
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The divergent effects of strong NHC donation in catalysis†
Justin A. M.
Lummiss
a,
Carolyn S.
Higman
a,
Devon L.
Fyson
a,
Robert
McDonald
b and
Deryn E.
Fogg
 *a
*a
aCenter for Catalysis Research & Innovation and Department of Chemistry, University of Ottawa, Ottawa, K1N 6N5, Canada. E-mail: dfogg@uottawa.ca
bDepartment of Chemistry, University of Alberta, Edmonton, T6G 2G2, AB, Canada
First published on 6th October 2015
Abstract
Strong σ-donation from NHC ligands (NHC = N-heterocyclic carbene) is shown to have profoundly conflicting consequences for the reactivity of transition-metal catalysts. Such donation is regarded as central to high catalyst activity in many contexts, of which the second-generation Grubbs metathesis catalysts (RuCl2(NHC)(PCy3)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh), GII) offer an early, prominent example. Less widely recognized is the dramatically inhibiting impact of NHC ligation on initiation of GII, and on re-entry into the catalytic cycle from the resting-state methylidene species RuCl2(NHC)(PCy3)(CH2), GIIm. Both GII and the methylidene complexes are activated by dissociation of PCy3. The impact of NHC donicity on the rate of PCy3 loss is explored in a comparison of s-GIIm, vs.u-GIIm, in which the NHC ligand is saturated H2IMes or unsaturated IMes, respectively. PCy3 loss is nearly an order of magnitude slower for the IMes derivative (a difference that is replicated, albeit smaller, for the benzylidene precatalysts GII). Proposed as an overlooked contributor to these rate differences is an increase in the Ru–PCy3 bond strength arising from π-back-donation onto the phosphine ligand. Strong σ-donation from the IMes ligand, coupled with the inability of this unsaturated NHC to participate in significant π-backbonding, amplifies Ru → PCy3 π-back-donation. The resulting increase in Ru–P bond strength greatly inhibits entry into the active cycle. For s-GII, in contrast, the greater π-acceptor capacity of the NHC ligand enables competing Ru → H2IMes back-donation (as confirmed by NOE experiments, which reveal restricted rotation about the Ru–NHC bond for H2IMes, but not IMes). Ru → PCy3 back-donation is thus attenuated in the H2IMes complexes, accounting for the greater lability of the PCy3 ligand in s-GIIm and s-GII. Similarly inhibited initiation is predicted for other metal–NHC catalysts in which a π-acceptor ligand L must be dissociated to permit substrate binding. Conversely, enhanced reactivity can be expected where such L ligands are pure σ-donors. These effects are expected to be particularly dramatic where the NHC ligand has minimal π-acceptor capacity (as in the unsaturated Arduengo carbenes), and in geometries that maximize NHC–M–L orbital interactions.
CHPh), GII) offer an early, prominent example. Less widely recognized is the dramatically inhibiting impact of NHC ligation on initiation of GII, and on re-entry into the catalytic cycle from the resting-state methylidene species RuCl2(NHC)(PCy3)(CH2), GIIm. Both GII and the methylidene complexes are activated by dissociation of PCy3. The impact of NHC donicity on the rate of PCy3 loss is explored in a comparison of s-GIIm, vs.u-GIIm, in which the NHC ligand is saturated H2IMes or unsaturated IMes, respectively. PCy3 loss is nearly an order of magnitude slower for the IMes derivative (a difference that is replicated, albeit smaller, for the benzylidene precatalysts GII). Proposed as an overlooked contributor to these rate differences is an increase in the Ru–PCy3 bond strength arising from π-back-donation onto the phosphine ligand. Strong σ-donation from the IMes ligand, coupled with the inability of this unsaturated NHC to participate in significant π-backbonding, amplifies Ru → PCy3 π-back-donation. The resulting increase in Ru–P bond strength greatly inhibits entry into the active cycle. For s-GII, in contrast, the greater π-acceptor capacity of the NHC ligand enables competing Ru → H2IMes back-donation (as confirmed by NOE experiments, which reveal restricted rotation about the Ru–NHC bond for H2IMes, but not IMes). Ru → PCy3 back-donation is thus attenuated in the H2IMes complexes, accounting for the greater lability of the PCy3 ligand in s-GIIm and s-GII. Similarly inhibited initiation is predicted for other metal–NHC catalysts in which a π-acceptor ligand L must be dissociated to permit substrate binding. Conversely, enhanced reactivity can be expected where such L ligands are pure σ-donors. These effects are expected to be particularly dramatic where the NHC ligand has minimal π-acceptor capacity (as in the unsaturated Arduengo carbenes), and in geometries that maximize NHC–M–L orbital interactions.
Introduction
The remarkable impact of N-heterocyclic carbene (NHC) ligands on transition-metal catalysis1–4 is due largely to their strong σ-donor character, a feature highlighted in even the earliest reviews.5–7 Strong NHC binding is believed to inhibit decomposition of molecular catalysts,1,8 and to stabilize the higher oxidation states essential in multiple catalytic contexts, including olefin metathesis and cross-coupling reactions.1–3 As well, however, emerging work points toward the potential for NHC donation to influence bonding interactions with other ligands present, both ancillary ligands and bound substrate.9–11In a leading recent example, the Neidig group reported evidence for ground-state weakening of the Fe–Cl bond by σ-donation from the NHC ligand in tetrahedral FeX2(NHC)2 complexes.9 The implied potential labilization of π-donor ligands by NHC ligands is of keen interest. The potentially broad implications of such behaviour in catalysis prompted us to explore the impact of NHC donicity on neutral, dative donor ligands, particularly in geometries that reinforce inter-ligand electronic communication. Here we demonstrate the impact of the NHC ligand on trans-ligand binding, in an important example drawn from olefin metathesis.
The breakthrough activity of the second-generation Grubbs catalysts,12,13 which greatly expanded the scope of the reaction relative to the parent system GI (Fig. 1), was originally attributed to labilization of the σ-donor PCy3 ligand by the strongly donating trans-NHC ligand.14 In a seminal kinetics study, however, Grubbs and co-workers demonstrated that PCy3 loss is in fact slower for GII than the first-generation catalyst GI.14
 | ||
| Fig. 1 The first and second-generation Grubbs catalysts, GI and GII. | ||
A leading explanation for this “inverse trans effect” highlights alkylidene rotation as a trigger for PCy3 dissociation, pointing out higher torsional barriers to such rotation in the NHC complexes.15 An alternative view emerges from Kennepohl's discovery, based on groundbreaking X-ray absorbance spectroscopy (XAS) studies, that the Ru center in s-GII is more electropositive than that in GI.16 This implies that the NHC ligand is a poor net charge donor, relative to PCy3. An increased electrostatic attraction between the more electron-deficient Ru center in GII and the strongly-donating PCy3 ligand was proposed to account for the reduced phosphine lability.
Adopting the majority view of NHC ligands as strong σ-donors, we speculated that NHC donation might itself be a factor: that strong σ-donation could in fact strengthen the trans Ru–PCy3 bond, by increasing Ru → PCy3 backbonding. In exploring this possibility, we focused on the methylidene species GIIm (Fig. 2), to eliminate steric or π-stacking effects associated with the benzylidene moiety, and electronic perturbation arising from benzylidene π-acidity. GIIm is, moreover, a key player in catalysis, as the resting-state species in most ring-closing and cross-metathesis reactions promoted by GII. That is, because GIIm is thermodynamically stable relative to both the benzylidene precatalyst GII, and other ruthenium species present in the catalytic cycle, its concentration builds up during metathesis. Recently-developed17 routes to the second-generation methylidene complexes enable their direct study.
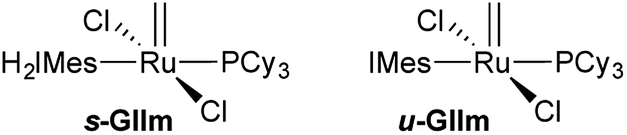 | ||
| Fig. 2 The off-cycle resting states for GII: methylidene complexes s-GIIm and u-GIIm. | ||
The availability of the closely related complexes u-GIIm and s-GIIm permits the effect of NHC donicity on trans-PCy3 bonding to be assessed with minimal extraneous perturbation.18,19 The π-acceptor capacity of saturated NHCs such as H2IMes, first proposed more than a decade ago, has seen much discussion.10,11,16,19–29 In recent years, the focus has shifted to means of deconvoluting NHC σ-donor and π-acceptor properties.23–26 While unsaturated Arduengo NHCs are generally viewed as poor π-acceptors, accumulating evidence suggests that their saturated analogues can exhibit significant π-acidity.10,11,16,19–28 If σ-donation from the H2IMes ligand in s-GIIm is countered by Ru–NHC backbonding, we considered that this should result in experimentally observable distinctions between the H2IMes and IMes complexes, which could potentially be correlated with differences in PCy3 lability.
Here we quantify the differences in PCy3 lability in GIIm; we demonstrate that strong σ-donation from the H2IMes ligand is indeed tempered by π-backbonding onto the NHC, as evidenced by restricted rotation about the Ru–H2IMes bond, and that PCy3 loss is dramatically slower for the IMes system, in which NHC σ-donation is unrelieved by NHC π-acidity (as confirmed by room-temperature rotation about the Ru–IMes bond). Based on these observations, we propose that enhanced backbonding onto the PCy3 ligand is a key, overlooked contributor to the low phosphine lability characteristic of the second-generation Grubbs catalysts. Such Ru → PCy3 backbonding relieves the heightened electron density at Ru that would otherwise result from strong NHC σ-donation, and consequently strengthens the Ru–P bond. The broader implications for catalysis are discussed.
Results and discussion
Assaying PCy3 lability for GIIm
Direct assessment of PCy3 lability for the second-generation methylidene complexes is hampered by a combination of strong phosphine binding and thermal instability. Even for the more labile benzylidene pre-catalysts, PCy3 loss from the IMes derivative u-GII was 640 times slower than from the first-generation complex GI.14 Qualitative evidence indicated drastically lower lability for the methylidene complexes GIIm, but attempts to measure rate constants were thwarted by decomposition at the temperatures required to induce PCy3 exchange (ca. 85 °C).14This underscores the point that the thermodynamic stability of GIIm relative to other catalytically relevant species does not equate to stability against decomposition. Indeed, the methylidene complexes are significantly more vulnerable than their benzylidene precursors, owing to their susceptibility to nucleophilic attack at the RuCH2 site.30–32
We considered that this vulnerability, which constituted a problem in the original exchange experiments, could offer a disguised opportunity to assess phosphine lability. Specifically, if decomposition of GIIm proceeds via rate-limiting loss of PCy3,30 then the rate of decomposition reports on the rate of PCy3 loss. To confirm that this reaction proceeds only via four-coordinate Ru-1, we examined the impact of added PCy3 on the reaction kinetics. If phosphine attack occurs on Ru-1 (Scheme 1a), the rate of decomposition should be unaffected, for the reasons discussed below. If, however, GIIm can react directly with PCy3 (Scheme 1b), decomposition should be accelerated.
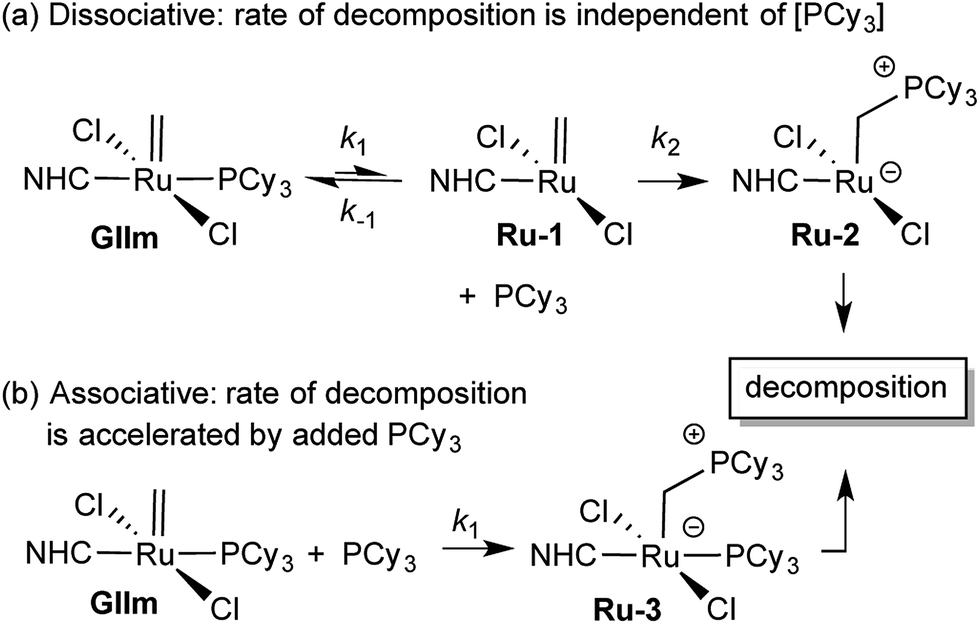 | ||
| Scheme 1 Predicted [PCy3] dependence for decomposition of GIImvia associative and dissociative pathways.33 For rate law derivations, see the ESI.† | ||
As seen from Fig. 3, the rate of decomposition is unaffected by added PCy3, indicating reaction via the dissociative pathway (Scheme 1a). The preference is unsurprising, given steric restrictions on the approach of PCy3 to the methylidene carbon in five-coordinate GIIm. The absence of an inverse dependence on [PCy3] may at first seem inconsistent with rate-determining loss of PCy3. This reflects the participation of PCy3 in the k2 step (i.e. the Ru-1 → Ru-2 transformation), as well as the k−1 step (the Ru-1 → GIIm back-reaction). If nucleophilic attack on Ru-1 is much faster than phosphine re-binding (i.e. k2 ≫ k−1), the rate expression reduces to k1 [GIIm] (see ESI†).
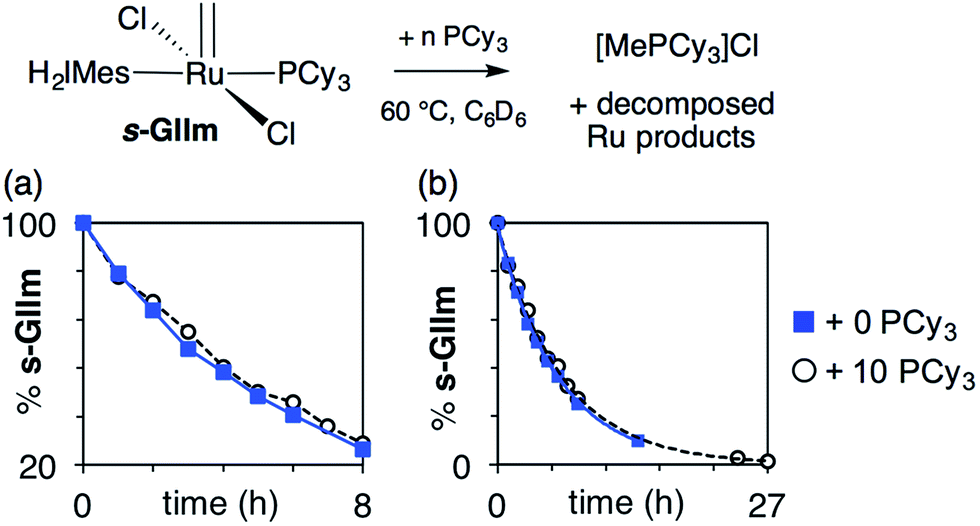 | ||
| Fig. 3 Assessing the rate of decomposition of s-GIIm in the presence and absence of added PCy3. (a) Over the first 8 h. (b) Over the full period of decomposition. | ||
(For completeness, it may be noted that even if k2 and k−1 were of comparable magnitude – or indeed if k2 ≪ k−1 – no phosphine inhibition would result. Because the rate of the k−1 step is k−1 [Ru-1][PCy3], and that of the k2 step is k2[Ru-1][PCy3], any change in [PCy3] alters both rates equivalently, and the phosphine concentration term cancels out. Thus the rate of reaction is independent of [PCy3], irrespective of the relative magnitudes of k2 and k−1).
To assess the rates of PCy3 loss from s-GIIm and u-GIIm, in the present case, where k2 ≫ k−1, we measured the rates of decomposition of these complexes in C6D6. Decreases in the proportion of GIIm over time were established by 1H NMR analysis. The integrated intensity of the methylidene singlet was measured relative to 1,3,5-trimethoxybenzene (TMB; δ CH 6.26 ppm) as internal standard. Decomposition was nearly eight times faster for s-GIIm than u-GIIm, as shown by the rate curves in Fig. 4. The relative rates show little change from 40–80 °C: in each case, loss of PCy3 from the IMes derivative was 7–8 times slower. DFT studies by the Jensen group reported an identical trend for the parent benzylidene catalysts, with k1 for u-GII being seven-fold lower than for s-GII.34
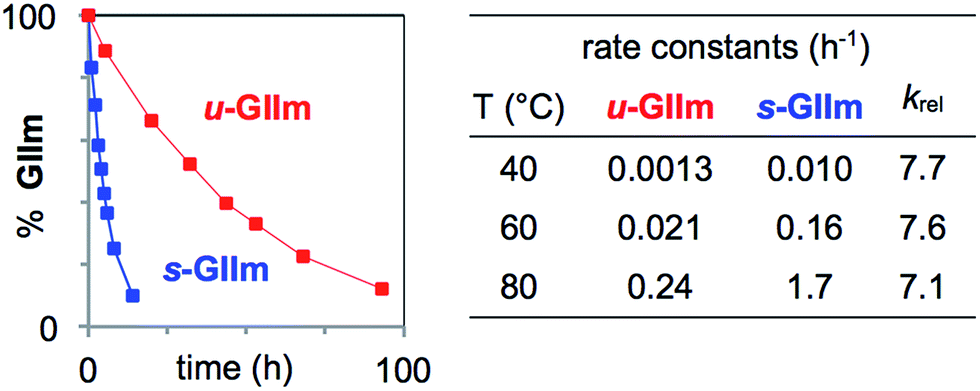 | ||
| Fig. 4 Assessing rates of PCy3 loss from the decomposition of s-GIIm and u-GIIm in C6D6. Left: Rate curves at 60 °C. Right: Initial rate constants and krel (normalized to u-GIIm) at 40 °C, 60 °C, and 80 °C. For half-lives and rate plots at other temperatures, see the ESI.† | ||
The lower phosphine lability of u-GIIm relative to s-GIIm was maintained in other solvents (Fig. 5). In these experiments, the proportion of GIIm remaining after 6 h at 60 °C was measured. Decomposition was marginally faster in chlorinated media than in aromatic solvents, and dramatically faster in the coordinating solvent THF. The solvent-dependence of PCy3 dissociation thus follows the trend C7H8 ∼ C6H6 < CH2Cl2 ∼ CHCl3 ≪ THF, for both the IMes and H2IMes methylidene complexes. This agrees with the trend previously established for initiation of the benzylidene precatalyst s-GII, for which the rate-determining step is likewise PCy3 loss.14
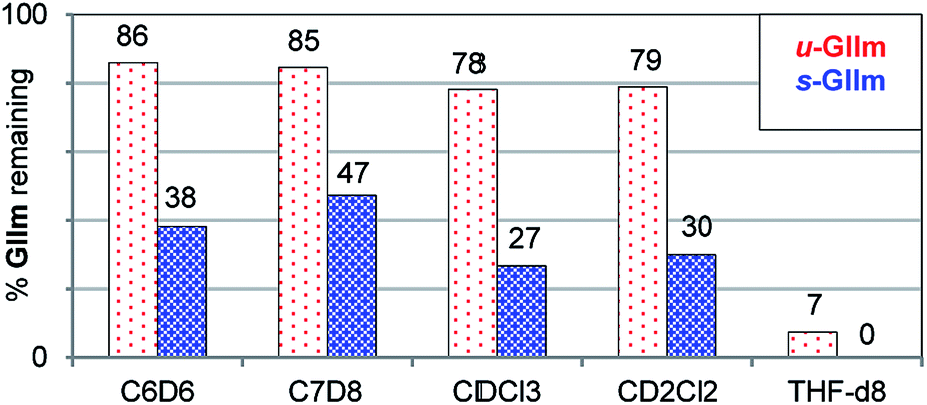 | ||
| Fig. 5 Assessing the relative stability of u-GIIm and s-GIIm in common solvents, as a proxy for PCy3 lability (6 h, 60 °C oil-bath; 1H NMR integration vs. TMB). Key chemical shift data for GII and GIIm in these solvents are tabulated in the ESI.† | ||
The consistency in these reactivity patterns, as well as the excellent agreement with the relative rate constants computed by Jensen (see above), validate the use of decomposition rates to quantify rates of PCy3 loss from GIIm. Also noteworthy is the close correlation between relative rates of initiation of GII in different solvents, and relative rates of decomposition of GIIm. This correlation accounts for the observation that increasing the rate of initiation does not improve reaction rates for the Grubbs catalysts.35 Instead, because productive metathesis generates an unprotected methylidene moiety, faster initiation is offset by faster methylidene abstraction by free PCy3.
Crystallographic analysis: comparison of u-GIIm with s-GIIm
In the hope of gaining insight into the bonding interactions that distinguish the IMes and H2IMes analogues, we undertook a crystallographic study of u-GIIm, for comparison with the reported structure of s-GIIm.36 The instability of these complexes in solution can be minimized by low-temperature handling, and X-ray quality crystals of u-GIIm deposited from concentrated solutions in toluene over days at −35 °C. The ORTEP plot is shown in Fig. 6; key bond lengths and angles are compared with those for s-GIIm in Table 1.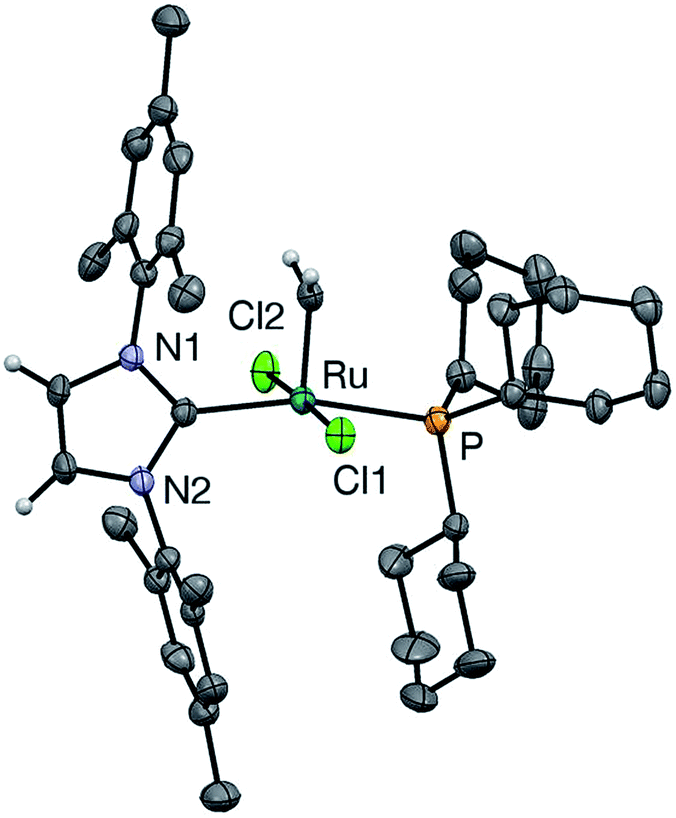 | ||
| Fig. 6 Perspective view of u-GIIm. Non-hydrogen atoms are represented by Gaussian ellipsoids at the 30% probability level. Hydrogen atoms on methylidene and NHC backbone carbons are shown with arbitrarily small thermal parameters; other hydrogens are not shown. | ||
| u-GIIm | s-GIIm 36 | |
|---|---|---|
| τ-parameter | 0.19 | 0.19 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Bond lengths (Å) | ||
| Ru–P | 2.4174(16) | 2.427(1) |
| RuC |
1.797(7) | 1.800(2) |
| Ru–CNHC | 2.077(5) | 2.065(2) |
| Ru–Cl(1) | 2.389(2) | 2.393(1) |
| Ru–Cl(2) | 2.381(2) | 2.379(1) |
|
||
| Bond angles (°) | ||
| Cl–Ru–Cl | 176.99(6) | 177.05(2) |
| P–Ru–CNHC | 165.63(16) | 165.81(5) |
| P–RuC |
97.2(2) | 96.90(7) |
| Cl(1)–RuC |
93.1(2) | 92.89(7) |
| Cl(2)-RuC |
89.9(2) | 89.77(7) |
| CNHC–RuC |
97.2(3) | 97.29(8) |
The geometry at Ru is square pyramidal in both cases, as indicated by the τ values of 0.19 (cf. τ = 0 for a perfect square pyramid, and τ = 1 for a perfect trigonal bipyramid).37 While the P–Ru–CNHC angle shows some distortion from the 180° ideal (ca. 166° in both u-GIIm and s-GIIm), excellent orbital communication is expected between the trans-disposed phosphine and NHC ligands. Importantly, however, the Ru–P bond distances in s-GIIm and u-GIIm are statistically indistinguishable, despite the nearly tenfold difference in phosphine lability. The absence of a correlation between Ru–PCy3 bond length and bond strength was pointed out for the parent benzylidene complexes,14 but has gone widely unnoticed. Frenking has pointed out that metal–ligand bond lengths are not reliable indicators of bond strength, where the ligand can function as an acceptor as well as a donor.38 The π-acceptor properties of the phosphine ligand in the NHC complexes are discussed below.
Molecular dynamics study: Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CNHC rotation and bond order
CNHC rotation and bond order
More direct insight emerged from a molecular dynamics study, in which 2D NOESY-NMR was used to assess rotational exchange between the mesityl rings above and below the basal plane (Fig. 7, top). Exchange cross-peaks were observed for all four unique mesityl methyl signals in u-GIIm and u-GII, indicating rotation about the Ru–IMes bond at room temperature (Fig. 7a). No such cross-peaks were evident for s-GIIm and s-GII (Fig. 7b), even for the well-resolved p-Me singlets (the o-Me singlets are less well resolved, perhaps due to [Ru]CHPh swiveling). Slower rotation of the H2IMes ligand in both the methylidene complex s-GIIm and its benzylidene parent s-GII is important in indicating that restricted rotation is unrelated to the steric demand of the [Ru]CHR substituent.
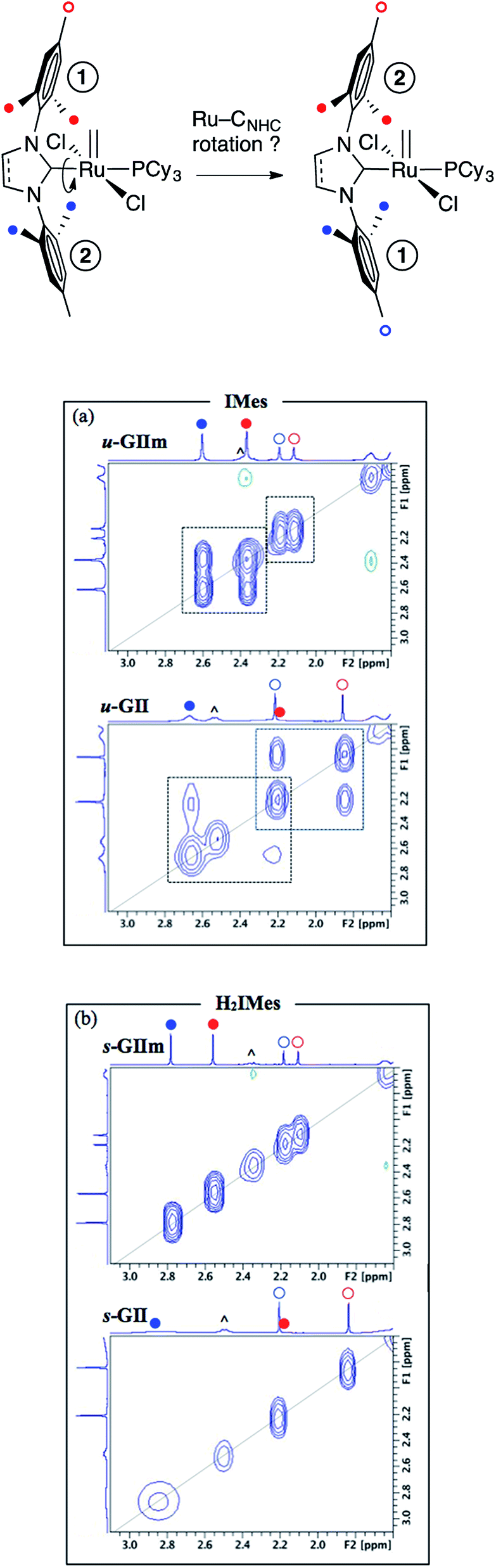 | ||
| Fig. 7 1H–1H NOESY spectra showing dependence of Ru–NHC rotation on NHC unsaturation. (a) Exchange correlations between mesityl methyl signals for the IMes derivatives. (b) Absence of correlations for the H2IMes analogues. (all in C6D6, 500.1 MHz, 25 °C, 1.5 s relaxation delay). Symbols: (^) = Cy; for others, see top. | ||
Restricted rotation about the Ru–H2IMes bond implies increased Ru–CNHC double-bond character, arising from π-back-donation from the metal onto the vacant p-orbital on the NHC carbon. Free rotation of the IMes ligand, in contrast, indicates a high proportion of single-bond character in the Ru–CNHC bond. This accords with the experimental and computational findings described above, showing stronger π-acceptor character for the H2IMes ligand than IMes. Bertrand and co-workers drew a similar conclusion in a comparative study of H2IPr–PPh and IPr–PPh adducts, also on the basis of a solution dynamics study (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene).23 Thus, the saturated H2IPr derivative was classified as a phosphaalkene species, and the unsaturated IPr adduct as a phosphinidene.
Origin of the inverse trans effect
As noted in the Introduction, the origin of the dramatically reduced phosphine lability in the second-generation Grubbs catalysts is a puzzle of long standing. Straub suggested that faster PCy3 loss from GI is due to repulsive interactions between the chloride ligands and the β-hydrogen atoms of the cyclohexyl rings.39 More recently, Yang, Truhlar and co-workers reported DFT evidence showing that alkylidene rotation functions as a toggle to trigger PCy3 dissociation, and that the torsional barriers to rotation are higher for s-GII.15Kennepohl's XAS study stands out, however, for the unexpected revelation that s-GII exhibits a higher 1s ionization potential for Ru – that is, a more electron-deficient metal center – than does the first-generation parent GI. We suggest that this is due to enhanced π-donation from Ru onto the NHC and PCy3 ligands. It should be noted that the Kennepohl study examined this possibility for s-GIIm. It was rejected, as calculations at the level of theory then available indicated limited Ru → PCy3 backbonding (in consequence of which, stronger PCy3 binding was attributed to an enhanced Ru/PCy3 electrostatic attraction). Importantly, however, consideration of dispersion forces has since emerged as critical to quantitative evaluation of the PCy3 dissociation step.40
The limited role heretofor assigned to Ru–PCy3 π-acceptor interactions in this system is perhaps unsurprising, given the widespread perception of alkylphosphines as strong σ-donors and weak π-acceptors (a situation also encountered in the context of NHC donicity; see above). Here too, however, a re-evaluation is in progress. In an analysis of electron density and structural effects, Leyssens, Harvey and co-workers demonstrated that π-backbonding from the metal atom onto the P–R σ*-antibonding orbitals can represent a significant component of metal–phosphine bonding, including for trialkylphosphine complexes.41 A recent leading review of computational approaches to the understanding of metal–phosphorus bonding likewise emphasizes that calculated ligand descriptors for phosphine ligands must consider their π-acceptor character.42
In light of these developments, we suggest that π-back-donation onto the phosphine is a significant, overlooked contribution to the low PCy3 lability in the second-generation Grubbs catalysts. The potent σ-donor properties of the NHC ligand constrain back-donation onto any π-acceptor ligands present. For precatalyst s-GII, three ligands can participate in π-backbonding: H2IMes, PCy3, and benzylidene.39 In the case of u-GIIm, the poor π-acceptor character of the IMes and methylidene ligands leaves the PCy3 ligand as the sole entity that can ameliorate the buildup of charge on the metal. We propose that this buildup is offset for u-GIIm by greater Ru → PCy3 back-donation (Fig. 8), and for s-GIIm, by greater Ru → H2IMes back-donation, accompanied by a lesser amount of Ru → PCy3 back-donation. This would account for the poor net charge donation from the saturated NHC ligand observed in the Kennepohl study. Also relevant in this context is an energy decomposition analysis by Poblet and co-workers, which suggested that the π-acceptor capacity of H2IMes reduces total charge donation to the metal for s-GIIm, relative to its IMes analogue.21
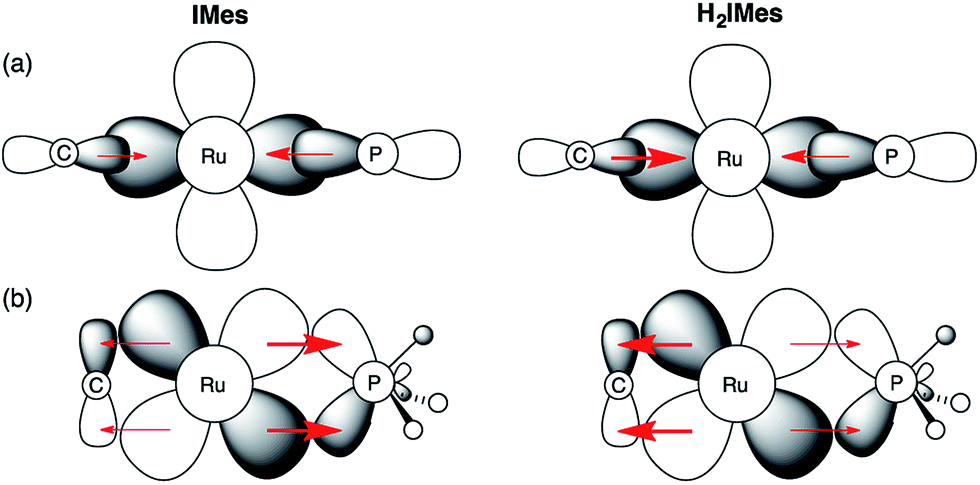 | ||
| Fig. 8 Impact of NHC π-acidity on PCy3 lability. (a) σ-Bonding interactions; (b) π–bonding interactions. Perspective down the RuCHR bond. | ||
Several consequences can be envisaged, which have a profound impact on catalytic behaviour. Most obviously, stronger Ru–P backbonding would account for the reduced lability of the PCy3 ligand in the IMes complexes, relative to their H2IMes analogues. Slower loss of PCy3 would in turn account for the 7–8-fold longer lifetime shown above for u-GIIm, relative to s-GIIm. Because phosphine dissociation is required for entry into the active catalytic cycle, however, the advantage of longer lifetime is offset by slower initiation for the precatalyst u-GII, and slower re-entry for the resting-state species u-GIIm. This proposal clarifies the greatly enhanced initiation efficiency of phosphine-free, Hoveyda-class metathesis catalysts,43 in which the π-accepting PCy3 ligand is replaced by a π-donating ether ligand, as well as the high latency of the Cazin catalysts, in which a much more strongly π-acidic phosphite ligand is present.44
In the Neidig study cited in the Introduction,9 the NHC ligands were shown to significantly reduce the binding strength of a chloride ligand in tetrahedral Fe–NHC complexes. The strengthening of the trans-PCy3 bond observed herein is a striking further manifestation of the impact of NHC donicity on M–L binding. Beyond the specific context of olefin metathesis, similar inhibition of uptake into catalysis may be expected whenever a π-acceptor ligand must be released in order to bind substrate, particularly where this ligand is trans to an NHC. Such effects are enhanced for systems in which the strong σ-donor character of the NHC ligand is undiminished by NHC π-acceptor capacity, as illustrated here for the IMes system.
Conclusions
Strong NHC donation is arguably the defining feature of the second-generation Grubbs catalysts, as the parameter that enables their high inherent reactivity. The foregoing reveals that such strong donation wears a Janus face. Enhancing the electron density at the metal center activates the Ru-olefin intermediate, and stabilizes the Ru(IV) metallacyclobutane intermediate. However, it also greatly amplifies Ru → PCy3 backbonding: Ru–P bond strengths are thereby increased, and loss of phosphine is severely inhibited. This inverse trans effect is manifested in retarded initiation of the benzylidene precatalysts GII, and very slow re-entry into the catalytic cycle from the resting-state methylidene complexes GIIm.Notwithstanding the central importance of the Grubbs catalysts and their descendents in olefin metathesis, the implications are considerably broader. The transformative impact of NHC ligands on homogeneous catalysis has long been assigned to their capacity to enhance the electron density at the metal. The influence of NHC donicity on the ancillary ligands, however, is now beginning to be examined more closely. The findings above contribute to emerging understanding of the profound impact of NHC donicity on M–L binding, and hence on catalytic behaviour. Specifically, inhibited initiation is predicted to be a general feature for M–NHC catalysts in which a π-acidic ancillary ligand occupies a latent substrate binding site, particularly where such ligands are trans to the NHC. The potential for activation of a π-accepting substrate located in this site is an obvious corollory. These findings complement recent work highlighting the labilizing effect of the NHC ligand on π-donor ligands in tetrahedral iron complexes. Differences in NHC π-acceptor capacity can thus either mitigate or reinforce trans-type M–L bonding interactions, with major consequences for catalyst conscription and activity.
Experimental
General procedures
Reactions were carried out under N2 using standard glovebox techniques, at ambient temperature (RT; 25–27 °C, unless otherwise noted). Dry, oxygen-free toluene was obtained using a Glass Contour solvent purification system. All NMR solvents (Cambridge Isotopes) were stored under N2 over Linde 4 Å molecular sieves for at least 6 h prior to use. Dimethyl terephthalate (DMT, >99%), 1,3,5-trimethoxybenzene (TMB, >99%), used as internal integration standards to support quantification in 1H NMR experiments, were obtained from Sigma-Aldrich. The methylidene complexes u-GIIm and s-GIIm were prepared by literature methods.17,45 X-ray quality crystals of u-GIIm were grown from toluene at −35 °C over 48 h.NMR spectra were recorded on Bruker Avance 300 and 500 spectrometers at 23 °C (unless otherwise noted), and referenced to the residual proton of the solvent. Signals are reported in ppm, relative to TMS (1H) or 85% H3PO4 (31P) at 0 ppm.
Representative procedure for measuring decomposition rates
In the glovebox, a J. Young NMR tube was charged with GIIm (10 mg, 0.013 mmol), TMB (ca. 0.5 mg), and C6D6 (660 μL). The sample was removed from the glovebox and a 1H NMR spectrum was measured to establish the initial ratio of s-GIIm to TMB. The NMR tube was then transferred to a 40 °C oil bath (thermocouple-equipped; ±1.5 °C). The rate was determined by collecting 1H NMR spectra at regular intervals. Rate profiles for u-GIIm and s-GIIm at 40 °C and 80 °C are given in the ESI.† To examine the [PCy3]-dependence of decomposition, a corresponding experiment was carried out with s-GIIm (9.2 mg, 0.0127 mmol), TMB (ca. 0.5 mg), and PCy3 (35.7 mg, 0.127 mmol, 10 equiv.) in C6D6 (635 μL) at 60 °C. Time-points were taken at regular intervals until decomposition was complete.Exploring the impact of solvent on decomposition of GIIm
These experiments were carried out as above at a bath temperature of 60 °C, with NMR analysis at a single time-point (6 h). Thermolysis experiments in CD2Cl2 (b.p. 40 °C) were carried out in thick-walled J. Young NMR tubes.Acknowledgements
Instructive comments on the kinetics analysis by Prof. Bob Bergman (Berkeley) are acknowledged with thanks. Magdalen College, Oxford, is thanked for a Visiting Fellowship to DEF. This work was funded by NSERC of Canada.Notes and references
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, ed. S. P. Nolan, Wiley, Weinheim, 2014 Search PubMed.
- N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Diez Gonzalez, Royal Society of Chemistry, Cambridge, 2011 Search PubMed.
- N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, New York, 2011 Search PubMed.
- A. J. Arduengo, Acc. Chem. Res., 1999, 32, 913–921 CrossRef CAS.
- D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef CAS PubMed.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS.
- C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS PubMed.
- K. L. Fillman, J. A. Przyojski, M. H. Al-Afyouni, Z. J. Tonzetich and M. L. Neidig, Chem. Sci., 2015, 6, 1178–1188 RSC.
- D. Marchione, L. Belpassi, G. Bistoni, A. Macchioni, F. Tarantelli and D. Zuccaccia, Organometallics, 2014, 33, 4200–4208 CrossRef CAS.
- A. Comas-Vives and J. N. Harvey, Eur. J. Inorg. Chem., 2011, 5025–5035 CrossRef CAS PubMed.
- M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 2247–2250 CrossRef CAS.
- J. Huang, E. D. Stevens, S. P. Nolan and J. L. Petersen, J. Am. Chem. Soc., 1999, 121, 2674–2678 CrossRef CAS.
- M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543–6554 CrossRef CAS PubMed.
- H.-C. Yang, Y.-C. Huang, Y.-K. Lan, T.-Y. Luh, Y. Zhao and D. G. Truhlar, Organometallics, 2011, 30, 4196–4200 CrossRef CAS.
- K. Getty, M. U. Delgado-Jaime and P. Kennepohl, J. Am. Chem. Soc., 2007, 129, 15774–15776 CrossRef CAS PubMed.
- J. A. M. Lummiss, N. J. Beach, J. C. Smith and D. E. Fogg, Catal. Sci. Technol., 2012, 2, 1630–1632 CAS.
- The change in the NHC ligand present in s-GIIm and u-GIIm results in limited steric perturbation, as indicated by the comparable % buried volume for the IMes and H2IMes ligands (26% vs. 27%, respectively). See: ref. 19.
- S. Diez-Gonzalez and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef CAS PubMed.
- H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef CAS PubMed.
- N. S. Antonova, J. J. Carbo and J. M. Poblet, Organometallics, 2009, 28, 4283–4287 CrossRef CAS . As DFT calculations were carried out on a model RuCl2(NHC)(PH3)(CH2) system, the computed Ru–NHC back-donation should be regarded as a lower limit.
- Y. Minenkov, G. Occhipinti, W. Heyndrickx and V. R. Jensen, Eur. J. Inorg. Chem., 2012, 1507–1516 CrossRef CAS PubMed.
- O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 2939–2943 CrossRef CAS PubMed.
- A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef CAS.
- G. Ciancaleoni, N. Scafuri, G. Bistoni, A. Macchioni, F. Tarantelli, D. Zuccaccia and L. Belpassi, Inorg. Chem., 2014, 53, 9907–9916 CrossRef CAS PubMed.
- S. V. C. Vummaleti, D. J. Nelson, A. Poater, A. Gomez-Suarez, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and L. Cavallo, Chem. Sci., 2015, 8, 1895–1904 RSC.
- For early computational or integrated experimental/computational studies showing evidence of backbonding onto NHC ligands, see: (a) M. Tafipolsky, W. Scherer, K. Öfele, G. Artus, B. Pedersen, W. A. Herrmann and G. S. McGrady, J. Am. Chem. Soc., 2002, 124, 5865–5880 CrossRef CAS PubMed; (b) D. Nemcsok, K. Wichmann and G. Frenking, Organometallics, 2004, 23, 3640–3646 CrossRef CAS; (c) X. Hu, I. Castro-Rodriguez, K. Olsen and K. Meyer, Organometallics, 2004, 23, 755–764 CrossRef CAS; (d) G. Occhipinti, H.-R. Bjorsvik and V. R. Jensen, J. Am. Chem. Soc., 2006, 128, 6952–6964 CrossRef CAS PubMed; (e) H. Jacobsen, A. Correa, C. Costabile and L. Cavallo, J. Organomet. Chem., 2006, 691, 4350–4358 CrossRef CAS PubMed; (f) E. F. Penka, C. W. Schlapfer, M. Atanasov, M. Albrecht and C. Daul, J. Organomet. Chem., 2007, 692, 5709–5716 CrossRef CAS PubMed.
- For early experimental evidence of the π-acceptor ability of NHC ligands, see: (a) A. A. D. Tulloch, A. A. Danopoulos, S. Kleinhenz, M. E. Light, M. B. Hursthouse and G. Eastham, Organometallics, 2001, 20, 2027–2031 CrossRef CAS; (b) L. Mercs, G. Labat, A. Neels, A. Ehlers and M. Albrecht, Organometallics, 2006, 25, 5648–5656 CrossRef CAS; (c) M. D. Sanderson, J. W. Kamplain and C. W. Bielawski, J. Am. Chem. Soc., 2006, 128, 16514–16515 CrossRef CAS PubMed; (d) S. Fantasia, J. L. Petersen, H. Jacobsen, L. Cavallo and S. P. Nolan, Organometallics, 2007, 26, 5880–5889 CrossRef CAS; (e) D. M. Khramov, V. M. Lynch and C. W. Bielawski, Organometallics, 2007, 26, 6042–6049 CrossRef CAS.
- M. Alcarazo, T. Stork, A. Anoop, W. Thiel and A. Fürstner, Angew. Chem., Int. Ed., 2010, 49, 2542–2546 CrossRef CAS PubMed.
- S. H. Hong, A. G. Wenzel, T. T. Salguero, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2007, 129, 7961–7968 CrossRef CAS PubMed.
- J. A. M. Lummiss, B. J. Ireland, J. M. Sommers and D. E. Fogg, ChemCatChem, 2014, 6, 459–463 CrossRef CAS PubMed.
- J. A. M. Lummiss, W. L. McClennan, R. McDonald and D. E. Fogg, Organometallics, 2014, 33, 6738–6741 CrossRef CAS.
- For derivation of the rate laws for the dissociative and associative pathways, see the ESI.† For a succinct derivation of the rate law for the dissociative pathway, see ref. 30.
- Y. Minenkov, G. Occhipinti and V. R. Jensen, Organometallics, 2013, 32, 2099–2111 CrossRef CAS.
- B. J. van Lierop, J. A. M. Lummiss and D. E. Fogg, in Olefin Metathesis-Theory and Practice, ed. K. Grela, Wiley, Hoboken, NJ, 2014, ch. 3, pp. 85–152 Search PubMed.
- T. M. Trnka, M. W. Day and R. H. Grubbs, Angew. Chem., Int. Ed., 2001, 40, 3441–3444 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- G. Frenking, K. Wichmann, N. Froehlich, J. Grobe, W. Golla, D. L. Van, B. Krebs and M. Laege, Organometallics, 2002, 21, 2921–2930 CrossRef CAS.
- B. F. Straub, Angew. Chem., Int. Ed., 2005, 44, 5974–5978 CrossRef CAS PubMed.
- See: ref. 22, 34 and background discussion in: Y. Zhao and D. G. Truhlar, Org. Lett., 2007, 9, 1967–1970 CrossRef CAS PubMed.
- T. Leyssens, D. Peeters, A. G. Orpen and J. N. Harvey, New J. Chem., 2005, 29, 1424–1430 RSC.
- N. Fey, A. G. Orpen and J. N. Harvey, Coord. Chem. Rev., 2009, 253, 704–722 CrossRef CAS PubMed.
- J. M. Bates, J. A. M. Lummiss, G. A. Bailey and D. E. Fogg, ACS Catal., 2014, 4, 2387–2394 CrossRef CAS.
- T. E. Schmid, X. Bantreil, C. A. Citadelle, A. M. Z. Slawin and C. S. J. Cazin, Chem. Commun., 2011, 47, 7060–7062 RSC.
- For further improvements in synthesis of GIIm, see: J. A. M. Lummiss, A. G. G. Botti and D. E. Fogg, Catal. Sci. Technol., 2014, 4, 4210–4218 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Rate profiles for decomposition of u-GIIm and s-GIIm; X-ray crystallographic details; NOESY spectra, and derivation of the [PCy3]-independence of decomposition. CCDC 1400077. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02592c |
| This journal is © The Royal Society of Chemistry 2015 |