
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromiscuous indolyl vinyl isonitrile synthases in the biogenesis and diversification of hapalindole-type alkaloids†
Kuljira
Ittiamornkul
a,
Qin
Zhu
a,
Danai S.
Gkotsi
b,
Duncan R. M.
Smith
b,
Matthew L.
Hillwig
a,
Nicole
Nightingale
a,
Rebecca J. M.
Goss
b and
Xinyu
Liu
*a
aDepartment of Chemistry, University of Pittsburgh, 219 Parkman Avenue, Pittsburgh, PA, USA 15260. E-mail: xinyuliu@pitt.edu; Tel: +1-412-624-6932
bSchool of Chemistry and BSRC, University of St. Andrews, St. Andrews, KY16 9ST, U.K
First published on 6th October 2015
Abstract
The hapalindole-type alkaloids naturally show striking late stage diversification of what was believed to be a conserved intermediate, cis-indolyl vinyl isonitrile (1a). Here we demonstrate enzymatically, as well as through applying a synthetic biology approach, that the pathway generating 1a (itself, a potent natural broad-spectrum antibiotic) is also dramatically flexible. We harness this to enable early stage diversification of the natural product and generation of a wide range of halo-analogues of 1a. This approach allows the preparatively useful generation of a series of antibiotics with increased lipophilicity over that of the parent antibiotic.
Introduction
Hapalindole-type natural products (Fig. 1a) are a large family of indole monoterpene alkaloids produced by fresh water and terrestrial cyanobacteria in the order of Stigonematales.1 They exhibit potent broad-spectrum cytotoxicities against multidrug resistant bacteria, fungi and human tumour cells.1 Recent studies in deciphering the genetic and molecular basis for the biogenesis of hapalindole-type alkaloids revealed cis-indolyl vinyl isonitrile 1a, itself, a natural broad-spectrum antibiotic,2 and geranyl pyrophosphate (GPP) are the common biosynthetic precursors to ambiguines, welwitindolinones and related hapalindoles and fischerindoles (Fig. 1b).3,4 Comparative analysis of the ambiguine and welwitindolinone biosynthetic gene clusters further suggests that the structural diversity of hapalindole-type alkaloids is likely to arise from a conserved indole monoterpene intermediate by the enzymatic unification of 1a and GPP, followed by sequential late stage oxidative diversifications by nonheme iron oxygenases (Fig. 1b).3,4 Recently, successful reconstitution of the first oxidative maturation step in welwitindolinone biogenesis provided direct evidence supporting this biosynthetic proposal, as WelO5, an α-ketoglutarate (α-KG)-dependent nonheme iron halogenase,5 was shown to selectively transform free 12-epi-fischerindole U and 12-epi-hapalindole C to 12-epi-fischerindole G and 12-epi-hapalindole E respectively.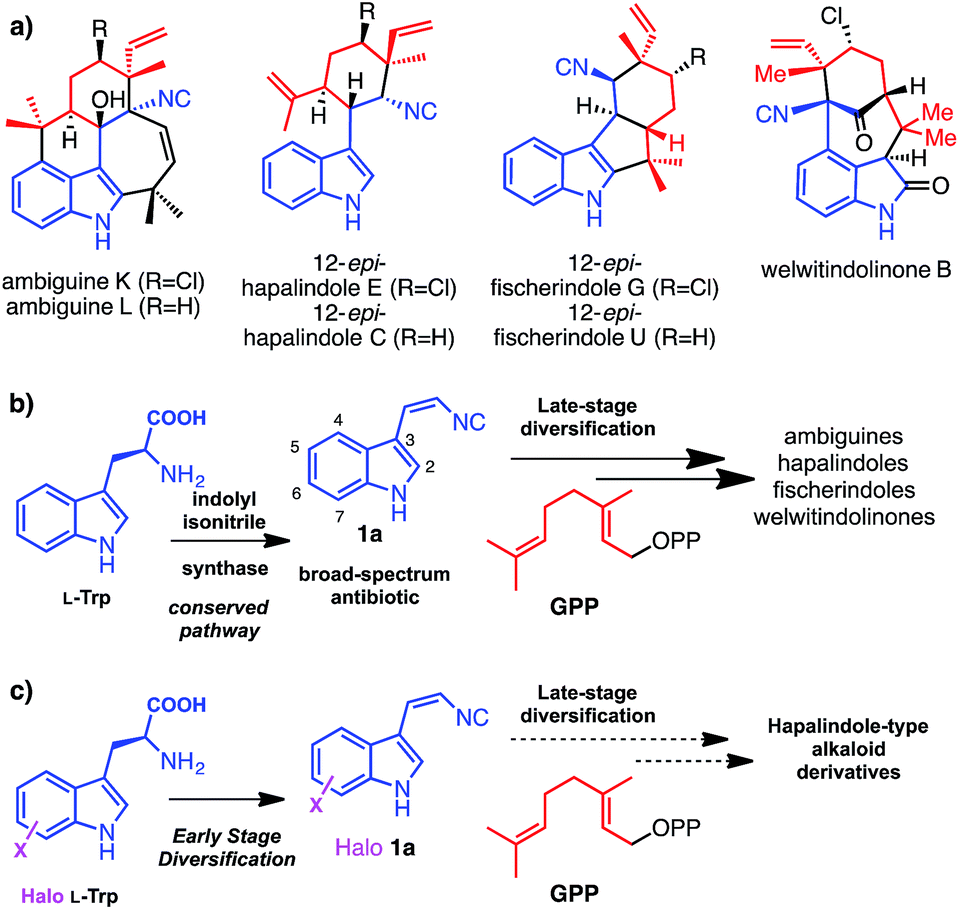 | ||
| Fig. 1 (a) Structures of selected hapalindole-type indole monoterpene alkaloids, (b) their biogenesis featuring convergent early stage pathways with shared precursors 1a and GPP and divergent late stage modifications, (c) the goal of this work to investigate the plasticity of early stage isonitrile biosynthetic enzymes. | ||
Biosynthetic natural product diversification is believed to confer evolutionary advantage, by being able to generate suites of compounds the producing organism is provided with the required degree of flexibility to cope with a diverse and changing environment. Many natural products including the hapalindole-type alkaloids, the pacidamycin and antimycin antibiotics,6,7 are observed to be produced as a family of related compounds. This diversification is usually, though not always, related to advantageous enzymatic flexibility and the ability of the organism to generate or acquire a suitable series of precursors. Having already observed striking late stage diversification of the hapalindole alkaloids, and with the expanding knowledge in the biogenesis of hapalindole-type alkaloids, we set out to explore whether early stage diversification of this important class of compounds could be effected. We envisaged that if we could leverage the promiscuity of the biosynthetic machinery in vitro or in vivo we would be able to incorporate unnatural functionalities into these cytotoxic natural products through precursor supply (Fig. 1c). Site-specific replacement of a C–H bond with a C–X (X = halogen) bond in a molecule of interest (MOI) is a common strategy to improve its bioactivity and bioavailability profiles.8 In particular, substitution of sp2-hybridized C–H group to C–X (X = Cl, Br, I) permits late-stage chemoselective modification by cross-coupling chemistry that represents a versatile strategy to expand the structural diversity of the parental MOI.9,10 In all hapalindole-type alkaloids, the indole C5–C7 carbon centres lack modifications and constitute ideal sites for introducing halogen functional groups.
We have previously demonstrated that the indole motifs in hapalindole-type molecules are derived from 1a, by the actions of the indolyl vinyl isonitrile synthases (AmbI1-3 and WelI1-3) using L-tryptophan (L-Trp) (2a) and ribulose 5-phosphate (Rub5P) as substrates (Fig. 2, scheme).4,5 Although the protein homologs of AmbI1-3, IsnA and IsnB for the biogenesis of trans-1a, were identified a decade ago,11,12 neither their enzymatic mechanism nor substrate promiscuity have been studied. An understanding of the enzymatic plasticity of indolyl vinyl isonitrile synthases in hapalindole biogenesis towards unnatural substrates is required before the feasibility of introducing a halogen functionality to the indole C5–C7 in hapalindole-type molecules, by either a precursor-directed biosynthetic or metabolic engineering approach, can be examined. Here, we report the first systematic study on the substrate promiscuity of AmbI1-3 and demonstrate the dramatic flexibility of the system; these enzymes readily accept L-Trp substituted with F, Cl or Br at positions 4, 5, 6 or 7 as substrates to generate halogenated 1 in practically useful yields both in vitro and in vivo. Strikingly, even the highly sterically demanding 7-iodo L-Trp is processed by this series of biosynthetic enzymes. The observation that F-, Cl-, Br-, I-substituted indolyl vinyl isonitriles show sequentially increased lipophilicity and 7-F/7-Cl substituted analogues retain the parental antibacterial activity, indicates that this type of antibiotic modification is likely to hold promise for enhanced bioavailability and therefore potency in vivo in animals and humans. Furthermore the inclusion of a Cl, Br and I provides a chemically orthogonal handle that could be used to enable further selective modification,9,10 and tuning of this promising family of compounds.
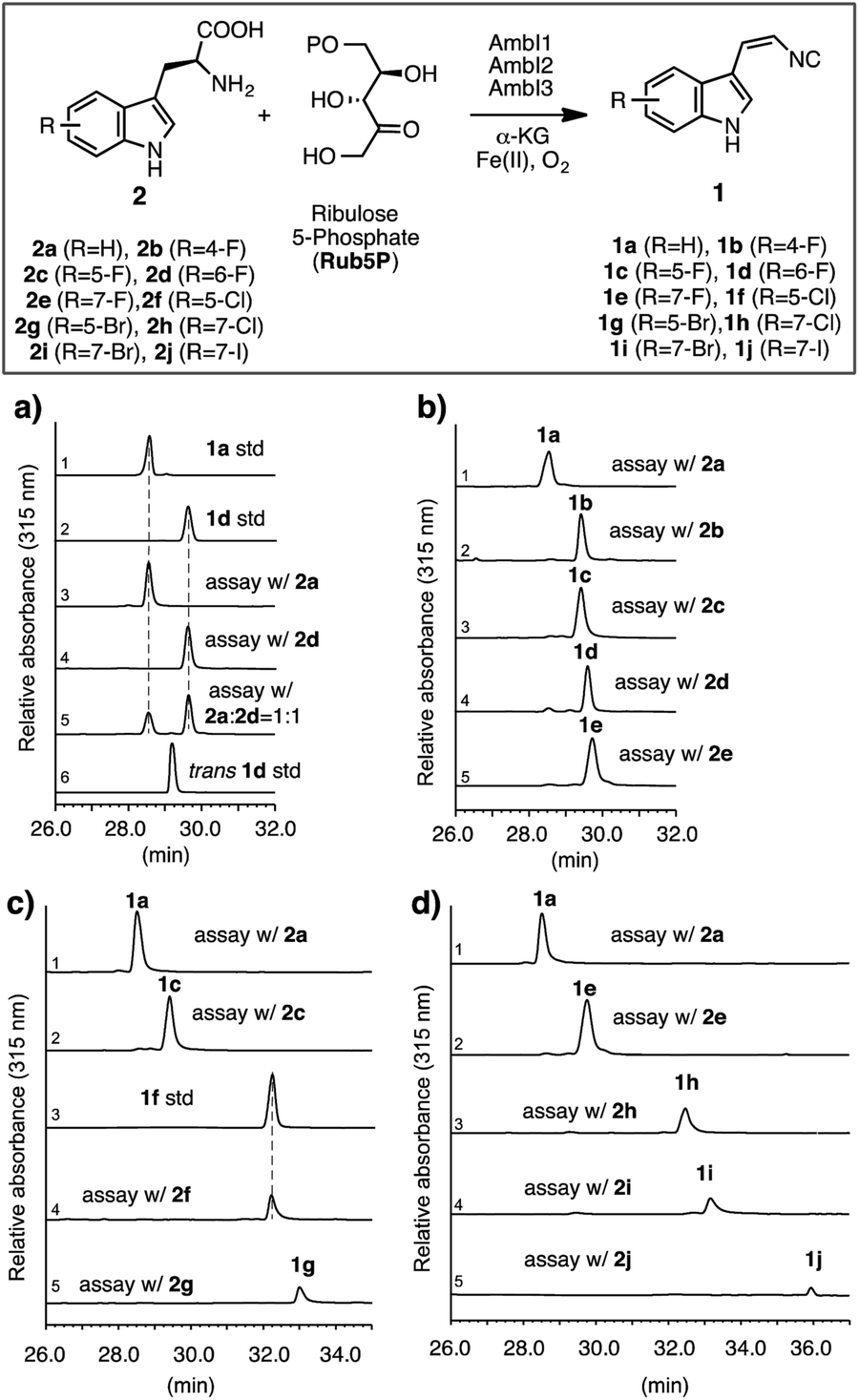 | ||
| Fig. 2 Investigation of the AmbI1-3 isonitrile synthase promiscuity in vitro, using differentially substituted halo tryptophans. Standard assay conditions: all assays were carried out in a 1 mL scale at pH 7.4 with 2 (2.5 mM), Rub5P (2.5 mM), α-KG (2.0 mM), (NH4)2Fe(SO4)2 (0.5 mM) and AmbI1, I2, I3 (20 μM each) for 4 h at 30 °C. All assays were stopped by extraction with ethyl acetate, processed and analysed by HPLC and HR-LCMS in an identical manner as detailed in ESI.† Individual assays were repeated at least in duplicate and representative results are shown. HPLC chromatographs with a UV detector at 315 nm that selectively detects indolyl vinyl isonitriles show: (a) 6-F-L-Trp 2d is a substrate for AmbI1-3 and is processed to give a compound with identical retention time to the synthetic standard 1d, and competitively preferred over the native substrate L-Trp 2a. L-Trp and halogen-substituted L-Trps starting materials, being insoluble in the ethyl acetate extract and not absorbing at 315 nm, are not present in the chromatogram; in all cases an excess of substrate was provided. The average conversion from L-Trp to 1a under standard assay conditions is estimated to be 30%, (b) The conversion of 4-F, 5-F, 6-F and 7-F L-Trp (2b–e) to the corresponding isonitriles by AmbI1-3 is equally effective as that of native substrate 2a, (c) the conversion of 5-F, 5-Cl, 5-Br L-Trp (2c, 2f–g) to the corresponding isonitriles by AmbI1-3 decreases with the increasing size of halogen substituent, (d) the conversion of 7-F, 7-Cl, 7-Br and 7-I L-Trp (2e, 2h–j) to the corresponding isonitriles by AmbI1-3 decreases with the increasing size of halogen substituent. The authenticity of each enzymatic product (1a–j) was ascertained by HRMS (Fig. SI 5–13, ESI†), comparative retention time and UV spectral analysis (Fig. SI 14, ESI†), in addition to HPLC co-elution with authentic standards applied to 1a, 1b, 1f. | ||
Results and discussion
To initiate the proposed study, we first examined the in vitro efficiency of AmbI1-3 to accept 6-F L-Trp (2d), a commercially available L-Trp analogue, as an alternative substrate (Fig. 2a). Incubation of AmbI1-3 with 2d, Rub5P, α-KG and Fe(II) under an aerobic atmosphere, led to the robust generation of a new product; the retention time and high resolution mass spectral (HRMS) data matched the synthetic standard 1d (Fig. 2a, lines 2 vs. 4 and SI-7†). Under the standard assay conditions, 2d appears a competent substrate for AmbI1-3 in comparison with native substrate 2a (Fig. 2a, lines 3 vs. 4) as AmbI1-3 enzyme mixture (20 μM each) was able to achieve 38 ± 2 turnover for 2a and 42 ± 4 for 2d (see ESI†). This observation was further validated by a competition assay with an equimolar amount of 2a and 2d subjected to the standard assay conditions, where 2d was marginally preferred (Fig. 2a, line 5). In addition, we did not detect any trans-isomer of 1d generated from 2d (Fig. 2a, lines 4 vs. 6) under the detection limit of our HPLC instrument with a UV detector set to 315 nm, a wavelength specific for indolyl vinyl isonitriles. This observation corroborates with our original observation that indolyl vinyl isonitrile synthases involved in hapalindole-alkaloid biogenesis are stereospecific,3,4 a common feature of all in vitro reconstituted isonitrile synthases.11,13,14 Encouraged by this initial result, we proceeded to test a series of halogen substituted L-Trp (2b–j) in vitro to gain a systematic understanding of the effect of the position and nature of the halogen substituent within L-Trp on the efficiency of AmbI1-3 enzymes to generate the corresponding indolyl vinyl isonitriles (1b–j). Each halo-L-Trp substrate (2b–j) was procured readily in sub-gram quantities using a robust chemoenzymatic approach recently developed by the Goss group.15 The effect of the position of halogen-substitution across the L-Trp indole backbone was first studied using C-4, C-5, C-6 and C-7 F-substituted L-Trp (2b–e). Under the standard assay conditions, these substrates (2b–e) were all converted effectively as the native substrate 2a to the corresponding F-substituted isonitriles (1b–e), as demonstrated by HPLC analysis (Fig. 2b). This result provides strong evidence that the F-substitution position across C4–C7 of L-Trp does not alter the efficiency of their conversion by AmbI1-3 to the corresponding isonitriles. Subsequently, the effect of the halogen substituent was probed using L-Trp containing Cl, Br or I at either C-5 (2c, 2f–g) or C-7 (2e, 2h–j). In changing the halogen substituent to chlorine from fluorine in 2c, substrate 2f showed reduced conversion to the corresponding isonitrile product 1f (Fig. 2c, lines 2 vs. 4) as AmbI1-3 enzymes (20 μM each) were only able to achieve 18 ± 2 turnover for 2f (see ESI†), indicating the levels of conversion decreased as the size of the halogen increased. This phenomenon was further evident with substrate 2g where the C-5 substituent is altered to bromine (Fig. 2c, lines 2/4 vs. 5), suggesting that the steric increase of halogen size at a specific position decreases the conversion by AmbI1-3. This trend was further examined using C-7 halogen-substituted L-Trp (2e, 2h–j) as substrates for AmbI1-3. With the increasing halogen size at C-7 of L-Trp, substrates 2e and 2h–j displayed decreasing product formation by AmbI1-3 to the corresponding isonitrile product 1e and 1h–j (Fig. 2d), corroborating with those observed for substrates 2c and 2f–g. Furthermore, cross comparison of the relative product formation from Cl-substituted 2fversus2h (Fig. 2c line 4 vs.Fig. 2d line 3) as well as Br-substituted 2gversus2i (Fig. 2c-line 5 vs.Fig. 2d-line 4), to the corresponding isonitriles (1f/1h and 1g/1i) reasserts the observation with F-substituted L-Trp 2b–e that halogen substitution position at C-5 or C-7 of L-Trp has no impact on their conversion by AmbI1-3. Strikingly, though the conversion is low, the sterically demanding 7-iodo L-Trp 2j is processed by AmbI1-3, further highlighting the promiscuous nature of this set of isonitrile synthase.Having an understanding on the substrate tolerance of isonitrile synthase AmbI1-3 towards differentially halo-substituted L-Trps, we next explored the possibility of generating the halo-substituted indole vinyl isonitrile in E. coli cells (Fig. 3a), by designing a simple synthetic biology system that employs a precursor directed biosynthesis strategy. We have previously demonstrated that the co-expression of ambI1-3 genes in an E. coli TOP10 cell chassis led to the robust production of 1a, using LB as the culture medium.5 As L-Trp, the native substrate for AmbI1-3, is present in LB and produced by E. coli cells, we envisioned an optimal approach to achieve the cell-based production of halo-substituted indole vinyl isonitrile could involve feeding the readily available halo-substituted L-Trp to a minimal culture medium that lacks L-Trp.
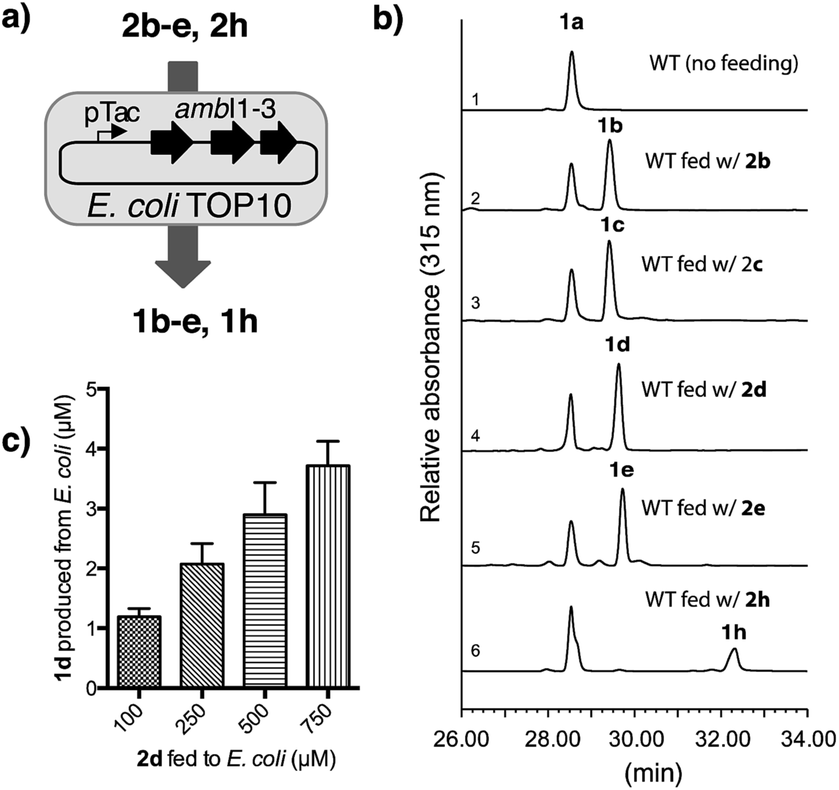 | ||
| Fig. 3 Precursor directed biosynthesis of halogen-substituted indolyl vinyl isonitrile in E. coli. (a) Schematic illustration of the directed biosynthesis of 1b–e and 1h from 2b–e and 2h, (b) HPLC chromatographs with a UV detector at 315 nm show 1b–e and 1h were readily generated from an E. coli strain overexpressing ambI1-3 and fed with 2b–e and 2h (100 μM) in M9 medium. The relative promiscuity of isonitrile synthase AmbI1-3 towards differentially halogen-substituted 2in vivo corroborates our results observed in the in vitro study, (c) quantification of 1d produced from ambI1-3-overexpressing E. coli strain in M9 medium supplemented with differential levels of 2d. | ||
To this end, we chose M9 minimal medium for culturing E. coli TOP10 cell that harbours ambI1-3 genes regulated by a pTac promoter. Upon induction with IPTG (1 mM final concentration), we were able to verify the production of isonitrile 1a without supplying exogenous L-Trp to the culture broth (Fig. 3b, line 1). Utilising the same culture medium, but supplemented individually with each of the fluorinated L-Trp regioisomers (2b–2e, 0.1 mM final concentration) and IPTG at the mid-log growth phase, the production of F-substituted 1b–1e was readily observed in the culture supernatant by HPLC analysis, of which production yield exceeded that of 1a (Fig. 3b, lines 2–5) by 0.3 fold. The production yield of 1b–1e in this engineered E. coli system is consistent with the in vitro data obtained with each substrate with AmbI1-3 enzyme. When 7-Cl L-Trp 2h was introduced, the corresponding Cl-substituted isonitrile 1h was also detected, albeit in a lower yield than the native 1a and its F-substituted counterpart (Fig. 3b, lines 6 vs. 5), consistent with the in vitro observation that Cl-substituted L-Trp is a poorer substrate for AmbI1-3. These results firmly established the feasibility to generate halogen-substituted indolyl vinyl isonitrile via precursor directed biogenesis in E. coli through harnessing the plasticity of the isonitrile synthase enzymes AmbI1-3. We were also able to quantify the production level of 1d by utilizing a concentration calibration curve of synthetic 1d (see ESI†). With the addition of 100 μM of exogenous 2d, 1.1 μM (0.20 mg L−1) of 1d was detected in the culture supernatant (Fig. 3c). By increasing the initial feeding concentration of 2d to 500–750 μM, the production level of 1d can be increased nearly 3-fold to 2.8–3.2 μM (up to 0.6 mg L−1) (Fig. 3c), highlighting the tunable feature of this expression system.
The establishment of a robust in vivo system for producing halo-substituted indole vinyl isonitrile in E. coli provided the opportunity to isolate structural analogues of 1a that differ with a single halogen substituent at the indole backbone to probe the effect of halogen modification. In particular, fluorine modification of aromatic rings is commonly employed to increase the metabolic profile of a drug-like molecule,16 while chlorine modification provides a unique chemical handle for further structural diversification by cross-coupling chemistry.9,10 We have noticed a significant increase in the lipophilicity of the newly generated analogues, as indicated by their HPLC retention times. Increasing the lipophilicity of a natural product often accompanies a change in its bioavailability or spectrum of activity. Though 1a is a natural intermediate in the biosynthesis of the hapalindole-type alkaloids, this compound itself is a broad-spectrum antimicrobial agent.2 To this end, we isolated 1e and 1h from E. coli (see ESI†) and accessed their antibacterial activities in comparison with the parent molecule 1a. Using disc diffusion agar assay, all three compounds (1a, 1e, 1h) showed antibacterial activity against bacterial pathogens V. cholera, E. coli and B. subtilis (Table 1). Notably, neither fluorine nor chlorine modification of 1a altered the antibacterial activity of the compounds, indicating the indole C-7 centre in 1a is likely distal from the antibiotic pharmacophore. This observation mirrors the previous study on glycosylated phenolic vinyl isonitrile natural product rhabduscin,13 where aglycosylated rhabduscin retains its bioactivity. These results collectively implicate the vinyl isonitrile motif in antibiotic 1a is likely the warhead and the peripheral indole motif constitutes an ideal derivatization site for introducing functional groups to enhance metabolic stability or enable downstream conjugation chemistry. Furthermore, even thought the indolyl vinyl isonitriles generated in this work lack specificities towards bacterial pathogens, the unique combination of increased lipophilicity that may enhance the absorption efficacy across mammalian cell membrane and retained antibacterial activity by F- and Cl-modification on antibiotic 1a suggests these modified analogues may gain increased in vivo potency, warranting future studies.
| Compound | lnhibition zone diameter (mm) | ||
|---|---|---|---|
| V. cholerae | E. coli | B. subtilis | |
| 1a | 19 | 38 | 29 |
| 1e | 19 | 36 | 31 |
| 1h | 20 | 39 | 31 |
Conclusions
In summary, our systematic investigation on the indolyl vinyl isonitrile synthase AmbI1-3 enzymatic activity towards a series of halogen substituted L-Trp revealed for the first time that this unique class of enzyme is promiscuous towards the L-Trp substrate. This study conclusively demonstrated that the substituent size but not the position at indole C4–C7 centres of L-Trp greatly impacts the substrate competency towards AmbI1-3. This work resulted in a series of 9 new indolyl vinyl isonitrile antibiotics all with increased lipophilicity. Reassuringly, introduction of the halogen did not have an adverse effect on antibiotic activity, indicating the strategic introduction of unique functional group at these positions, by the methods outlined in this work, may find important application in tuning the antibiotic uptake and potency. The promiscuous nature of indolyl isonitrile synthase, as revealed in this study for hapalindole-type alkaloid biogenesis, parallels that of strictosidine synthase,17 a conserved early-stage enzyme crucial for the biosynthesis of plant-derived complex indole monoterpene alkaloids.18 The fundamental studies on strictosidine synthase ultimately paved the way for the recent successful generation of modified antitumor vinca alkaloids in planta.19 Though the hapalindole alkaloid producers (subsection V cyanobacteria) are not yet genetically tractable, the striking flexibility of the early stage enzymes, coupled to the existing understanding of late stage enzymatic diversification of cis-indolyl vinyl isonitrile hold promise for future exciting studies toward reengineering the full biosynthetic pathway to this family of complex bioactive natural products. Furthermore it may be envisioned that the approach described in this study could open the way to generating indolyl vinyl isonitrile substrate probes that can be applied for elucidating the enigmatic enzymatic logic of isonitrile synthase, a crucial point for the rational engineering of this unique enzyme family or for further probing the mechanism of action. These investigations are currently being pursued.Acknowledgements
The research leading to these results has received funding from the Department of Chemistry, University of Pittsburgh (to X.L) and the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013/ERC grant agreement no 614779 (to R.J.M.G).References
- V. Bhat, A. Dave, J. A. MacKay and V. H. Rawal, Alkaloids Chem Biol, 2014, 73, 65–160 CrossRef CAS PubMed.
- J. R. Evans, E. J. Napier and P. Yates, J. Antibiot., 1976, 29, 850–852 CrossRef CAS.
- M. L. Hillwig, Q. Zhu and X. Liu, ACS Chem. Biol., 2014, 9, 372–377 CrossRef CAS PubMed.
- M. L. Hillwig, H. A. Fuhrman, K. Ittiamornkul, T. J. Sevco, D. H. Kwak and X. Liu, ChemBioChem, 2014, 15, 665–669 CrossRef CAS PubMed.
- M. L. Hillwig and X. Liu, Nat. Chem. Biol., 2014, 10, 921–923 CrossRef CAS PubMed.
- Y. Yan, J. Chen, L. Zhang, Q. Zheng, Y. Han, H. Zhang, D. Zhang, T. Awakawa, I. Abe and W. Liu, Angew. Chem., Int. Ed., 2013, 52, 12308–12312 CrossRef CAS PubMed.
- S. Gruschow, E. J. Rackham and R. J. M. Goss, Chem. Sci., 2011, 2, 2182–2186 RSC.
- H. Sun, C. E. Keefer and D. O. Scott, Drug Metab. Lett., 2011, 5, 232–242 CrossRef CAS.
- A. D. Roy, S. Grüschow, N. Cairns and R. J. M. Goss, J. Am. Chem. Soc., 2010, 132, 12243–12245 CrossRef CAS PubMed.
- W. Runguphan and S. E. O'Connor, Org. Lett., 2013, 15, 2850–2853 CrossRef CAS PubMed.
- S. F. Brady and J. Clardy, Angew. Chem., Int. Ed. Engl., 2005, 44, 7063–7065 CrossRef CAS PubMed.
- S. F. Brady and J. Clardy, Angew. Chem., Int. Ed. Engl., 2005, 44, 7045–7048 CrossRef CAS PubMed.
- J. M. Crawford, C. Portmann, X. Zhang, M. B. Roeffaers and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10821–10826 CrossRef CAS PubMed.
- M. F. Clarke-Pearson and S. F. Brady, J. Bacteriol., 2008, 190, 6927–6930 CrossRef CAS PubMed.
- D. R. Smith, T. Willemse, D. S. Gkotsi, W. Schepens, B. U. Maes, S. Ballet and R. J. Goss, Org. Lett., 2014, 16, 2622–2625 CrossRef CAS PubMed.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- E. McCoy, M. C. Galan and S. E. O'Connor, Bioorg. Med. Chem. Lett., 2006, 16, 2475–2478 CrossRef CAS PubMed.
- S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC.
- W. Runguphan, X. Qu and S. E. O'Connor, Nature, 2010, 468, 461–464 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: In vitro, in vivo assay conditions, 1H and 13C NMR spectra of 1d/1f, HRMS and UV spectra of 1a–1j. See DOI: 10.1039/c5sc02919h |
| This journal is © The Royal Society of Chemistry 2015 |