
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective acceleration of disfavored enolate addition reactions by anion–π interactions†
Yingjie
Zhao‡
,
Sebastian
Benz
,
Naomi
Sakai
and
Stefan
Matile
*
Department of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: http://www.unige.ch/sciences/chiorg/matile/ Fax: +41 22 379 5123; Tel: +41 22 379 6523
First published on 25th August 2015
Abstract
In chemistry and biology, cation–π interactions contribute significantly to many important transformations. In sharp contrast, reactions accomplished with support from the complementary anion–π interactions are essentially unknown. In this report, we show that anion–π interactions can determine the selectivity of the enolate chemistry of malonate half thioesters. Their addition to enolate acceptors is central in natural product biosynthesis but fails without enzymes because non-productive decarboxylation dominates. The newly designed and synthesized anion–π tweezers invert this selectivity by accelerating the disfavored and decelerating the favored process. The discrimination of anionic tautomers of different planarization and charge delocalization on π-acidic surfaces is expected to account for this intriguing “tortoise-and-hare catalysis.” Almost exponentially increasing selectivity with increasing π acidity of the catalyst supports that contributions from anion–π interactions are decisive.
Cation–π interactions play a central role in molecular recognition, translocation and transformation.1–5 Arguably the most spectacular manifestation of cation–π catalysis in biology is found in the biosynthesis of steroids, in which cascade cyclization occurs via carbocation hopping on a stabilizing cluster of π-basic amino acid residues (Fig. 1).2 Cation–π interactions are also increasingly recognized in organocatalysis.3–5 Contributions from the complementary but much younger6 anion–π interactions7 have been reported for anion binding8–11 and transport.11,12 In sharp contrast, explicit considerations of anion–π interactions in catalysis are extremely rare and very recent.13–15
 | ||
| Fig. 1 In nature, carbocation chemistry in the biosynthesis of terpenes and steroids is accomplished with cation–π interactions (red circles indicate the position of π-basic amino-acid residues in the cation–π enzyme for substrate 1). The complementary enolate chemistry in polyketide biosynthesis and the beginning of both pathways fails in solution because decarboxylation of 2 (solid arrows) dominates over enolate addition (dashed arrows). In this report, selective acceleration of this disfavored but relevant process is achieved with anion–π interactions (blue background) and explained with the discrimination between non-planar tautomers (2) and planar tautomers (4/5; BH+: protonated base, E = electrophilic carbon). | ||
Looking for more significant transformations that could benefit from anion–π interactions, we considered malonate half thioester (MHT) 2, which is obtained by deprotonation of malonyl-CoA, a malonic acid half thioester (MAHT), under mildest conditions (Fig. 1).16 Claisen condensation with acetyl-CoA 3 yields acetoacetyl-CoA as entry into both biosynthetic pathways, terpenoids and polyketides. Catalyzed by polyketide synthases, repeated decarboxylative enolate addition of the same substrate provides access to more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 natural products as important as fatty acids and lipids, macrolactones and higher aromatics.16 In solution, MHTs have been shown to add to nitroolefins, enones, aldehydes, imines or (thio)esters as in polyketide biosynthesis.17–22
000 natural products as important as fatty acids and lipids, macrolactones and higher aromatics.16 In solution, MHTs have been shown to add to nitroolefins, enones, aldehydes, imines or (thio)esters as in polyketide biosynthesis.17–22
However, under unoptimized conditions, the non-productive decarboxylation to 3 dominates. Recent mechanistic studies indicate that for addition to occur, it should precede decarboxylation.21,22 This should be possible with tautomers 4 or 5, whereas tautomer 2 should favor decarboxylation. Control over the selectivity between addition and decarboxylation thus calls for the discrimination between planar tautomers in which the negative charge is delocalized by resonance and tautomers in which planarity and resonance are disrupted by the tetrahedral sp3 carbon between the two carbonyl groups. Anion recognition on π-acidic aromatic planes appeared just ideal to feel these subtle structural differences.
The addition of aromatic18 and aliphatic MAHTs 6 and 7 to aromatic and aliphatic nitroolefins9–12,208 and 9 was selected to elaborate on this hypothesis (Fig. 2). In bifunctional anion–π catalysts, π-acidic 1,4,5,8-naphthalenediimide (NDI) derivatives12–14,23–25 are appended to an amine base to provide stabilizing π surfaces for the enolate intermediates as soon as they are produced. All substrates and catalysts were synthesized from commercially available starting materials in a few steps (Scheme S1†).26 The reaction of MAHTs 6 and 7 with nitroolefins 8 and 9 was followed by 1H NMR spectroscopy, in which the evolution of the intrinsically disfavored addition products d (10) and the favored decarboxylation products f (3) was recorded with time against internal standards (Fig. S1–S4†). Results were quantified with ηd/f values, that is the yield ηd of the intrinsically disfavored divided by ηf for the favored product (Table 1).
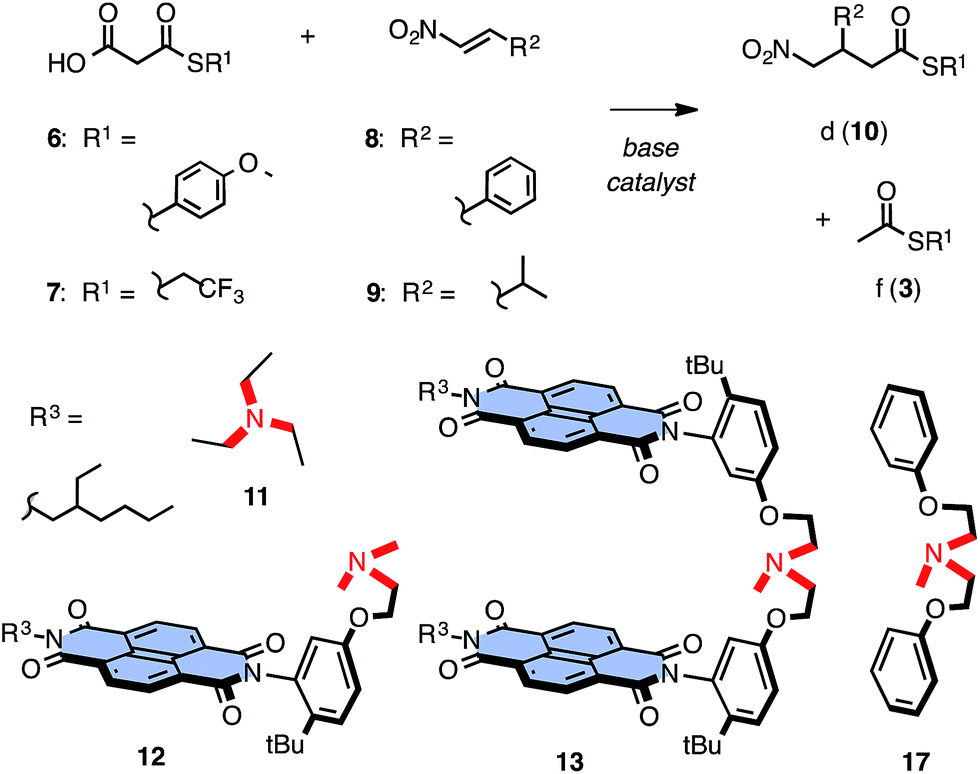 | ||
| Fig. 2 Structure of substrates (6–9), favored product f (3), disfavored product d (10), the minimalist bifunctional catalyst 12, anion–π tweezer 13 and control bases 11 and 17. | ||
| Cb | E LUMO (eV) | S1b | S2b | S1/Cd | T (°C)e | t (h)f | η d (%) | η f (%) | η d/f | ΔEdaj (kJ mol−1) | ΔEfak (kJ mol−1) | ΔΔEd–fal (kJ mol−1) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reactions were conducted in THF, with 4–40 mM catalyst C, 200 mM substrate S1 (6 and 7), 2 M S2 (8 and 9), results were analyzed by 1H NMR spectroscopy, compare Fig. 3a and 4 for data analysis. b See Fig. 2 and 3 for structures. c LUMO energy levels in eV relative to −5.1 eV for Fc+/Fc, approximated from cyclic voltammetry data.13,23,25 d Catalyst C per substrate S1 used in the reaction. e Reaction temperature, RT = room temperature. f Reaction time for >95% conversion. g Yield of intrinsically disfavored product d (10). h Yield of intrinsically favored product f (3). i η d/f = ηd/ηf. j Difference in activation energy of the disfavored (d) reaction compared to control 11 (or 17)m, from initial velocity of formation of product d (10). k Same for favored (f) reaction vs.11 (or 17)m, from vini of f (3). l Selective catalysis: ΔEda − ΔEfa. m Measured against 17. | |||||||||||||
| 1 | 11 | — | 6 | 8 | 0.2 | RT | 1.5 | 36 | 62 | 0.6 | — | — | — |
| 2 | 12 | −4.2 | 6 | 8 | 0.2 | RT | 6 | 46 | 54 | 0.8 | +2.8/−3.5m | +3.8/−2.7m | −0.9/−0.8m |
| 3 | 13 | −4.2 | 6 | 8 | 0.2 | RT | 15 | 48 | 51 | 0.9 | +5.5/−0.8m | +6.6/+0.1m | −1.1/−0.9m |
| 4 | 14 | −3.9 | 6 | 8 | 0.2 | RT | 12 | 50 | 48 | 1.0 | +4.5/−1.8m | +5.6/−0.9m | −1.1/−0.9m |
| 5 | 15 | −4.4 | 6 | 8 | 0.2 | RT | 12 | 59 | 36 | 1.6 | +3.9/−2.4m | +6.1/−0.4m | −2.2/−2.0m |
| 6 | 16 | −4.6 | 6 | 8 | 0.2 | RT | 12 | 59 | 31 | 1.9 | +3.7/−2.7m | +6.6/+0.1m | −2.9/−2.6m |
| 7 | 17 | — | 6 | 8 | 0.2 | RT | 24 | 37 | 53 | 0.7 | — | — | — |
| 8 | 11 | — | 6 | 8 | 0.2 | 5 | 8 | 57 | 40 | 1.4 | — | — | — |
| 9 | 12 | −4.2 | 6 | 8 | 0.2 | 5 | 20 | 69 | 30 | 2.3 | +1.3/−3.3m | +2.5/−2.6m | −1.2/−0.7m |
| 10 | 13 | −4.2 | 6 | 8 | 0.2 | 5 | 40 | 77 | 20 | 3.8 | +3.7/−0.9m | +5.8/+0.7m | −2.1/−1.6m |
| 11 | 14 | −3.9 | 6 | 8 | 0.2 | 5 | 40 | 71 | 23 | 3.1 | +3.7/−0.9m | +5.4/+0.3m | −1.8/−1.2m |
| 12 | 15 | −4.4 | 6 | 8 | 0.2 | 5 | 40 | 80 | 14 | 5.7 | +3.1/−1.4m | +6.6/+1.5m | −3.5/−2.9m |
| 13 | 16 | −4.6 | 6 | 8 | 0.2 | 5 | 40 | 80 | 11 | 7.3 | +2.8/−1.7m | +6.9/+1.8m | −4.1/−3.5m |
| 14 | 16 | −4.6 | 6 | 8 | 0.02 | 5 | 672 | 84 | 14 | 6.0 | — | — | — |
| 15 | 17 | — | 6 | 8 | 0.2 | 5 | 75 | 60 | 30 | 2.0 | — | — | — |
| 16 | 11 | — | 6 | 9 | 0.2 | 5 | 9 | 40 | 54 | 0.7 | — | — | — |
| 17 | 13 | −4.2 | 6 | 9 | 0.2 | 5 | 70 | 59 | 40 | 1.5 | +3.1 | +4.3 | −1.3 |
| 18 | 13 | −4.2 | 6 | 9 | 0.2 | RT | 20 | 31 | 65 | 0.5 | +5.1/−1.0m | +8.1/+0.9m | −3.0/−1.9m |
| 19 | 14 | −3.9 | 6 | 9 | 0.2 | RT | 20 | 28 | 62 | 0.4 | +5.3/−0.8m | +7.6/+0.5m | −2.3/−1.3m |
| 20 | 15 | −4.4 | 6 | 9 | 0.2 | RT | 20 | 39 | 53 | 0.7 | +4.7/−1.4m | +8.4/+1.2m | −3.7/−2.6m |
| 21 | 16 | −4.6 | 6 | 9 | 0.2 | RT | 20 | 45 | 48 | 0.9 | +4.4/−1.8m | +8.5/+1.4m | −4.2/−3.2m |
| 22 | 17 | — | 6 | 9 | 0.2 | RT | 25 | 23 | 70 | 0.3 | — | — | — |
| 23 | 13 | −4.2 | 7 | 8 | 0.2 | RT | 9 | 41 | 57 | 0.7 | −0.7 | +0.5m | −1.2m |
| 24 | 17 | — | 7 | 8 | 0.2 | RT | 9 | 33 | 66 | 0.5 | — | — | — |
Catalyzed with TEA 11 at room temperature, ηd/f = 0.6 confirmed that the undesired decarboxylation is indeed favored under these conditions (Fig. 2, Table 1, entry 1). In comparison, the simplest possible anion–π catalyst, i.e., catalyst 12 composed of a π-acidic NDI surface next to a tertiary amine base, already gave rise to a slightly better ηd/f = 0.8 (Table 1, entry 2). The number of π-acidic surfaces in the catalyst was doubled next to increase the effective molarity of π-acidic surfaces or to even act from two sides on the reaction. The resulting ηd/f = 0.9 demonstrated that with anion–π tweezer 13, addition became almost as good as decarboxylation (Table 1, entry 3).
These encouraging results called for a systematic assessment of the contribution from anion–π interactions. The reversible oxidation of sulfide donors to sulfoxide and sulfone acceptors has been introduced and validated previously as unique approach to vary π acidity with minimal structural changes.18 Anion–π tweezer 14 with two sulfides in the core of each NDI was prepared as a mixture of axial stereoisomers (Fig. 3b). The temperature-controlled stepwise sulfide oxidation was unproblematic as long as the tertiary amine was protected first against oxidation by protonation with TFA. Although insufficient,7 the energy levels of the LUMOs are used as an approximative measure for π acidity. They decrease from ELUMO = −3.9 eV for NDIs in 14 with two sulfide donors to ELUMO = −4.4 eV for NDIs in 15 with sulfoxide acceptors and ELUMO = −4.6 eV for NDIs in 16 with sulfones.12,13 With decreasing ELUMO of the catalyst, the selectivity increased almost exponentially14b from ηd/f = 1.0 for 14 to ηd/f = 1.6 for 15 and ηd/f = 1.9 for the strongest π acid 16 (Fig. 3a, ●; Table 1, entries 4–6). Enolate addition became more dominant at lower temperatures. At 5 °C, selectivity perfectly followed π acidity, increasing from ηd/f = 3.1 for anion–π tweezers 14 with donating sulfides to ηd/f = 3.8 for tweezers 13 with unsubstituted NDIs and ηd/f = 5.7 and ηd/f = 7.3 for tweezers 15 and 16 with withdrawing sulfoxides and sulfones, respectively (Fig. 3a, ♦; Table 1, entries 10–13).
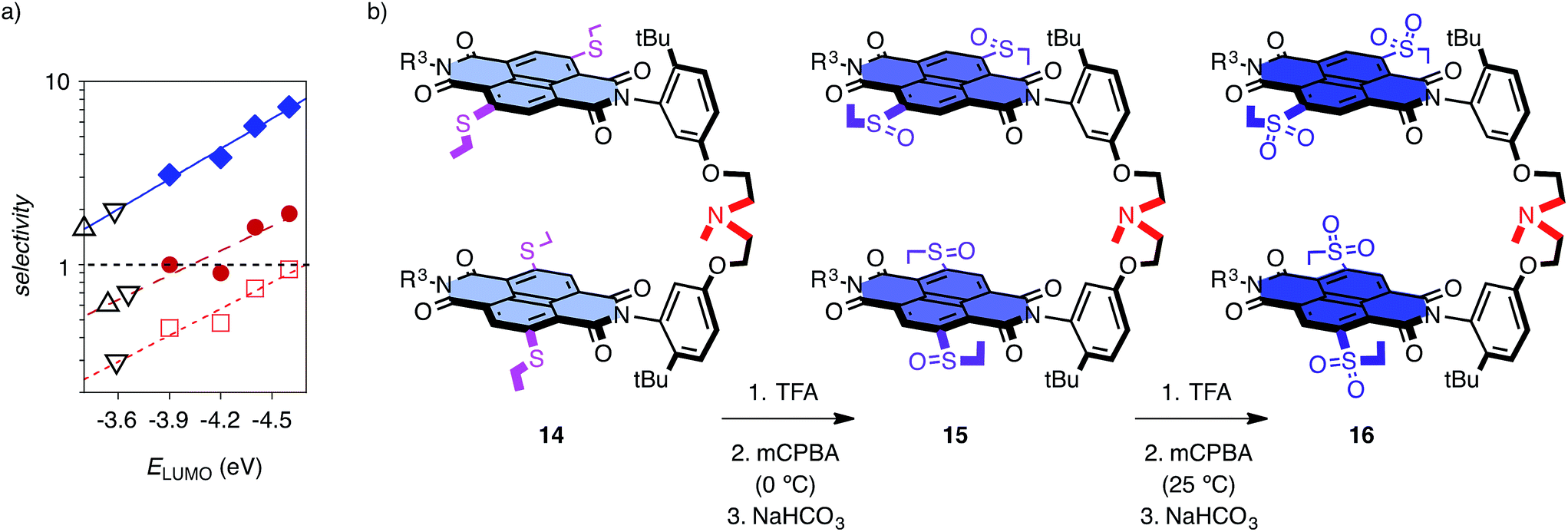 | ||
| Fig. 3 (a) Dependence of ηd/f, i.e. the yield ηd of the intrinsically disfavored product (10) divided by ηf of the favored product (3), on the energy of the LUMO of tweezers 13–16 at RT (red, ●, □) and 5 °C (blue, ♦) for substrates 6 (●, ♦) and 7 (□) with 8; with exponential curve fit. Controls 11 (△) and 17 (▽) select below ELUMO = −3.7 eV of π-neutral NDIs. (b) Stepwise oxidation of the core substituents of anion–π tweezers 14 gradually increases the π acidity of the catalyst without global structural changes. All tweezers used are mixtures of stereoisomers (axial chirality, sulfoxides).25 | ||
All reactions proceeded to completion, with little formation of other side products (Table 1). The nitroolefin acceptor 8 was used in excess to maximize the probability of addition once the reactive enolate is formed on the π-acidic surface. For comparative evaluation, 20 mol% catalyst was used with regard to the MAHT substrate 6. With the best anion–π tweezer 16, selectivity ratios were with ηd/f = 6.0 nearly preserved at reduced catalyst loading (Table 1, entries 13 and 14). With 2 mol% 16, full conversion within 30 days at 5 °C gave a turnover number TON = 50 (Fig. S2†).
Replacement of the π-basic phenyl substituents in substrates 6 and 8 by alkyl groups in 7 and 9 did not disturb the observed trends (Fig. 2, Table 1, entries 16–24). With 6 and 9 at low temperature, a clean inversion of selectivity was obtained from control 11 with preference for decarboxylation (ηd/f = 0.7) to anion–π tweezers 13 with preference for addition (ηd/f = 1.5, Table 1, entries 16 and 17). Measured at RT, ηd/f values increased with increasing π acidity of the catalyst from ηd/f = 0.4 for 14 with sulfide donors to ηd/f = 0.5 for original 13 and ηd/f = 0.7 and ηd/f = 0.9 for 15 and 16 with increasing π acidity (Fig. 3a, □; Table 1, entries 18–21). As a final control, we replaced TEA 11 by a standard more similar to the operational anion–π tweezers 12–16. With substrates 6 and 8 at room temperature, control 17 afforded ηd/f = 0.7 (Fig. 3a, ▽; Table 1, entry 7), better than TEA 11 (ηd/f = 0.6, Fig. 3a, △) but clearly inferior to the original tweezers 13 (ηd/f = 0.9, Table 1, entry 3) and far off the best performing anion–π tweezers 16 (ηd/f = 1.9, Table 1, entry 6; Fig. 3a, ●).
The dependence of selectivity on π acidity, expressed as ELUMO of the catalysts, was close to exponential,14b independent of temperature and substrates (Fig. 3a, ●, ♦, □). The compared to the perfect sulfur series 14–16 somewhat underperforming unsubstituted NDI tweezers 13 indicated the presence of minor, supportive as well as constant contributions from the ethyl sidechains to catalysis and thus confirmed the importance of the isostructural variation of π acidity provided by stepwise sulfide oxidation in the series 14–16 (Fig. 3a and 4). The selectivities obtained for controls 11 and 17 at different temperatures clustered below a virtual ELUMO = −3.7 eV (Fig. 3a, △, ▽). This value corresponds to a nearly π-neutral NDI with two alkylamino donors in the core.12,23 Selectivity values of controls coinciding with those extrapolated for π-neutral NDI surfaces provided corroborative support that anion–π interactions indeed account for the selective acceleration of disfavored reactions.
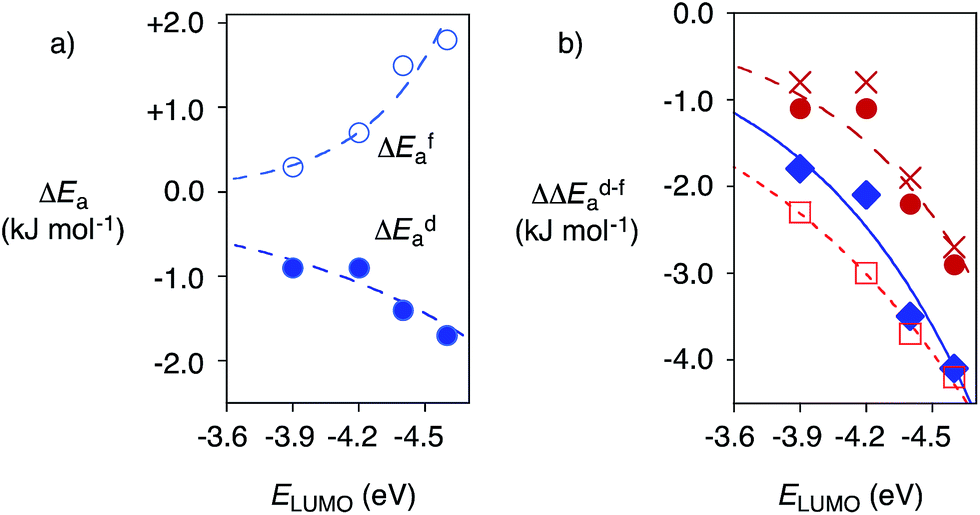 | ||
| Fig. 4 (a) Dependence of the changes in activation energy ΔEa for substrate 6 for the favored decarboxylation (ΔEfa, ○) and the disfavored addition (to 8, ΔEda, ●) on the π acidity of anion–π tweezers 13–16 (ELUMO), relative to control 17, at 5 °C, with exponential curve fit (Table 1, entries 10–13). (b) Selective acceleration of a disfavoured reaction: dependence of ΔΔEd–fa, i.e., ΔEda − ΔEfa, for 6 on the π acidity of 13–16 (ELUMO) compared to control 11 (●, ♦, □) or 17 (X) at RT (●, X, □) and 5 °C (♦) for 6 (●, X, ♦) and 7 (□) with 8, with exponential curve fit (compare Table 1). | ||
Reactions with anion–π catalysts were slower than with TEA 11 but still much faster than without any amine catalyst. The initial velocities of product formation were used to determine activation energies Efa and Eda, that is the energy difference between ground state and transition state for the favored decarboxylation (f) and the disfavored addition (d, Fig. S5†). Subtraction of activation energies of controls 11 or 17 from those of anion–π catalysts gave ΔEfa and ΔEda (deceleration: ΔEa > 0, acceleration: ΔEa < 0). Positive ΔEfa and ΔEda revealed that compared to control 11, anion–π tweezers 13–16 slowed down both processes (Table 1). Compared to the more revealing control 17, anion–π catalysts 13–16 always accelerated the disfavored (ΔEda < 0) and mostly decelerated the favored process (ΔEfa > 0, Table 1). Most impressive the trends at low temperatures: without exception, acceleration of disfavored and deceleration of favored reaction both increased with increasing π acidity of anion–π tweezers 13–16 (Fig. 4a, Table 1, entries 10–13).
Selective acceleration of a disfavored reaction is given as ΔΔEd–fa = ΔEda − ΔEfa < 0, valid for both deceleration or acceleration of the competing processes. Close to exponential14b increase of the negative ΔΔEd–fa with increasing π acidity of the catalyst was found, independent of conditions (Fig. 4b, ● (warm) vs. ○ (cold)), substrates (Fig. 4b, ● (6) vs. □ (7)) and controls (Fig. 4b, ● (11) vs. X (17), Table 1). This consistent kinetic response to increasing π acidity supported that the inversion of selectivity indeed originates from anion–π interactions.
Selective deceleration of the favored decarboxylation and selective acceleration of the disfavored addition were both in agreement with the envisioned discrimination of differently planarized and delocalized tautomers by anion–π interactions (Fig. 1). It might be important to recall that direct experimental evidence for the ground-state stabilization of enolates on π-acidic surfaces is available from covalent model systems.14 Transition-state destabilization for decarboxylation (by immobilizing the localized negative charge in tautomer 2 on the carboxylate oxygens on the π-acidic surface) could contribute as well. The same is true for transition-state stabilization for addition (by stabilizing the formation of the topologically matching nitronate9–12 on the π-acidic surface). More explicit comments on mechanisms, applications and perspectives26,13c would be premature. Such concluding remarks are also not needed to appreciate the main lesson learned from this study: selective “tortoise-and-hare catalysis”27 of enolate chemistry provides experimental support that anion–π catalysis13 not only exists but also matters.
Acknowledgements
We thank J. Gajewy, G. Huang and D.-H. Tran for contributions to synthesis, the NMR and the Sciences Mass Spectrometry (SMS) platforms for services, and the University of Geneva, the European Research Council (ERC Advanced Investigator), the Swiss National Centre of Competence in Research (NCCR) Chemical Biology, the NCCR Molecular Systems Engineering and the Swiss NSF for financial support.References
- D. A. Stauffer, R. E. Barrans Jr and D. A. Dougherty, Angew. Chem., Int. Ed., 1990, 29, 915–918 CrossRef.
- (a) K. U. Wendt, G. E. Schulz, E. J. Corey and D. R. Liu, Angew. Chem., Int. Ed., 2000, 39, 2812–2833 CrossRef CAS; (b) J. A. Faraldos, A. K. Antonczak, V. Gonzalez, R. Fullerton, E. M. Tippmann and R. K. Allemann, J. Am. Chem. Soc., 2011, 133, 13906–13909 CrossRef CAS PubMed.
- (a) Q. Zhang and K. Tiefenbacher, Nat. Chem., 2015, 7, 197–202 CrossRef CAS PubMed; (b) M. C. Holland, J. B. Metternich, C. Mück-Lichtenfeld and R. Gilmour, Chem. Commun., 2015, 51, 5322–5325 RSC.
- R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030–5032 CrossRef CAS PubMed.
- S. Yamada and J. S. Fossey, Org. Biomol. Chem., 2011, 9, 7275–7281 Search PubMed.
- (a) D. Quinonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deya, Angew. Chem., Int. Ed., 2002, 41, 3389–3392 CrossRef CAS; (b) M. Mascal, A. Armstrong and M. D. Bartberger, J. Am. Chem. Soc., 2002, 124, 6274–6276 CrossRef CAS PubMed; (c) I. Alkorta, I. Rozas and J. Elguero, J. Am. Chem. Soc., 2002, 124, 8593–8598 CrossRef CAS PubMed.
- In analogy to cation–π interactions, the term anion–π interactions is used herein to refer to the site of the interaction (i.e., on the aromatic π surface, orthogonal to the plane with distances around or preferably shorter than the sum of the WdV radii) without any implications on the nature of the interaction (Qzz quadrupoles, in-plane multipoles, orbital overlap, etc). Complementary to the HOMO chemistry with cation–π interactions, anion–π interactions relate to LUMO chemistry, “too strong” anion–π interactions afford charge-transfer complexes and radicals (comparable to proton transfer and conjugate acids and bases with “too strong” hydrogen bonds), and to nucleophilic aromatic substitution.6–12 (a) A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564–9583 CrossRef CAS PubMed; (b) H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2013, 46, 894–906 CrossRef CAS PubMed; (c) L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed; (d) S. T. Schneebeli, M. Frasconi, Z. Liu, Y. Wu, D. M. Gardner, N. L. Strutt, C. Cheng, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 13100–13104 CrossRef CAS PubMed; (e) S. Kumar, M. R. Ajayakumar, G. Hundal and P. Mukhopadhyay, J. Am. Chem. Soc., 2014, 136, 12004–12010 CrossRef CAS PubMed; (f) C. Estarellas, A. Frontera, D. Quiñonero and P. M. Deyà, Angew. Chem., Int. Ed., 2011, 50, 415–418 CrossRef CAS PubMed; (g) K. Fujisawa, C. Beuchat, M. Humbert-Droz, A. Wilson, T. A. Wesolowski, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2014, 53, 11266–11269 CrossRef CAS PubMed; (h) B. P. Hay and V. S. Bryantsev, Chem. Commun., 2008, 44, 2417–2428 RSC; (i) H. T. Chifotides, B. L. Schottel and K. R. Dunbar, Angew. Chem., Int. Ed., 2010, 49, 7202–7207 CrossRef CAS PubMed; (j) D.-X. Wang, Q. Y. Zheng, Q. Q. Wang and M.-X. Wang, Angew. Chem., Int. Ed., 2008, 47, 7485–7488 CrossRef CAS PubMed; (k) D.-X. Wang, Q.-Q. Wang, Y. Han, Y. Wang, Z.-T. Huang and M.-X. Wang, Chem.–Eur. J., 2010, 44, 13053–13057 CrossRef PubMed; (l) D.-X. Wang and M.-X. Wang, Chimia, 2011, 65, 939–943 CrossRef CAS PubMed.
- P. Ballester, Acc. Chem. Res., 2013, 46, 874–884 CrossRef CAS PubMed.
- D.-X. Wang and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 892–897 CrossRef CAS PubMed.
- (a) M. M. Watt, L. N. Zakharov, M. M. Haley and D. W. Johnson, Angew. Chem., Int. Ed., 2013, 52, 10275–10280 CrossRef CAS PubMed; (b) M. Giese, M. Albrecht, G. Ivanova, A. Valkonen and K. Rissanen, Supramol. Chem., 2011, 24, 48–55 CrossRef.
- L. Adriaenssens, C. Estarellas, A. Vargas Jentzsch, M. Martinez Belmonte, S. Matile and P. Ballester, J. Am. Chem. Soc., 2013, 135, 8324–8330 CrossRef CAS PubMed.
- A. Vargas Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791–2800 CrossRef CAS PubMed.
- (a) Y. Zhao, Y. Domoto, E. Orentas, C. Beuchat, D. Emery, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2013, 52, 9940–9943 CrossRef CAS PubMed; (b) Y. Zhao, C. Beuchat, Y. Domoto, J. Gajewy, A. Wilson, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 2101–2111 CrossRef CAS PubMed; (c) Y. Zhao, Y. Cotelle, A.-J. Avestro, N. Sakai and S. Matile, submitted.
- (a) Y. Zhao, N. Sakai and S. Matile, Nat. Commun., 2014, 5, 3911 CAS; (b) F. N. Miros, Y. Zhao, G. Sargsyan, M. Pupier, C. Besnard, C. Beuchat, J. Mareda, N. Sakai and S. Matile, submitted.
- A. Berkessel, S. Das, D. Pekel and J. M. Neudörfl, Angew. Chem., Int. Ed., 2014, 53, 11660–11664 CrossRef CAS PubMed.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- Y. Kobuke and J. Yoshida, Tetrahedron Lett., 1978, 19, 367–370 CrossRef.
- N. Sakai, N. Sordé and S. Matile, Molecules, 2001, 6, 845–851 CrossRef CAS.
- (a) L. Bernardi, M. Fochi, M. Comes Franchini and A. Ricci, Org. Biomol. Chem., 2012, 10, 2911–2922 RSC; (b) H. Y. Bae, J. H. Sim, J.-W. Lee, B. List and C. E. Song, Angew. Chem., Int. Ed., 2013, 52, 12143–12147 CrossRef CAS PubMed.
- A. Bahlinger, S. P. Fritz and H. Wennemers, Angew. Chem., Int. Ed., 2014, 53, 8779–8783 CrossRef CAS PubMed.
- N. Blaquiere, D. G. Shore, S. Rousseaux and K. Fagnou, J. Org. Chem., 2009, 74, 6190–6198 CrossRef CAS PubMed.
- Y. Pan, C. W. Kee, Z. Jiang, T. Ma, Y. Zhao, Y. Yang, H. Xue and C.-H. Tan, Chem.–Eur. J., 2011, 17, 8363–8370 CrossRef CAS PubMed.
- N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225–4237 RSC.
- (a) S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC; (b) S. L. Suraru and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 7428–7448 CrossRef CAS PubMed; (c) M. R. Molla and S. Ghosh, Chem.–Eur. J., 2012, 18, 9860–9869 CrossRef CAS PubMed; (d) G. J. Gabriel and B. L. Iverson, J. Am. Chem. Soc., 2002, 124, 15174–15175 CrossRef CAS PubMed; (e) Y. S. Chong, B. E. Dial, W. G. Burns and K. D. Shimizu, Chem. Commun., 2012, 48, 1296–1298 RSC; (f) N. Ponnuswamy, G. D. Pantoş, M. M. J. Smulders and J. M. K. Sanders, J. Am. Chem. Soc., 2012, 134, 566–573 CrossRef CAS PubMed.
- Y. Zhao, G. Huang, C. Besnard, J. Mareda, N. Sakai and S. Matile, Chem.–Eur. J., 2015, 21, 6202–6207 CrossRef CAS PubMed.
- See ESI†.
- L. Gibbs, Aesop's Fables, Oxford University Press, 2002, Perry Index 226 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed procedures and results for all reported experiments. See DOI: 10.1039/c5sc02563j |
| ‡ Present address: Institute of Polymers, ETH Zurich, Switzerland and Qingdao University of Science and Technology, China. |
| This journal is © The Royal Society of Chemistry 2015 |