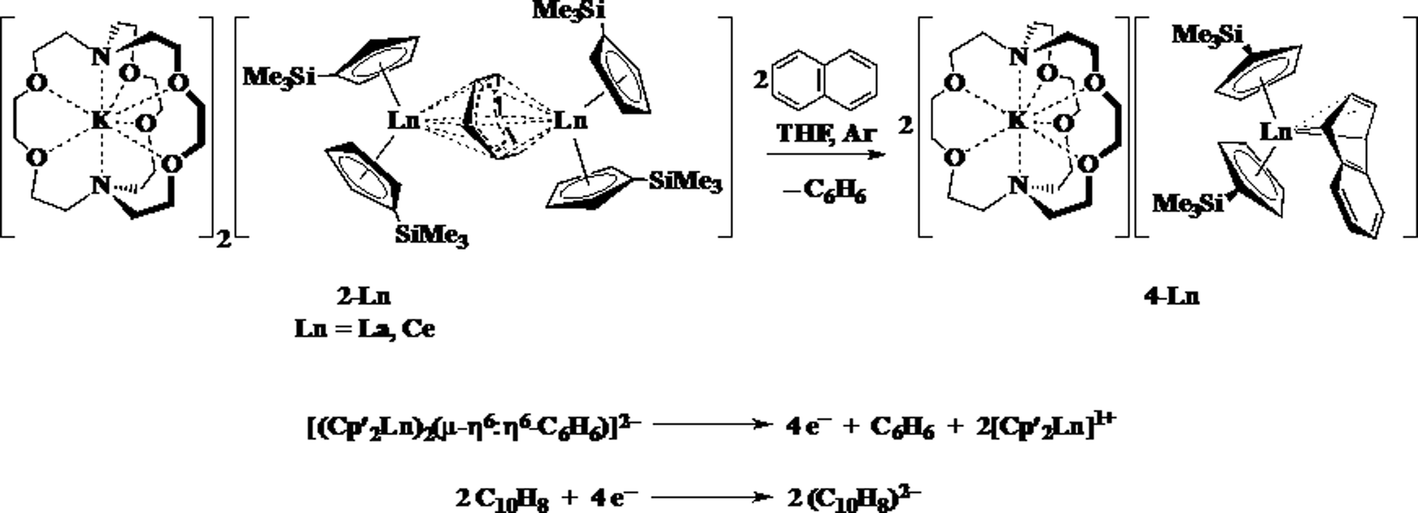 | ||
| Scheme 5 Reduction of naphthalene by 2-Ln to produce 4-Ln. | ||
Complex 2-Ln does not appear to react with N2, but it does reduce 1,3,5,7-cyclooctatetraene in a reaction that is more complicated than Scheme 5. [K(2.2.2-crypt)][(C8H8)2La] was identified as the main product by independent synthesis from K[(C8H8)2La] made in 1973.38 Attempts to reduce 2-Ln further did not give isolable organometallic products.
In summary, a new type of rare earth compound has been identified that is best described as a bimetallic complex of two Ln2+ ions bridged by (C6H6)2−. This result constitutes an intriguing variation of the arene reduction reactions in Schemes 2 and 3 in which complexes are found with either (C6H6)1− and two Ln2+ ions or with (C6H6)2− and one Ln3+ ion. Clearly, small variations in these reactions and in the substituents on the cyclopentadienyl rings can have a significant effect on the product isolated in the tris(cyclopentadienyl)rare earth/alkali metal reductions of arenes.
The isolation of 2-Ln demonstrates the generality of isolating Ln2+ ions with three anionic carbocyclic rings beyond the (Cp′′3)3− and (Cp′3)3− coordination environments. In 2-Ln, a (C6H6)2− dianion shared between two metals takes the place of a cyclopentadienyl anion in the monometallic Ln2+ systems.1–3 This is not a common substitution in organometallic chemistry since arene anions are only formed under highly reducing conditions. DFT studies on 2-La show that this heteroleptic three ring ligand system differs from the tris(cyclopentadienyl) complexes in that the HOMOs have much more ligand character than in the (Cp′′3)3− and (Cp′3)3−-ligated complexes, in which the HOMOs are primarily dz2. These results suggest that other variations of three carbocyclic rings with orbital character beyond that found in tris(cyclopentadienyl) ligand environments could also stabilize unusual Ln2+ ions.
It is important to note that 2-La displays some unexpected properties, i.e. the long La–(Cp′ ring centroid) distances in the crystal structure, the long La–C(C6H6) distances in the DFT calculations, and the extremely high (for a rare earth) extinction coefficients. Determining the origin of these properties will require the isolation of more examples of other ligand systems for these Ln2+ ions. Although the electronic nature of these Ln2+ complexes may not be completely understood, it is clear they can function as multi-electron reductants that can provide four electrons from a single molecule. Hence, these complexes demonstrate a new approach using f element chemistry to multi-electron reducing systems, which are not very common.
Acknowledgements
We thank the U.S. National Science Foundation for support of the magnetic susceptibility studies (CHE-1464841 to J. R. L.), the theoretical studies (CHE-1213382 to F. F.), and all the other experimental studies (CHE-1265396 to W. J. E). We also thank NSF for providing graduate fellowship support for Lucy E. Darago and Chad T. Palumbo.Notes and references
- P. B. Hitchcock, M. F. Lappert, L. Maron and A. V. Protchenko, Angew. Chem., Int. Ed., 2008, 47, 1488–1491 CrossRef CAS PubMed.
- M. R. MacDonald, J. W. Ziller and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 15914–15917 CrossRef CAS PubMed.
- M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 9857–9868 CrossRef CAS PubMed.
- M. E. Fieser, M. R. MacDonald, B. T. Krull, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2015, 137, 369–382 CrossRef CAS PubMed.
- M. R. MacDonald, J. E. Bates, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2012, 134, 8420–8423 CrossRef CAS PubMed.
- J. W. Lauher and R. Hoffmann, J. Am. Chem. Soc., 1976, 98, 1729–1742 CrossRef CAS.
- B. E. Bursten, L. F. Rhodes and R. J. Strittmatter, J. Am. Chem. Soc., 1989, 111, 2756–2758 CrossRef CAS.
- B. E. Bursten, L. F. Rhodes and R. J. Strittmatter, J. Am. Chem. Soc., 1989, 111, 2758–2766 CrossRef CAS.
- R. J. Strittmatter and B. E. Bursten, J. Am. Chem. Soc., 1991, 113, 552–559 CrossRef CAS.
- W. W. Lukens Jr and R. A. Andersen, Organometallics, 1995, 14, 3435–3439 CrossRef.
- R. G. Denning, J. Harmer, J. C. Green and M. Irwin, J. Am. Chem. Soc., 2011, 133, 20644–20660 CrossRef CAS PubMed.
- K. C. Nicolaou, S. P. Ellery and J. S. Chen, Angew. Chem., Int. Ed., 2009, 48, 7140–7165 CrossRef CAS PubMed.
- D. J. Procter, R. A. Flowers II and T. Skrydstrup, Organic Synthesis Using Samarium Diiodide: A Practical Guide, RSC Publishing, Cambridge, U. K., 2010 Search PubMed.
- M. C. Cassani, D. J. Duncalf and M. F. Lappert, J. Am. Chem. Soc., 1998, 120, 12958–12959 CrossRef CAS.
- Y. K. Gun'ko, P. B. Hitchcock and M. F. Lappert, Organometallics, 2000, 19, 2832–2834 CrossRef.
- M. D. Fryzuk, J. B. Love and S. J. Rettig, J. Am. Chem. Soc., 1997, 119, 9071–9072 CrossRef CAS.
- M. D. Fryzuk, L. Jafarpour, F. M. Kerton, J. B. Love, B. O. Patrick and S. J. Rettig, Organometallics, 2001, 20, 1387–1396 CrossRef CAS.
- M. N. Bochkarev, Chem. Rev., 2002, 102, 2089–2117 CrossRef CAS PubMed.
- W. Huang, F. Dulong, T. Wu, S. I. Khan, J. T. Miller, T. Cantat and P. L. Diaconescu, Nat. Commun., 2013, 4, 1448–1455 CrossRef PubMed.
- M. C. Cassani, Y. K. Gun'ko, P. B. Hitchcock, A. G. Hulkes, A. V. Khvostov, M. F. Lappert and A. V. Protchenko, J. Organomet. Chem., 2002, 647, 71–83 CrossRef CAS.
- P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108–6109 CrossRef CAS.
- W. J. Evans, S. A. Kozimor, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2004, 126, 14533–14547 CrossRef CAS PubMed.
- P. L. Arnold, S. M. Mansell, L. Maron and D. McKay, Nat. Chem., 2012, 4, 668–674 CrossRef CAS PubMed.
- D. Patel, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2013, 42, 5224–5227 RSC.
- C. Camp, V. Mougel, J. Pécaut, L. Maron and M. Mazzanti, Chem.–Eur. J., 2013, 19, 17528–17540 CrossRef CAS PubMed.
- M. C. Cassani, Y. K. Gun'ko, P. B. Hitchcock and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1996, 1987–1988 RSC.
- M. C. Cassani, Y. K. Gun'ko, P. B. Hitchcock, M. F. Lappert and F. Laschi, Organometallics, 1999, 18, 5539–5547 CrossRef CAS.
- M. D. Walter, C. H. Booth, W. W. Lukens and R. A. Andersen, Organometallics, 2009, 28, 698–707 CrossRef CAS.
- K. R. Meihaus, M. E. Fieser, J. F. Corbey, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2015, 137, 9855–9860 CrossRef CAS PubMed.
- F. N. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- J. K. Peterson, M. R. MacDonald, J. W. Ziller and W. J. Evans, Organometallics, 2013, 32, 2625–2631 CrossRef CAS.
- F. Furche, R. Ahlrichs, C. Hattig, W. Klopper, M. Sierka and F. Weigend, WIREs Computational Molecular Science, 2014, 4, 91–100 CrossRef CAS PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS PubMed.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- C. M. Kotyk, M. R. MacDonald, J. W. Ziller and W. J. Evans, Organometallics, 2015, 34, 2287–2295 CrossRef CAS.
- K. O. Hodgson, F. Mares, D. F. Starks and A. Streitwieser, J. Am. Chem. Soc., 1973, 95, 8650–8658 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details; converged structure data; crystallographic data collection, structure solution, and refinement; and crystallographic data and complete bond distances and angles for compound 2-La. CCDC 1409845. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02486b |
| This journal is © The Royal Society of Chemistry 2015 |