
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective photocatalytic C–C bond cleavage under ambient conditions with earth abundant vanadium complexes†
Sarifuddin
Gazi
ab,
Wilson Kwok
Hung Ng
a,
Rakesh
Ganguly
 a,
Adhitya Mangala
Putra Moeljadi
a,
Hajime
Hirao
*a and
Han Sen
Soo
*ab
a,
Adhitya Mangala
Putra Moeljadi
a,
Hajime
Hirao
*a and
Han Sen
Soo
*ab
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: hirao@ntu.edu.sg; hansen@ntu.edu.sg
bSingapore-Berkeley Research Initiative for Sustainable Energy (SinBeRISE), 1 Create Way, Singapore 138602
First published on 14th September 2015
Abstract
Selective C–C bond cleavage under ambient conditions is a challenging chemical transformation that can be a valuable tool for organic syntheses and macromolecular disassembly. Herein, we show that base metal vanadium photocatalysts can harvest visible light to effect the chemoselective C–C bond cleavage of lignin model compounds under ambient conditions. Lignin, a major aromatic constituent of non-food biomass, is an inexpensive, accessible source of fine chemical feedstocks such as phenols and aryl ethers. However, existing lignin degradation technologies are harsh and indiscriminately degrade valuable functional groups to produce intractable mixtures. The selective, photocatalytic depolymerization of lignin remains underexplored. In the course of our studies on lignin model compounds, we have uncovered a new C–C activation reaction that takes place under exceptionally mild conditions with high conversions. We present our fundamental studies on representative lignin model compounds, with the aim of expanding and generalizing the substrate scope in the future. Visible light is employed in the presence of earth-abundant vanadium oxo catalysts under ambient conditions. Selective C–C bond cleavage leads to valuable and functionally rich fine chemicals such as substituted aryl aldehydes and formates. Isotope labeling experiments, product analyses, and intermediate radical trapping, together with density functional theory studies, suggest a unique pathway that involves a photogenerated T1 state during the C–C bond cleavage reactions. Our study demonstrates a sustainable approach to harvest sunlight for an unusual, selective bond activation, which can potentially be applied in organic transformations and biomass valorization.
Introduction
Photoredox catalysis has recently emerged as a versatile methodology to harvest visible light as a sustainable source of energy to facilitate chemical conversions in organic reactions.1–6 Pioneering studies have been reported in the construction of complex organic molecules through C–C bond formation,1–6 but the application of photocatalytic processes in selective C–C bond activation reactions has been largely neglected. Related to this context, our team has been developing (photo)catalysts for bioinspired oxidation reactions,7,8 proton reduction, and degradation of pollutants,9 as part of a common theme to achieve a sustainable, integrated artificial photosynthetic system.10–13 Intrigued by the dearth of research on photoredox catalysis in selective C–C bond cleavage reactions, we endeavored to explore the development of base-metal photocatalyzed reactions to be potentially applied in the disassembly and valorization of macromolecules such as non-food biomass.The increasingly severe effects of global climate change and environmental pollution due to fossil fuel combustion have led to growing interest in the development of non-food biomass as feedstocks for fuels and chemicals.14,15 In particular, the conversion of biomass into fine chemicals and pharmaceutical precursors is attractive because of the inherent chemical complexity of biopolymers. One of the most under-utilized renewable resources is non-food plant biomass, the major constituents of which are biomacromolecules such as cellulose, hemicelluloses, and lignin.1 Lignin (Fig. 1), which constitutes up to 30% by weight in biomass, is especially appealing as a source of aromatic small molecules such as phenols and aryl ethers.16 However, unlike cellulose and hemicelluloses that have already been utilized in biorefineries for biofuel production,17 lignin is often regarded as waste since it is notoriously resistant to oxidative, hydrolytic, photochemical, and even biological degradation.15,16 Consequently, lignin is typically burned for energy production despite the presence of rich chemical functionalities that would otherwise require energy-intensive multi-step chemical syntheses.18 The chemoselective photochemical depolymerization of lignin remains an underexplored field.19
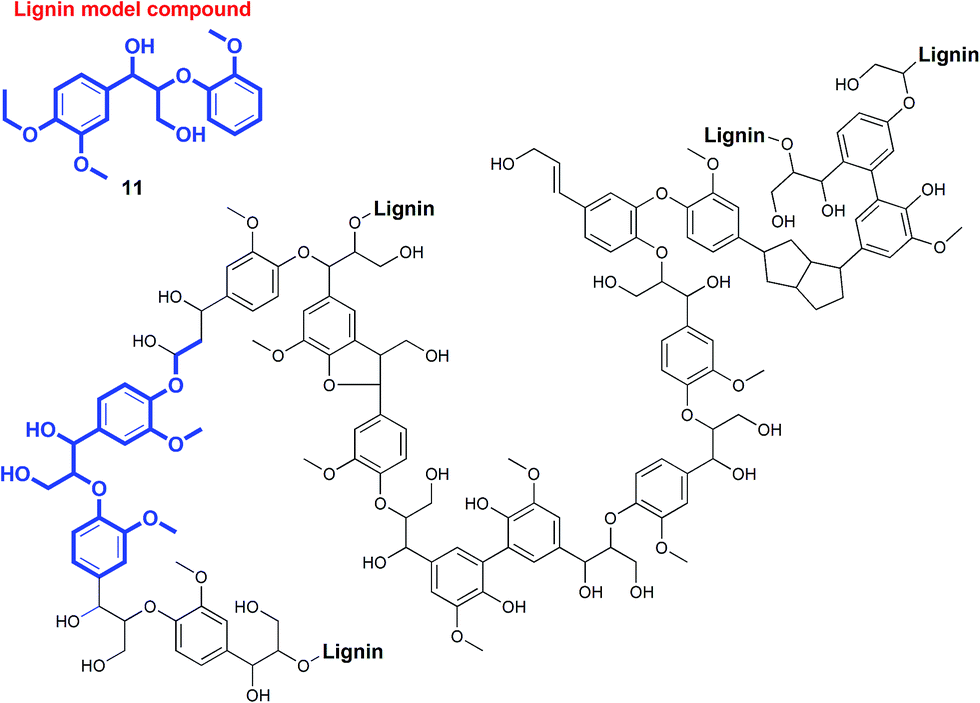 | ||
| Fig. 1 Representative fragment of biomass lignin and one of the model compounds used in our studies. | ||
The exclusive utilization of lignin as a biofuel has been considered a waste of its potential.14–16,18,20 Biomass lignin (Fig. 1) is a complex polymer that can realistically be harnessed as the largest source of inexpensive, easily accessible precursors for the production of numerous aromatic building blocks through judicious disasssembly.18 Current technology for the valorization of lignin involves harsh solvothermal or pyrolytic processes in the presence of excess reagents like acids or H2.15,16,21,22 This causes the unselective decomposition of lignin into many products with low yields, or even mineralization and charring. Patently, a cost-effective, environmentally benign protocol for lignin depolymerization through chemoselective bond cleavage will be critical for the sustainable production of chemical feedstocks from biomass lignin.19,23–25
Recently, the selective bond cleavage of lignin model compounds and even lignin in some cases, by molecular catalysts, has been reported in a few seminal papers. This includes the use of molecular catalysts based on Ru,26,27 Ni,24 and V28–33 under thermal conditions. For instance, Bergman and Ellman adopted a tandem dehydrogenative-reductive ether cleavage approach with Ru complexes,26 whereas Hartwig reported efficient Ni complexes for the reductive hydrogenation of aryl ether linkages commonly found in lignin. On the other hand, Toste et al. and Hanson and Silks independently demonstrated the promise of earth-abundant V complexes in catalyzing selective C–O and C–C bonds in lignin model compounds with high yields at moderately elevated temperatures.28–33 Stahl and Westwood also separately described novel two-step thermal depolymerization of lignin itself (Fig. 2), through the initial oxidation of benzylic alcohols, followed by C–O bond cleavage via acid-promoted or reductive pathways.23,25 The first successful degradation of lignin and lignin model compounds under ambient conditions was reported by Stephenson and coworkers, involving a tandem TEMPO-oxidation and photocatalytic C–O bond cleavage using visible light irradiation (Fig. 2).19 Their protocol is notable for the high yields and preservation of valuable alcohol, aldehyde, and ether functional groups in the products. However, the second step includes the use of expensive Ir-based photosensitizers and sacrificial amine-formate reductants,19 which can be more sustainable if earth-abundant (photo)catalysts and reagents like air are utilized instead.34,35
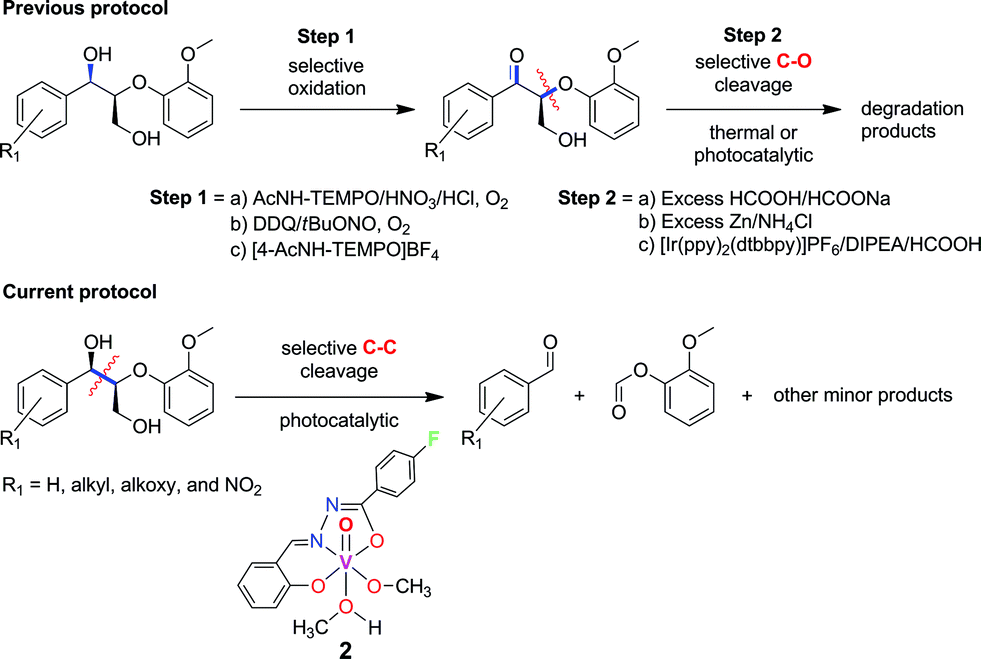 | ||
| Fig. 2 Previously reported strategies for selective bond cleavage of lignin model compounds include initial oxidation, followed by thermal or noble-metal driven photocatalytic conditions.19,23,25 Our current procedure involves mild, photocatalytic C–C cleavage with earth-abundant vanadium oxo catalysts. | ||
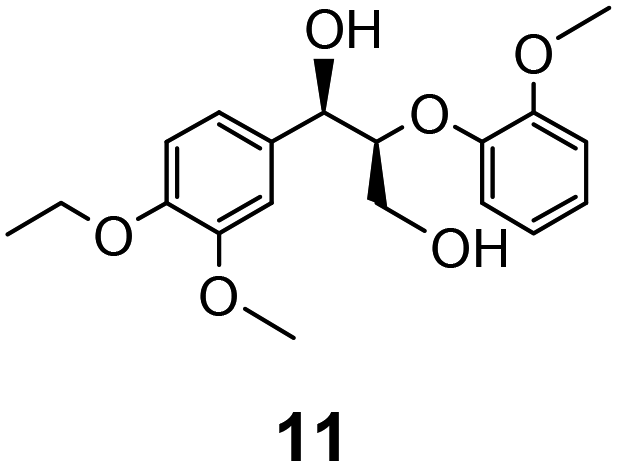
In the course of our studies on the disassembly of lignin model compounds, we have uncovered a photocatalytic earth-abundant vanadium oxo complex that can effect selective C–C bond cleavage of specific bonds in some aliphatic alcohols. Herein, we describe the fundamental studies on the reactivity and mechanistic understanding of the degradation of lignin model compounds, with the intention of future expansion and generalization of the substrate scope. A representative lignin model compound (11, Fig. 1) with a β-O-4 linkage is employed as the substrate,25–36 since this moiety constitutes over 50% of the inter-monomer functionalities in lignin, and has been proposed as a vulnerable link for chemical cleavage.15,16,18,21,22,25,37 We show that in the presence of our vanadium catalyst, upon irradiation with visible light (>420 nm) under AM1.5 solar intensity and ambient temperature and pressure, a solution of the model compound 11 is converted into an aryl aldehyde and an aryl formate (Fig. 2). Mechanistic studies with isotope labeling, product analyses, and density functional theory (DFT) calculations suggest a distinct, unique, and functional-group tolerant C–C bond activation pathway for lignin substrates under exceptionally mild reaction conditions.
Results and discussion
Thermal and photocatalytic lignin model degradation by monomeric vanadium(V) oxo complexes
The detailed synthetic procedures of the vanadium(V) oxo complexes 2 and 5 are described in the ESI.† Complex 2 is a new compound synthesized from a hydrazone benzohydroxamate ligand 1, which is redox non-innocent. The ligand 1 was synthesized in two steps from commercial reagents by adapting a previously published procedure.38 In the presence of stoichiometric equivalents of V(V) oxytripropoxide and 1, complex 2 was prepared in high yields and characterized thoroughly. Complex 5 has been prepared for control experiments to represent the V(V) oxo catalyst reported previously by Toste and coworkers.30,36The single crystal X-ray diffraction structures of complexes 2 and 5 are depicted in Fig. 3. Complex 2 displays an octahedral geometry in the solid state supported by ligand 1, with a characteristic V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distance of 1.59 Å. An equatorial methoxide (O5) and an axial methanol ligand (O4) are bound, with the V–O bond for the methanol ligand (2.34 Å) being dramatically elongated compared to the methoxide (1.76 Å). Other salient parameters of 2 are summarized in the ESI† (Tables S1–S5). Complex 5 crystallizes in two different polymorphs from methanol (MeOH), depending on the temperature. At low temperatures (−20 °C), the monomeric polymorph (Fig. 3b) was isolated, featuring a square pyramidal V center possessing an axial oxo ligand. On the other hand, the dimeric, centrosymmetric polymorph (Fig. S1 in the ESI†) crystallized at room temperature, with an octahedral V geometry and the alkoxide group of ligand 4 behaving as the bridging donor. In solution with acetonitrile (CH3CN) as the solvent, both 2 and 5 are believed to be monomeric or in equilibrium, with no perceptible effects on the catalytic activity.
O bond distance of 1.59 Å. An equatorial methoxide (O5) and an axial methanol ligand (O4) are bound, with the V–O bond for the methanol ligand (2.34 Å) being dramatically elongated compared to the methoxide (1.76 Å). Other salient parameters of 2 are summarized in the ESI† (Tables S1–S5). Complex 5 crystallizes in two different polymorphs from methanol (MeOH), depending on the temperature. At low temperatures (−20 °C), the monomeric polymorph (Fig. 3b) was isolated, featuring a square pyramidal V center possessing an axial oxo ligand. On the other hand, the dimeric, centrosymmetric polymorph (Fig. S1 in the ESI†) crystallized at room temperature, with an octahedral V geometry and the alkoxide group of ligand 4 behaving as the bridging donor. In solution with acetonitrile (CH3CN) as the solvent, both 2 and 5 are believed to be monomeric or in equilibrium, with no perceptible effects on the catalytic activity.
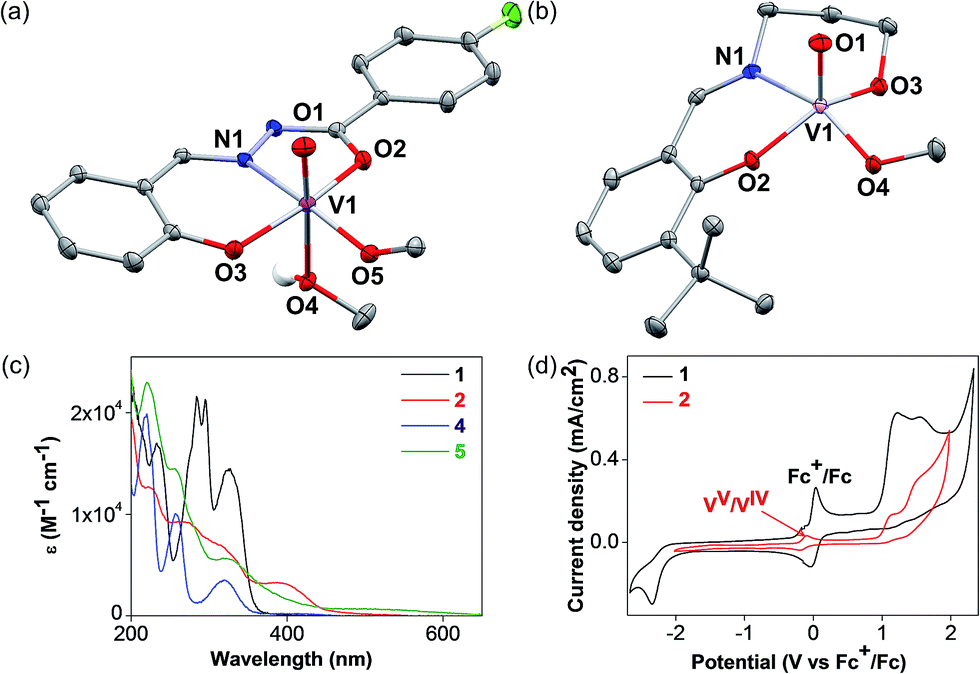 | ||
| Fig. 3 Oak Ridge Thermal Ellipsoid Plots (ORTEPs) from single crystal X-ray diffraction experiments of (a) complex 2, and (b) complex 5. (c) UV-vis absorption spectra of 1 (red), 2 (red), 4 (blue), and 5 (green) in CH3CN (0.10 mM). (d) CV of 1.0 mM 1 (black line with Fc+/Fc as internal standard), and 2 (red) using 0.10 M n-Bu4NPF6 as electrolyte in CH3CN at a scan rate of 100 mV s−1. | ||
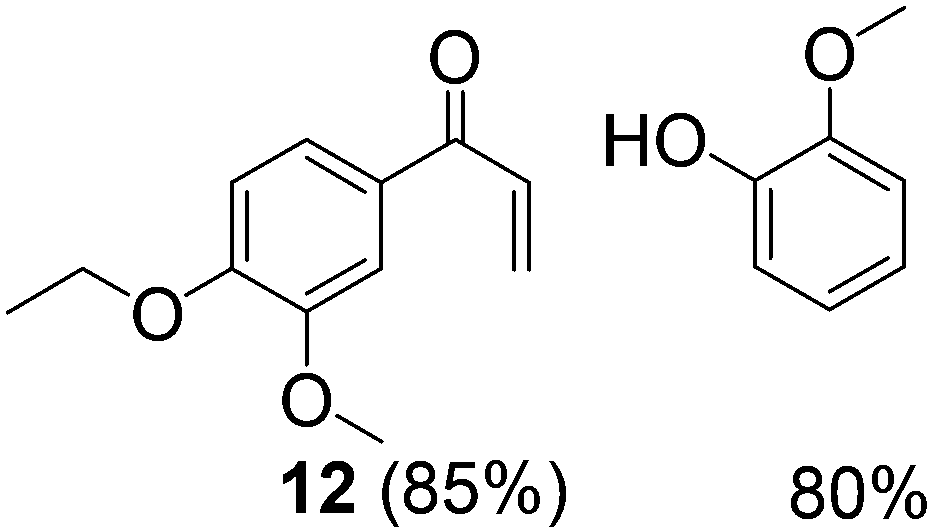
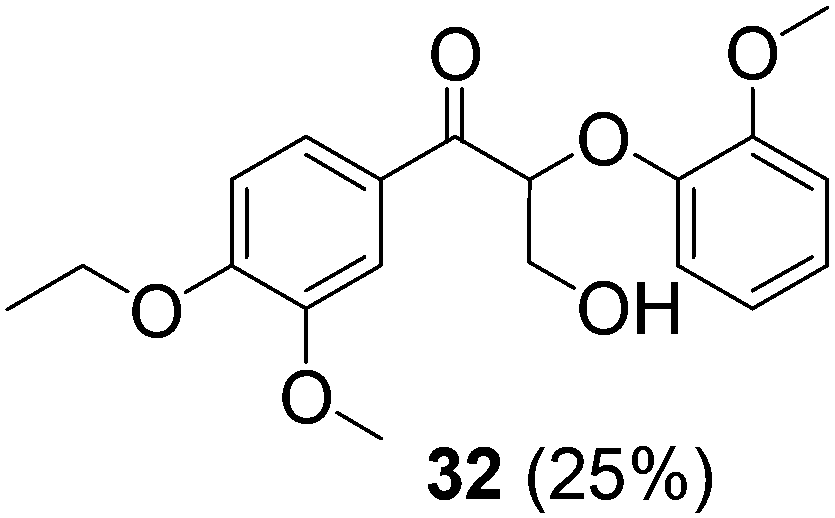
With complexes 2 and 5 in hand, we investigated their reactivity with lignin model compound 11 under thermal conditions (Table 1). As expected, 5 showed catalytic reactivity towards lignin degradation through selective C–O bond cleavage similar to the prior report by the Toste group. At room temperature, 5 showed slow reactivity with 11 (18% conversion, entry 1), but gave >95% conversion with good selectivity under reflux conditions (entry 2). The products arise from C–O bond cleavage and give predominantly α,β-unsaturated ketones. In contrast, catalytic amounts of 2 resulted in only about 25% conversion of 11 to the ketone (32), after oxidation of the benzylic alcohol under similar reaction conditions over 36 h (entry 4). The product selectivity is patently different under thermal conditions. However, 2 has a brown while 5 has a red color, alluding to the ligand-to-metal charge transfer (LMCT) transitions occurring in these d0 V(V) oxo complexes. Accordingly, the UV-visible (UV-vis) spectrum of 2 exhibits an absorption band in the near-UV region with λmax at 396 nm (Fig. 3c) tailing to 500 nm, which can be attributed to LMCT based on DFT calculations (vide infra).
| Entry | Substrate | Vanadium catalyst | Amount (mol%) | Temperature (°C) | Reaction time (h) | Conversion (%) | Productsa |
|---|---|---|---|---|---|---|---|
| a The yields are derived from the 1H NMR spectra with 1,1,2,2-tetrachloroethane as an internal standard. All the reactions were carried out in CH3CN in the dark under ambient air. | |||||||
| 1 |

|
5 | 10 | ∼25 | 24 | 18 |
|
| 2 | 11 | 5 | 10 | ∼80 | 24 | 100 |
|
| 3 | 11 | 2 | 10 | ∼25 | 24 | 0 | — |
| 4 | 11 | 2 | 10 | ∼80 | 36 | 25 |
|
The UV region consists of more intense absorptions typical of salicylaldimine intra-ligand charge transfers. Similarly, 5 also displays a near-UV absorption band with a blue-shifted λmax at 330 nm that tails into the visible region (Fig. 3c). The UV-vis spectra of ligands 1 and 4 are illustrated in Fig. 3c for comparison. Both ligands evidently do not exhibit absorption above 380 nm, lending credence to the assignment of the visible absorption bands as arising from LMCT transitions. The UV-vis spectra of VO(acac)2 and VO(OPr)3 are shown in Fig. S2a† on the same scale, and both complexes absorb less light in the visible region relative to 2, consistent with the low photodegradation products yields (Table 2, entries 1 and 2). Notably, 2 also has a photoluminescence λmax at 480 nm with lifetimes of 9.9 ns (59%), 2.0 ns (29%), and <110 ps (12%) as determined by time-correlated single photon counting (TCSPC, Fig. S2 in the ESI†). Additional photophysical studies are underway to probe this intriguing phenomenon, which has not been commonly observed among molecular vanadium systems, and will be reported in a separate manuscript.
| Entry | Substrate | Vanadium catalyst | Amount (mol%) | Irradiation time (h) | Conversion (%) | Identifiable products |
|---|---|---|---|---|---|---|
| a The yields are calculated based on 1H NMR spectra with 1,1,2,2-tetrachloroethane as an internal standard. Unless otherwise mentioned, the reactions were carried out in CH3CN under aerobic conditions with visible light irradiation (λ > 420 nm, AM1.5 solar irradiation). During irradiation, the temperature was maintained below 30 °C by water circulation through a transparent glass jacket. b Reaction performed in the dark. c Reaction performed under an Ar atmosphere. d Reaction carried out with AM1.5 solar irradiation without visible light filter. | ||||||
| 1 |
|
VO(acac)2 | 10 | 24 | 11 |
|
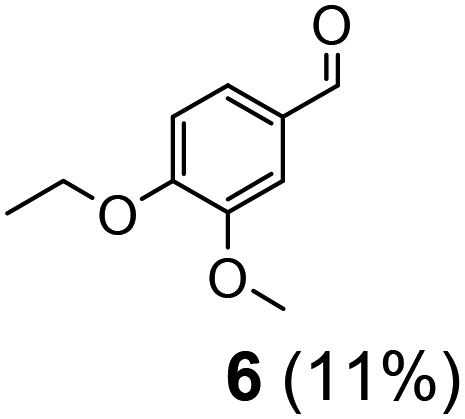
| 2 | 11 | VO(OPr)3 | 10 | 24 | 7 | 6 (7%) |
| 3 | 11 | 5 | 10 | 24 | >50 |
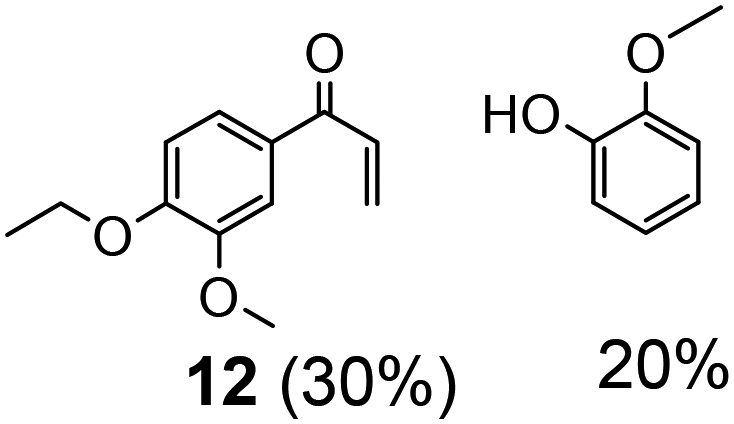
|
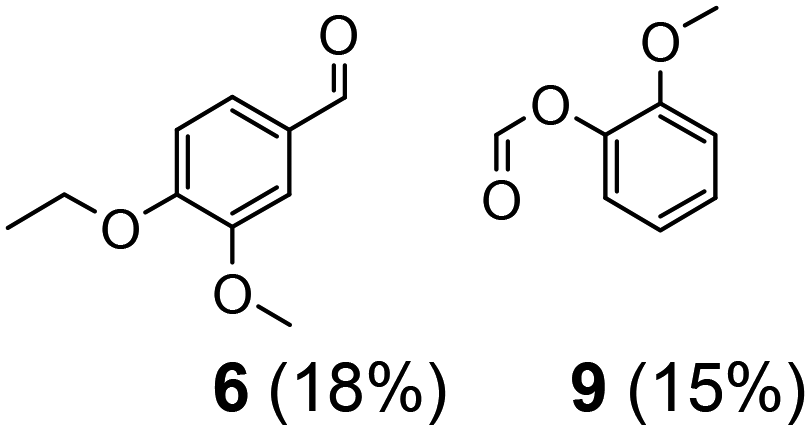
|
||||||
| 4b | 11 | 2 | 5 | 24 | 0 | — |
| 5 | 11 | 2 | 5 | 24 | ∼41 | 6 (39%), 6a (7%), 7 (9%), 9 (34%), HCOOH (10%) |
| 6 | 11 | 2 | 10 | 24 | 100 | 6 (70%), 6a (24%), 7 (6%), 9 (61%), HCOOH (32%) |
| 7c | 11 | 2 | 10 | 24 | ∼3 | 6 (3%) |
| 8d | 11 | 2 | 10 | 16 | 100 | 6 (50%), 6a (49%), 7 (30%), 9 (54%), HCOOH (45%) |
| 9 | 11 | 2 | 20 | 24 | ∼35 | 6 (32%), 6a (3%), 7 (10%), 9 (25%), HCOOH (10%) |
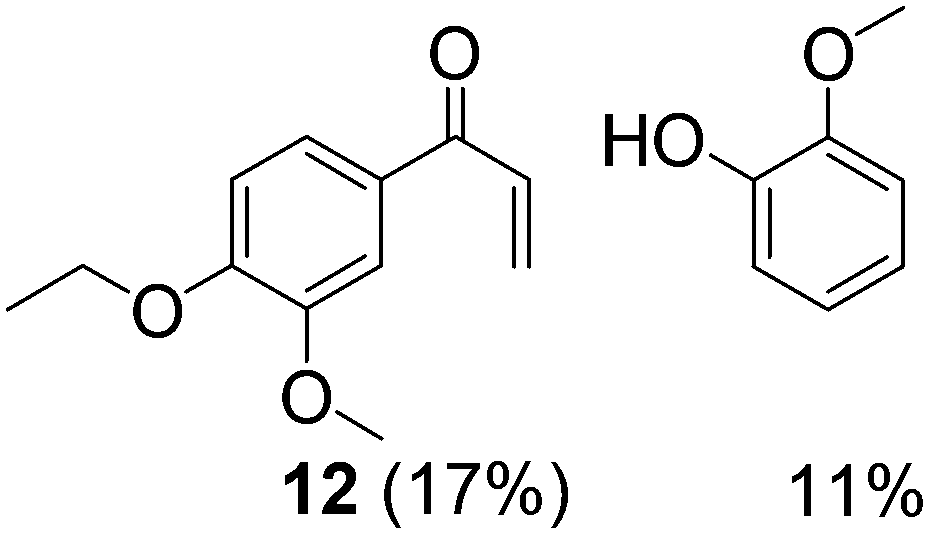
Prompted by the absorption and emission properties of these complexes, we probed their photocatalytic behavior in the degradation of 11. Remarkably, when a solution of 11 and 5 mol% of 2 was irradiated under ambient conditions by a solar simulator with a visible light filter (>420 nm) for 24 h, >40% of 11 were chemoselectively converted (Table 2, entry 5), in contrast to the lower thermal activity of 2. The products have been identified as 4-ethoxy-3-methoxybenzaldehyde (6) and 2-methoxyphenylformate (9), arising from selective aliphatic C–C bond cleavage reactions under ambient conditions in the preserve of enzymatic processes.37,39 These products were identified by NMR spectroscopy, high-resolution mass spectrometry (HRMS), isotope labeling studies (vide infra), and also confirmed by independent syntheses. The product selectivity and reaction conditions are distinct from all previous reports about degradation of lignin model compounds. A catalyst concentration of 10 mol% 2 gave the best results with >95% consumption of 11 and high yields of 6 (70%) and 9 (61%, entry 6). However, further increase in the concentration of 2 led to lower conversion and product yields, possibly due to reduced light transmission through the sample (entry 9). Under the optimized conditions for visible light photodegradation of 11, formic acid, 4-ethoxy-3-methoxybenzoic acid (6a), and guaiacol were also observed by 1H NMR spectroscopy or isolated by preparatory thin layer chromatography (TLC), as illustrated in Fig. 4. The clean and controlled transformation of 11 into chemical products that retain valuable functional groups occurs under exceptionally mild conditions, with the major energy inputs being light and the water circulation needed to maintain temperatures below 30 °C. A large scale photolysis of 11 (0.300 g, 0.86 mmol) was carried out with 10 mol% of 2. Due to the poor light transmission properties of the reaction solution, the photolysis experiment was conducted with a rudimentary homemade flow reactor (Fig. S71†) with a residence time of around 3 min. After 24 h of irradiation under visible light, the products 6 (67%) and 9 (56%) were isolated by column chromatography over silica.
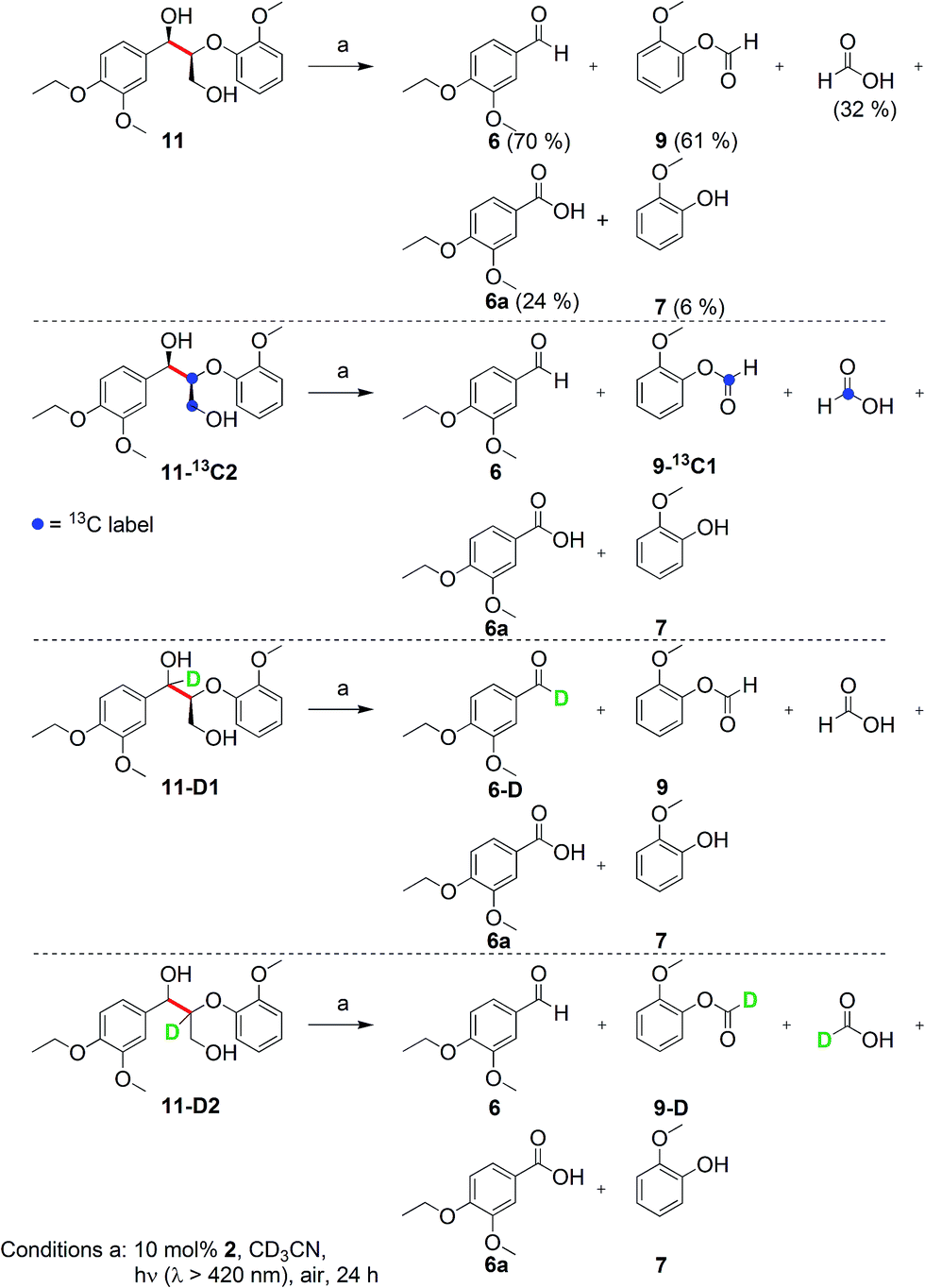 | ||
| Fig. 4 Product distribution from photocatalytic degradation of lignin model 11. Compounds 6a, 7, and formic acid are secondary products from 6 and 9. Labeling experiments with 13C isotopes indicate that the label is incorporated into aryl formate 9-13C1 and formic acid. Labeling experiments with deuterium provide evidence that no isotope scrambling occurs prior to or after C–C bond cleavage, suggesting that benzylic alcohol oxidation is not part of the mechanism. Moreover, the deuterium label on formic acid suggests that formic acid is a hydrolysis secondary product from the aryl formate. | ||
To elucidate the nature of photocatalysis by 2, a series of control experiments were conducted to ascertain the role of reagents and reaction conditions. In the absence of 2, lignin model compound 11 did not react when irradiated with visible light (λ > 420 nm) for 24 h at ambient temperature, since 11 does not absorb visible light. Likewise, no reaction was observed when 11 was stirred in the dark with 2 for 24 h in air (entry 4). Under an Ar atmosphere, 2 surprisingly remained photocatalytically active with an approximately 3% yield of 6 whereas 9 was not observed in the NMR spectra (entry 7). Compound 6 could have formed from a stoichiometric reaction, whereas 9 can still be produced in the absence of O2 since 2 can act as a stoichiometric VV oxidant, as supported by our DFT calculations (vide infra). When the reaction was conducted with AM1.5 solar irradiation without the visible light filter, the rate accelerated resulting in complete consumption of 11 within 16 h (entry 8). However, the yields of 6 and 9 decreased vis-à-vis the optimized conditions (entry 6), with increased amounts of 6a and guaiacol. Compound 6a is believed to have arisen by aerial oxidation of 6, whereas guaiacol and the formic acid could be derived from 9 by hydrolysis, as supported by isotope labeling studies below. The quantum efficiency of the photocatalytic degradation of 11 was measured with the use of ferrioxalate as the chemical actinometer, and was found to be 1.3% at 436 nm under visible light irradiation with a 420 nm filter, and 1.5% at 334 nm using AM1.5 irradiation without a 420 nm filter (in the ESI†).
Inspired by these findings, we examined the photocatalytic activity of 10 mol% of catalyst 5 under visible irradiation and observed 50% conversion of 11 under similar reaction conditions. Two products are identical with those obtained under thermal conditions, namely 12 and guaiacol, while 6 and 9 are different compared to the thermal experiments and similar to the results with photocatalyst 2 (entry 3). This experiment suggests that two simultaneous catalytic pathways may operate for 5, with the thermal reaction predominant over the photochemical reaction.36 When the colored complexes VIVO(acac)2 and VVO(OPr)3 were used under photocatalytic conditions, only trace amounts of 6 were identified by 1H NMR spectroscopy (entries 1 and 2). These experiments support the critical role of LMCT for visible light absorption, and redox non-innocent ligands in mediating the photocatalysis, as verified by DFT calculations (vide infra). Moreover, an unprecedented, general pathway for the selective photodriven C–C bond cleavage adjacent to aliphatic alcohol groups may be accessible for other V(V) oxo catalysts that absorb visible light.
Mechanistic understanding from substrate screening and isotope-labeling
To ascertain the identity of the products and obtain additional mechanistic insights, we carried out isotope labeling studies.Three different isotopically labeled lignin model compounds 11-13C2, 11-D1, and 11-D2 were synthesized and used as substrates in NMR tube experiments with CD3CN as the solvent under the optimal conditions from Table 2 entry 6. NMR experiments for 1H, 2H, and 13C were conducted before and after irradiation. With the 13C labeled 11-13C2 as substrate, 13C-labeled formic acid (blue hexagons) and 2-methoxyphenylformate (9-13C1, blue triangles) were observed by 1H NMR spectroscopy, with the formate protons split into doublets due to coupling with 13C (Fig. 5c).
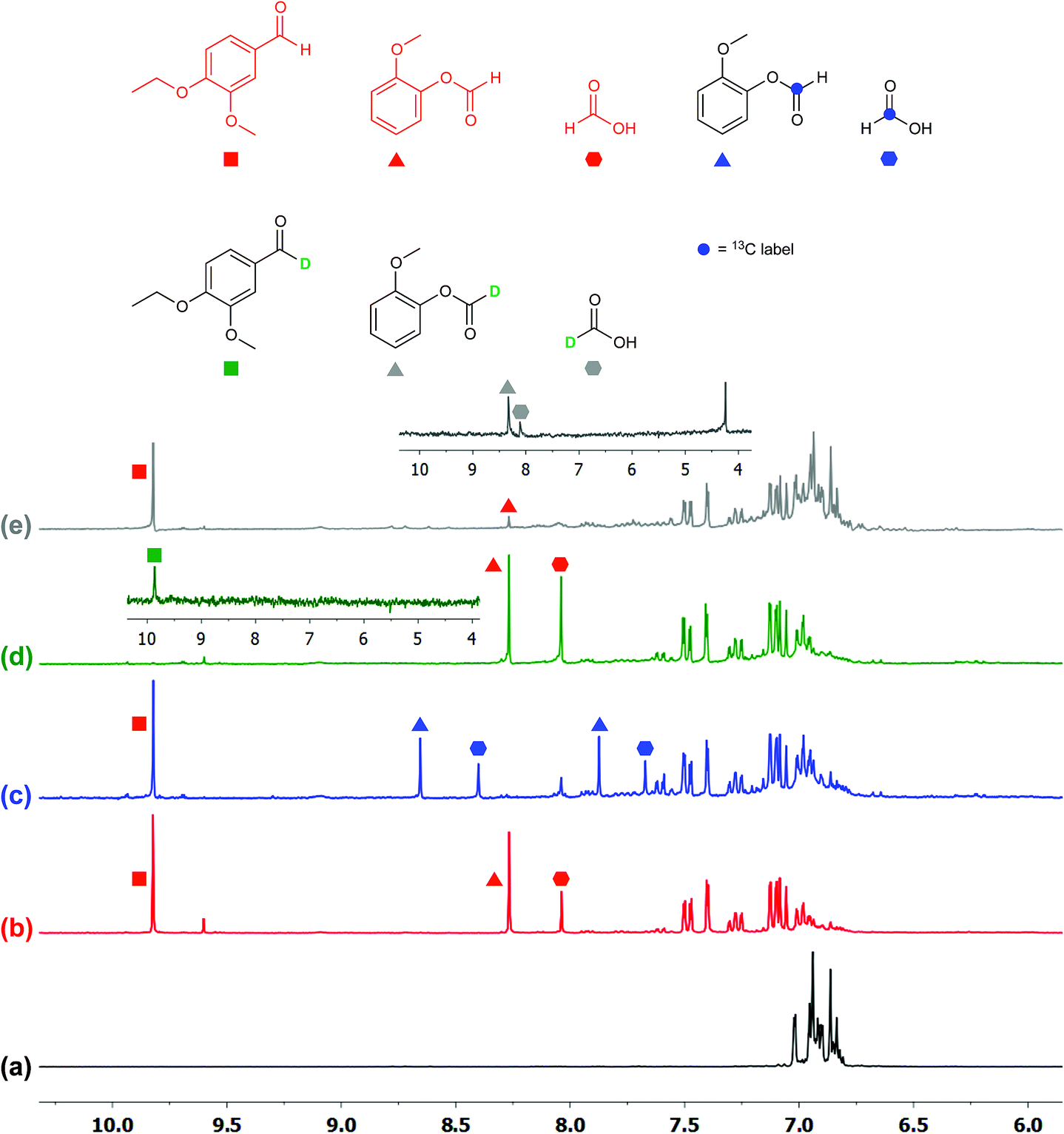 | ||
| Fig. 5 NMR (300 MHz, CD3CN) spectra of lignin model compounds before and after photocatalytic reactions. (a) Compound 11; (b) 11 after photolysis; (c) 11-13C2 after photoreaction; (d) 11-D1 after photoreaction (inset: 1H-decoupled 2H NMR spectrum of 6-D); and (e) 11-D2 after photoreaction (inset: 1H-decoupled 2H NMR spectrum after partial conversion. The peak at 4.27 ppm is from unreacted 11-D2). The unlabeled products 6 (red square), 9 (red triangle), and formic acid (red hexagon) are indicated at their distinctive chemical shifts. The 13C labeled 9-13C1 (blue triangle) and 13C labeled formic acid (blue hexagon) have been split due to the 1H coupling with 13C. The deuterated 6-D (green square), 9-D (grey triangle), and formic acid (grey hexagon) have been detected by 2H NMR spectroscopy. | ||
The 1H (Fig. 5c) and 13C NMR spectra of both products (Fig. S3 in ESI†) confirm the assignment of the 1H NMR chemical shifts and the regioselective aliphatic C–C bond cleavage sites. When 11 is deuterated at the benzylic position (11-D1), the 1H NMR spectrum of the product mixture no longer displays the aldehyde proton signal at 9.8 ppm (Fig. 5d), while the 2H NMR spectrum (inset) clearly shows the corresponding deuterium label (green square). The product 6-D was isolated by preparatory TLC and characterized by 1H and 2H NMR spectroscopy as well as by HRMS. The spectroscopic data suggested that the benzylic hydrogen is not scrambled or exchanged during the photocatalytic C–C bond cleavage reactions. When another deuterated lignin model 11-D2 was subjected to photocatalytic degradation, we found that the reaction rate was slower and it generated deuterated 2-methoxyphenylformate (9-D, grey triangle, Fig. 5e) and almost exclusively deuterated formic acid (grey hexagon, inset), further confirming the absence of isotope scrambling. The absence of formic acid in the 1H NMR spectrum with 11-D2 as the substrate also suggests that the formic acid produced during this photocatalytic reaction originated from the 2-methoxyphenylformate fragment, likely via hydrolysis (Fig. 5e and inset). This proposal is confirmed by the identification of guaiacol by NMR spectroscopy and liquid chromatography-mass spectrometry (LC-MS), although isolation by preparatory TLC proved challenging possibly due to decomposition of guaiacol.
These isotope labeling studies confirm that 2 is a photocatalyst for the unique and selective C–C bond cleavage of 11, ruling out the benzylic hydrogen abstraction and oxidation pathways proposed in prior reports.23,25–27,30,36 The product distribution of the isotope labeling experiments is summarized in Fig. 4. A systematic series of various lignin model compounds have been investigated under the same photocatalytic reaction conditions to explore the generality and selectivity of the C–C cleavage reactions (Fig. 6). Model compound 13 was degraded to yield 6 and 9, albeit with lower yields than for 11. Similarly, compound 15 was photocatalytically converted into 6 and 9 cleanly (Fig. 6), with a dramatically lower yield. These results suggest that the primary alcohol in 11 is important for facilitating the chemical reactivity, although our DFT calculations indicate that the lower yields from degradation of 13 and 15 over 24 h may have kinetic origins (vide infra). The results for substrates 13 and 15, as well as the DFT studies also indicate that a reaction path involving the tandem oxidation of the primary alcohol to an aldehyde, followed by a retro-aldol reaction, is probably not operational.23,27,32,33
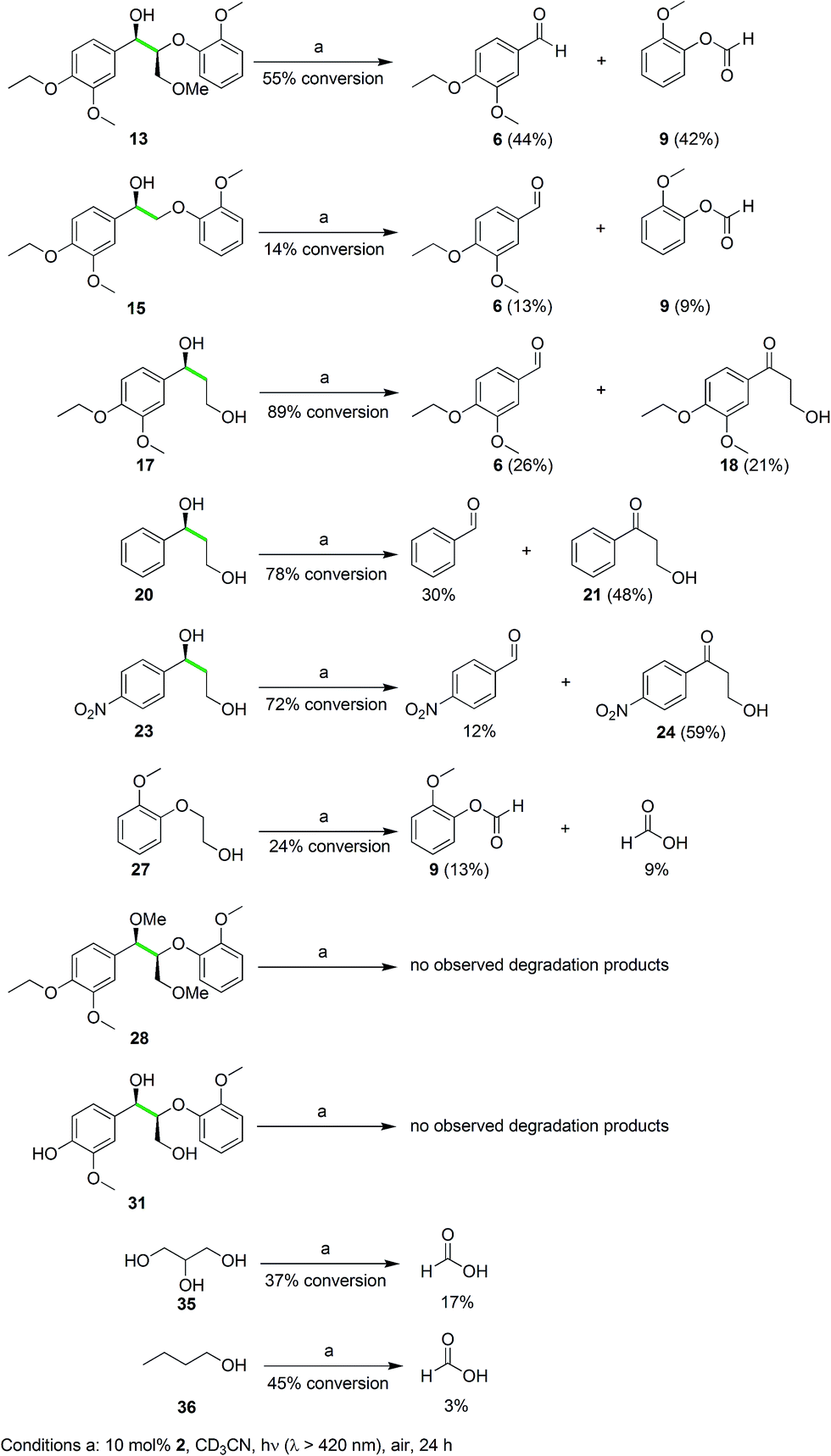 | ||
| Fig. 6 Lignin model compounds and aliphatic alcohols subjected to the photocatalytic conditions. Substrates 13 and 15 demonstrate that the benzylic alcohol is sufficient for C–C bond cleavage activity. Substrates 17, 20, and 23 show that electron-rich and electron-deficient aryl rings react, although benzylic alcohol oxidation instead of C–C bond cleavage dominates with electron-deficient aryl rings. Substrates 28 and 31 illustrate the necessity of aliphatic alcohol groups and the incompatibility with phenolic functions respectively. Substrates 35 and 36 demonstrate the potential versatility of this photocatalytic protocol for C–C bond cleavage reactivity under ambient conditions. | ||
A set of mono-aryl aliphatic alcohol substrates was also subjected to the same photocatalytic conditions, and the product distribution gave additional insights into the catalytic mechanism. After photolysis, 17 was oxidized to 6 and formic acid as well as 6a, but the ketone 18 was also observed as a minor product (Fig. 6), with no aldehyde derived from primary alcohol oxidation. Interestingly, the ratio of the oxidized ketones (21 and 24) to the corresponding aldehyde originating from C–C bond cleavage (benzaldehyde and 4-nitrobenzaldehyde, respectively) was found to rise when increasingly electron-deficient aryl groups were introduced at the benzylic alcohol (20 and 23 in Fig. 6). This phenomenon suggests a vital role of the aryl ring in dictating the selectivity of our unusual photocatalytic reactions. On the other hand, protection of the hydroxyl group of the benzylic alcohol as a methoxy ether provided 25, and the reaction yielded a more complex mixture including aldehydes and formic acid upon photodegradation (Fig. S4 in the ESI†).
When 27 (Fig. 6) was photolyzed with the same protocol, 9 and formic acid were observed in poor yields at low conversion. Furthermore, irradiation of the ether-protected model 28 and the phenolic substrate 31, in the presence of 2, resulted in no degradation (Fig. 6). Alcohol functionalities appear to be essential to direct the selective C–C bond cleavage, but binding of the aryl ring to the vanadium center as a phenolate shuts down the catalytic activity. To probe whether unactivated alcohols can undergo this remarkable photocatalyzed C–C cleavage, both glycerol (a by-product of biodiesel production)40 and 1-butanol were used as substrates. Gratifyingly, formic acid was observed as one of the products for both substrates as anticipated (Fig. 6), although the identification of the remaining products will require additional studies. The photocatalytic cleavage of C–C bonds in alcohols shows promise for further generalization. Electrochemical experiments were then initiated to probe the feasibility and thermodynamics of this photochemical reaction.
Electrochemical measurements and intermediate trapping
Cyclic voltammetry (CV) measurements conducted on photocatalyst 2 shows a reversible wave at −0.14 V relative to ferrocene (Fc, Fig. 3d). This can be attributed to the VV/VIV redox reaction. By assuming a LMCT band edge of ∼550 nm (Fig. 3c), which corresponds to an optical band gap of at least 2.25 eV, the absorbed photon energy far exceeds the dark T1 excited state that mediates the C–C bond cleavage, as supported by DFT calculations below.The CV of 11 is illustrated in Fig. S5a (in the ESI†), with two irreversible oxidation waves observed at +0.94 and +1.40 V. Alkoxy and alkyl groups are known to be electron-donating moieties for aromatic rings; the oxidation wave at +0.94 V likely corresponds to oxidation of the tri-substituted aryl ring, whereas the wave at +1.40 V corresponds to oxidation of the other di-alkoxy aryl ring. However, these redox potentials arise from the outer-sphere single electron transfer processes only. On the other hand, when 11 binds to VV, the C–C bond adjacent to the alkoxide becomes activated and will require substantially lower potentials for cleavage, which has been substantiated by our DFT calculations. To further support the hypothesis of a LMCT-initiated photocatalytic reaction, we attempted to synthesize the one-electron reduced congener of 2, the vanadium(IV) oxo 3. However, the product obtained by one-electron reduction using cobaltocene was poorly soluble and could not be fully characterized. Nonetheless, the EPR spectrum (Fig. S5b in the ESI†) confirms the presence of a paramagnetic VIV species. With 3 as the photocatalyst, irradiation of 11 with >420 nm light led to no degradation of 11 in the absence of air, confirming that LMCT from the supporting ligand 1 to the VV center in 2 is required to trigger the intramolecular radical oxidative processes. Since organic radicals are expected after the C–C cleavage reaction, the stable radical (2,2,6,6-tetramethylpiperidin-1-yl)oxy (TEMPO) has been employed as a trap to capture intermediates during the degradation of the model compounds. TEMPO (5 equivalents) was introduced during the photocatalytic reaction of stoichiometric amounts of 11 with 2 under an Ar atmosphere to maximize yields of the intermediate. The aldehyde 6 was still observed but an additional new product (34) derived from the radical coupling between the other fragment of 11 and TEMPO was detected by LCMS (Fig. S6 in the ESI†). The compound 34 was isolated by preparatory TLC and confirmed with 1H, 13C, and DEPT NMR spectroscopy, NOESY as well as HRMS. The proton-decoupled 13C NMR spectrum confirms the number of distinct carbons in the molecule, of which five carbons are CH (DEPT 90, Fig. S63 in the ESI†), four carbons are CH2, and five carbons are CH3 (DEPT 135, Fig. S62 in the ESI†). The NOESY data indicated correlations between the TEMPO methyl protons with the aliphatic CH, CH2, and aromatic CH protons, consistent with our structural formulation of compound 34 (Fig. S64 in the ESI†). The isolation of 34 and 6 supports the intermediacy of a controlled radical cleavage pathway.
In addition, transmission electron microscopy (TEM) measurements have been conducted before and after the photocatalytic reactions to explore the operation of heterogeneous catalysis. The TEM images of the reaction mixture before irradiation showed small amounts of amorphous residue, whereas sporadic amounts of some irregular particles appeared after irradiation (Fig. S7 in the ESI†). These amorphous materials could have arisen due to precipitation of the catalyst or substrate after solvent evaporation. Dynamic light scattering (DLS) experiments conducted before photodegradation showed the presence of small amounts of particles with sizes between 30 to 60 nm, whereas larger aggregates between 200 nm to 1 μm were detected after 24 h irradiation. To rule out the possibility of heterogeneous catalysis, an experiment was performed under the conditions of Table 2 entry 6, and the reaction mixture was centrifuged at the end of 24 h. After a fresh batch of 11 was added to the supernatant that was decanted and the solution was irradiated over 24 h, the product mixture contained 6 (40%), 9 (34%), and formic acid (21%). On the other hand, when 11 was added to the little residue that was collected from centrifugation, no reaction was observed. Moreover, experiments have been performed with suspended V2O5, and no photodegradation of 11 was observed after irradiation for 24 h under the same ambient conditions as the experiments with catalyst 2. We believe that the small amounts of particles are unlikely to explain the remarkable selectivity of our C–C bond cleavage reactions, especially since 11 displays no reactivity when irradiated with V2O5 particles under the typical catalytic conditions. Based on the experimental studies, a cogent mechanistic pathway involving LMCT in 2, followed by selective C–C cleavage of the bound aliphatic alcohol function, is proposed. Theoretical studies have been pursued to gain a deeper understanding of the exceptionally mild, unique, and potentially general photocatalytic reaction.
TD-DFT calculation of 2
The electronic structure of 2 was examined by a time-dependent DFT (TD-DFT) calculation using the B3LYP functional and the 6-31G* basis set.41,42 Gaussian 09 was used for the calculations.43 The TD-DFT calculation was performed at the ground-state (S0) geometry of 2 without the labile methanol ligand. At the valence edge, the occupied molecular orbitals are composed mostly of π orbitals of the ligand, whereas the unoccupied molecular orbitals always involve large contributions from the d orbitals at the vanadium center (Fig. S10 in the ESI†). As such, the six low-lying excited states are all associated with LMCT (Table S16 in the ESI†).The lowest-energy (singlet) exited state S1 excitation is of a π(L) → dπ*(M) LMCT type (Fig. 7a–c) and has an excitation energy of 536 nm (53.4 kcal mol−1), which falls within the range of wavelengths for light absorption by 2 observed by UV-vis spectroscopy (Fig. 3c). The visible absorption band with a λmax at 396 nm should be due to a higher-energy excitation, most probably to the S3 state that lies 401 nm above S0 and is also of a π(L) → dπ*(M) LMCT type (Fig. 7a).
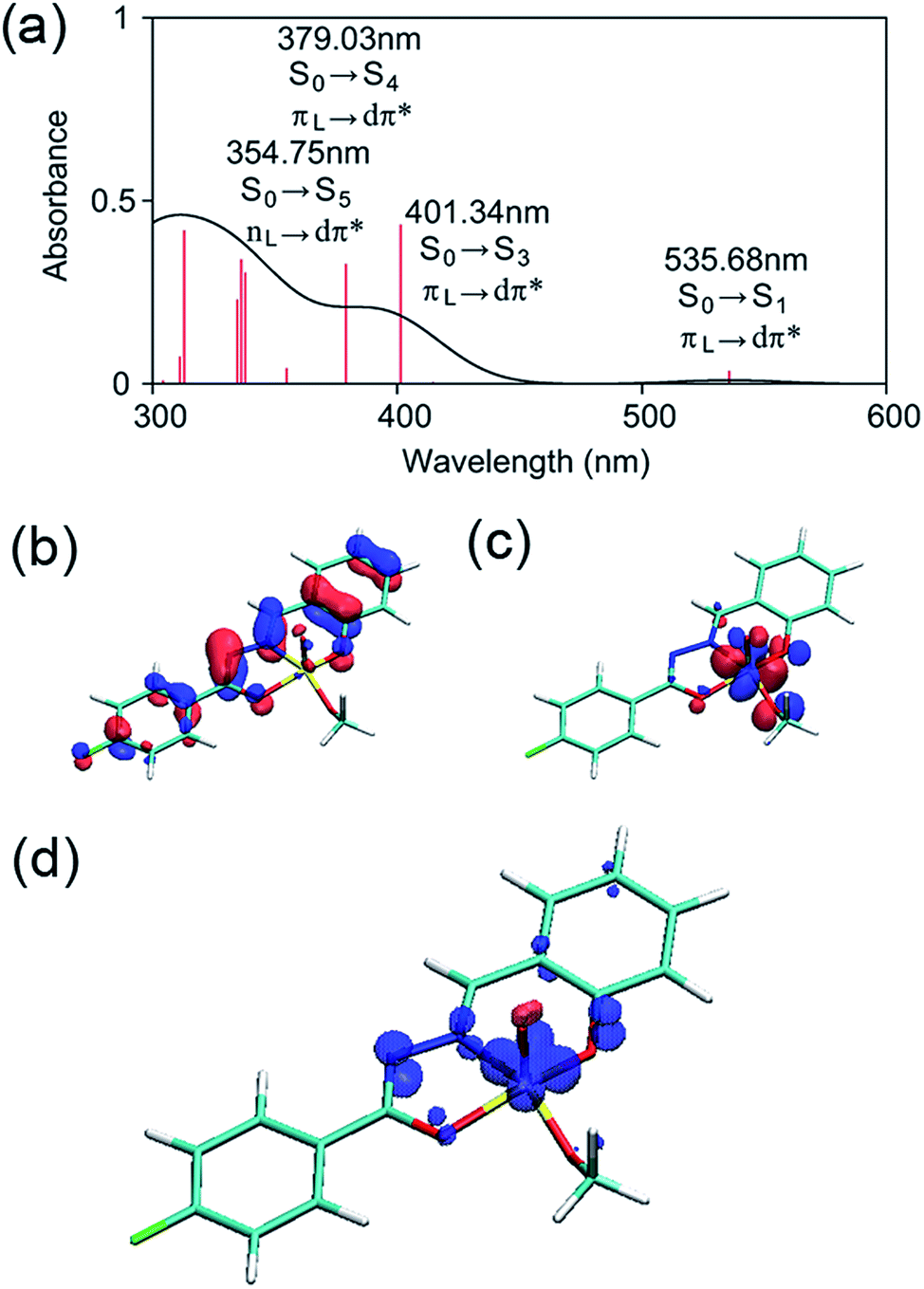 | ||
| Fig. 7 (a) Absorption spectrum obtained from a TD-DFT/B3LYP/6-31G* calculation. To compare the experimental and theoretical spectra, the theoretical absorption curve was scaled such that the absorbance at 396 nm became 0.2. Oscillator strengths for TD-DFT data are shown in an arbitrary unit. The spatial distributions of HOMO (b) and LUMO (c) indicate that the S0–S1 transition is characterized as a LMCT. ΔE(HOMO–LUMO) (=3.21 eV) is much larger than the S0–S1 excitation energy obtained by TDDFT (2.31 eV). The spin density distribution for the T1 state (d) indicates that the spin is localized at the vanadium center. | ||
The TD-DFT calculation also suggested that the triplet T1 state is less stable than S0 by 49.6 kcal mol−1 and is thus slightly lower in energy than the S1 state. Intersystem crossing from S1 to T1 may be facile since the two excited states should be in thermal equilibrium at room temperature. As illustrated in Fig. 7d, the spin density of 2 in the T1 state is distributed in the diazo and oxygen donors of the ligand 1 and the V-based dxy orbital; therefore, the T1 state is also viewed as an LMCT-type excited state. However, we note that these TD-DFT calculations correspond to the allowed vertical transitions into the vibronically excited states according to the Franck–Condon principle. Subsequently, intramolecular relaxation, internal conversion, and intersystem crossing should take place on ultrafast time-scales, which may lead to dark states in which the C–C bond cleavage occurs. Therefore, we sought a feasible reaction mechanism for this unusual reaction from a geometry-optimized, dark T1 excited state using DFT calculations.
Exploration of reaction mechanisms using DFT calculations
Having established that 2 has photo-accessible S1 and T1 excited states, we proceeded to explore viable reaction pathways for the selective C–C bond cleavage reaction of the V-bound 11. Our isotope labelling experiments suggest that C–C bond cleavage takes place without activation of any C–H bonds. Therefore, we sought a pathway that included direct C–C cleavage via a single electron transfer (SET) process as frequently observed in natural lignin peroxidase enzymes.34,35,37,39 The substrate we used for calculations is similar to 11, but the ethoxy group on the phenyl group on the left-hand side and the methoxy group on the phenyl group on the right-hand side in Fig. 1 were replaced with methoxy and hydrogen, respectively.It was found that the C–C cleavage in the S0 state is a very unfavourable process (Fig. S11†). This corroborates the slow thermal reactivity of 2 even at 80 °C, with the major observed product being the ketone 32 instead of 6 and 9 (Table 1, entry 4). It is therefore more probable that the reaction is triggered by photoexcitation. In the reactant complex (RC) for the reaction involving 2 and a model of 11, the benzylic oxygen of 11 is bound to the vanadium of 2. A TD-DFT calculation determined the S0–S1 excitation energy for this species at the ground-state geometry as 48.9 kcal mol−1, which is similar to the excitation energy calculated for 2 (53.4 kcal mol−1).
Although TD-DFT appears to describe the photoexcitation of 2 reasonably well, geometry optimization of various electronically excited species with TD-DFT is computationally too demanding. We have seen above that T1, which can be treated by ordinary DFT calculations, has an LMCT state. In addition, many of the intermediates and transition states on the S1 excited-state pathway can be described using broken-symmetry (BS) DFT calculations. Therefore, we decided to use (BS-)DFT calculations to explore the subsequent photochemical reactions of 2. The methanol ligand of 2 was removed. Geometry optimization and frequency calculations were then performed at the B3LYP/6-31G* level in the gas phase, and the energies were refined by performing single-point calculations with the 6-311+G(d,p) basis set, while including the effect of acetonitrile solvent with the IEFPCM-SCRF model.44
Geometry relaxation of T1 led to a somewhat smaller S0–T1 energy gap (20.2 kcal mol−1) for RC (Fig. 8a). It should be noted that B3LYP might possibly underestimate the energies of excited-states.45–47 Intriguingly, DFT calculations identified low-energy-barrier, exothermic pathways on the excited S1 and T1 energy surfaces (Fig. 8a). This suggests that the photoexcited 2 possesses an exceptionally mild, thermally accessible pathway for aliphatic C–C bond cleavage by inner-sphere SET from 11 to the ligand-centered cation radical, reminiscent of enzymes like lignin peroxidases and cytochromes P450.34,35,37,39,48,49 The facile LMCT-driven homolytic C–C bond cleavage in our reaction is due to an efficient SET to the ligand's π orbital that is lower in energy than the vanadium's d orbital (Fig. 9). Although the oxidation of 11 ostensibly requires at least +0.94 V by electrochemical measurements (Fig. S5a†), binding of the benzylic alkoxide to VV activates the adjacent C–C bond (red bond in Fig. 9). Furthermore, photoexcitation of 2 eventually results in an energetic, dark T1 excited state after non-radiative relaxation processes. The additional impetus from the T1 excited state together with formation of the CO double bond in the aldehyde, more than compensates for cleavage of the C–C single bond to produce Int 1 (Fig. 8a). In other words, the dark T1 excited state, which may be accessible only via photoexcitation and relaxation, combined with the Lewis acidic V center, allowed us to target proximal C–C bonds that are otherwise not electrochemically oxidizable. Our additional DFT calculations also suggested that when the substrate 11 is bound to the vanadium center via the primary alcohol, the C–C bond cleavage is not as favourable as the substrate bound via the benzylic alcohol (Fig. S12†); this is presumably due to the lower stability of the resultant substrate radical. The resulting substrate radical is trapped by O2 (see Fig. S13 in the ESI†) under aerobic conditions, while the aldehyde 6 dissociates. Subsequently, heterolytic cleavage of the peroxide O–O and aliphatic C–C bonds, coupled with proton transfer occurs on the S0 surface viaTS3 (Fig. 8b). This concerted process has a thermally accessible barrier of 19.7 kcal mol−1, generating formaldehyde and 9 (Fig. 8b), the latter of which has been isolated experimentally.
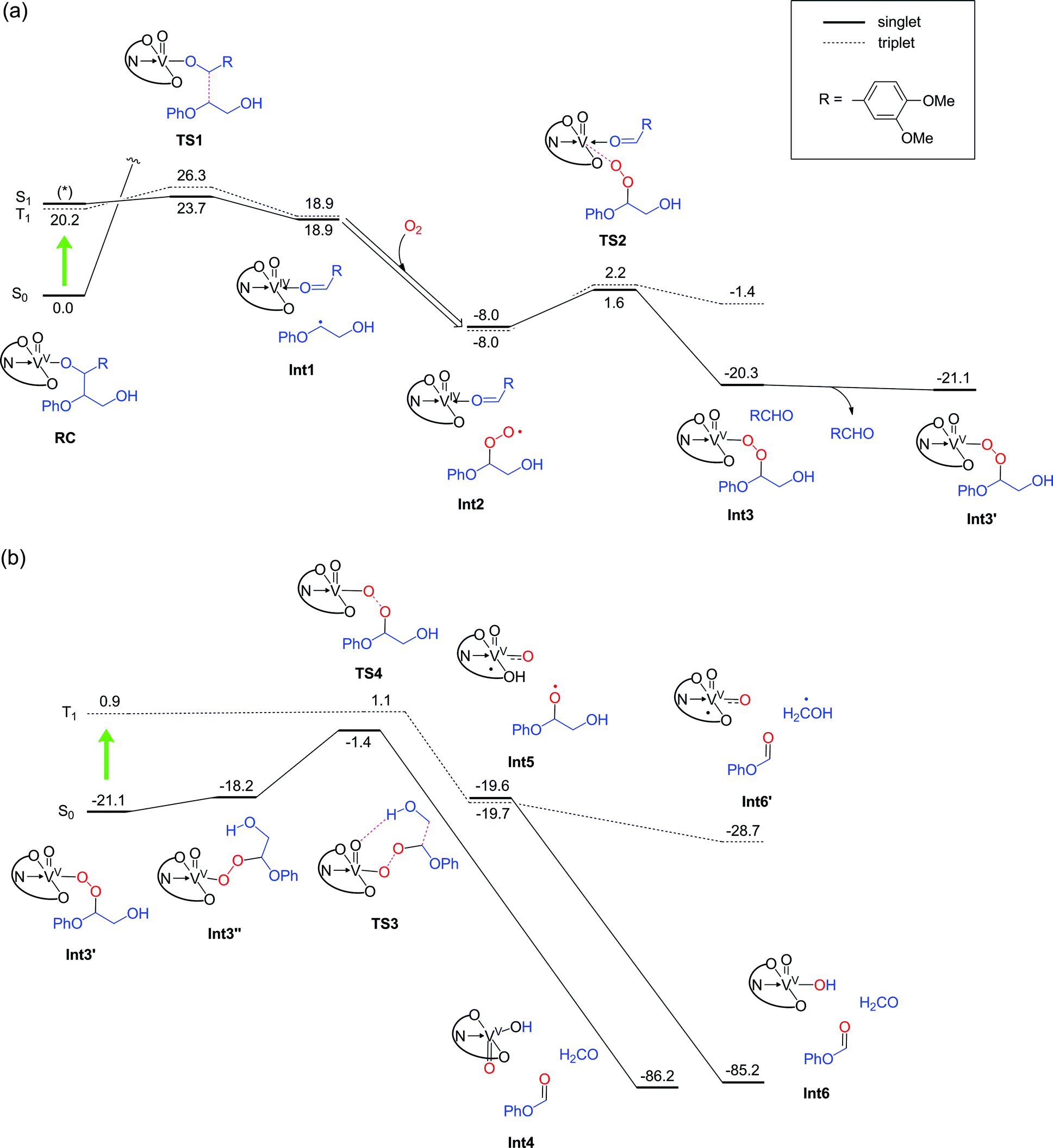 | ||
| Fig. 8 (a) Energy profile (kcal mol−1) for the first half of the reaction under aerobic conditions, determined at the B3LYP(SCRF)/6-311+G(d,p)//B3LYP/6-31G* level with zero-point energy corrections. Biradical-type singlet states were obtained by broken-symmetry (BS) calculations. BS calculations did not allow us to determine the S1 state for RC (*), because the calculation converged back to the stable S0 state. Nonetheless, as shown by the TDDFT calculations for 2, it should lie close in energy to the T1 state. A light-green arrow indicates a photoexcitation. (b) The second half of the reaction. | ||
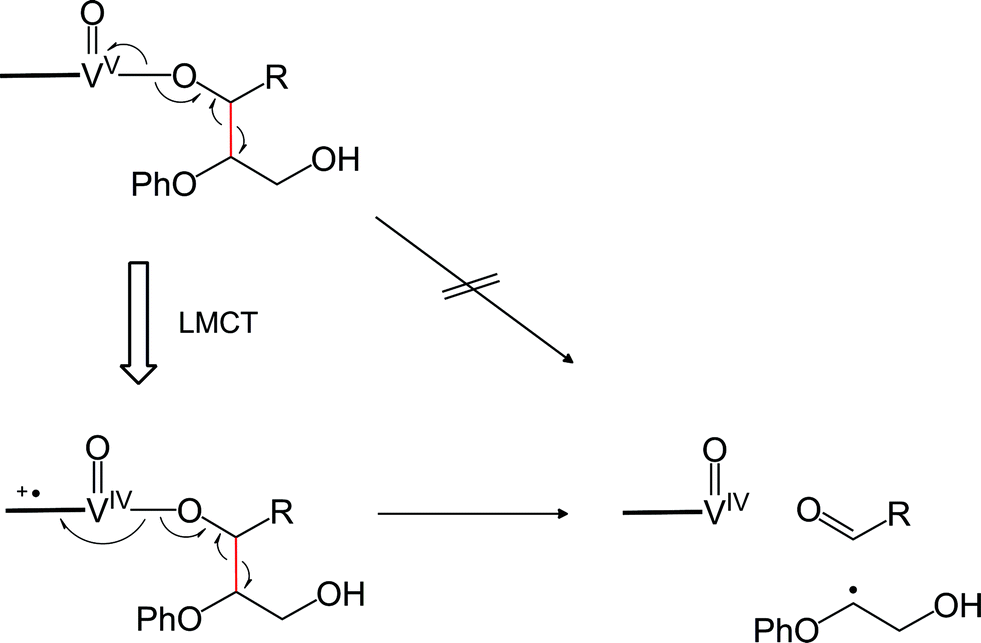 | ||
| Fig. 9 Illustration of LMCT-driven homolytic C–C bond cleavage. | ||
Alternatively, in analogy to the first step, photoexcitation of the VV peroxide may occur to generate the T1 excited state to complete the reaction. The transient peroxide in the excited state undergoes homolytic O–O bond cleavage and subsequent homolytic C–C bond cleavage in a virtually barrierless fashion, to produce 9 and formaldehyde (or a hydroxymethyl radical). Importantly, although the vanadium is temporarily reduced to VIV in the midst of the reaction under aerobic conditions (Fig. 8), it is oxidized back to the VV state, thereby permitting repeated usage of the vanadium complex for the catalytic reactions.
In the absence of air, the photocatalytic reactions can still proceed, since 2 is nominally a moderate VV oxidant. Although O2 is not present in the system, after the first C–C bond cleavage (RC to Int1 in Fig. 8a), the substrate radical can be trapped by another VV oxo to produce a VIV alkoxide complex; the barrier height for this process is only 3.2 kcal mol−1 (Fig. 10). This process will lead to consumption of 2 in the system, thus decreasing the amount of 2 that can be used for homolytic C–C bond cleavage of the substrate to produce Int1. The resultant VIV alkoxide will then exchange ligands with another molecule of 2 in the system to form a VV oxo alkoxide complex (Int3A). We found two possible pathways for photo-excitation-driven reactions of this VV complex.
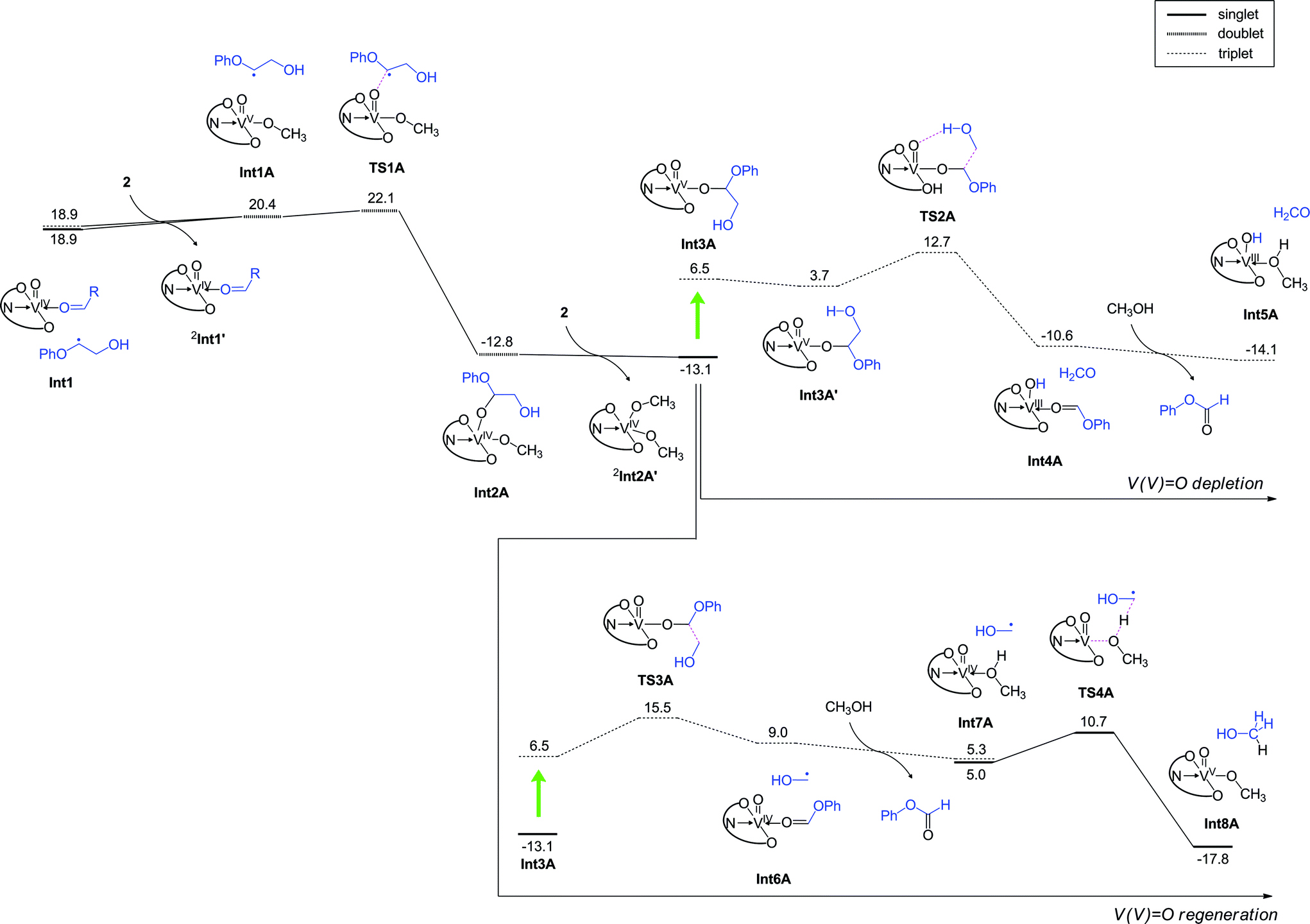 | ||
| Fig. 10 DFT calculated energy profile (kcal mol−1) for the reaction under anaerobic conditions, determined at the B3LYP(SCRF)/6-311+G(d,p)//B3LYP/6-31G* level with zero-point energy corrections. Note that the step from RC to Int1 is the same as that in the aerobic reaction (Fig. 8a). | ||
In one of the two mechanisms, homolytic C–C bond cleavage on the excited state energy surface is accompanied by hydrogen abstraction from the hydroxyl group of glycol viaTS2A, with an energy barrier of 8.9 kcal mol−1. Consequently, formate 9 and a VIII complex are generated (Fig. 10, top right). Thus, this pathway leads to further depletion of VV in the system. The other pathway proceeds with an energy barrier of 9.0 kcal mol−1 in the excited state, and a transiently formed VIV species is oxidized back to VV, thus allowing the catalytically useful VV species to be regenerated for subsequent use. Both of these processes lead to the formation of 9, but their energy barriers are somewhat higher than the barrier for LMCT-driven C–C cleavage of 11 (Fig. 8a). Therefore, after the formation of the VIV complex, a next round of the C–C cleavage of 11 should occur more favourably than these formate generation processes. A set of C–C cleavage and formation of the VIV species Int2A requires consumption of two molecules of 2, ultimately resulting in depletion of the available 2 in the system. These arguments explain why the reaction is not efficient and negligible amounts of 9 are formed under anaerobic conditions (Table 2). Our DFT studies of the aerobic and anaerobic reactions therefore suggest that the high conversion efficiency of the aerobic reaction considered here originates from the LCMT-type photoexcitation of the vanadium complex as well as the efficient regeneration of VV complexes in the presence of O2. Attempts were also made to understand the selectivity of C–C bond cleavage over C–H oxidation. Calculations were performed for the H-abstraction pathway of the ground state VV oxo proposed by Toste and coworkers.30,36 The activation barrier was calculated as 36.1 kcal mol−1 on the S0 surface (Fig. S14†). Even on the photoexcited T1 energy surface, H-abstraction from the benzylic hydrogen to the V oxo has an activation barrier of 20.2 kcal mol−1, which is much higher than the barrier for excited-state C–C bond cleavage (Fig. 8a). An alternative H-abstraction reaction from the C–H bond adjacent to the hydroxymethyl group also has a high energy barrier of 29.2 kcal mol−1 (Fig. S15†) and is endothermic. These calculations are consistent with the fact that the thermal catalytic reactions of 2 exhibit low yields, and oxidation of the benzylic alcohol of 11 is slow even at 80 °C.
Since 2 can be easily photoexcited, outer sphere mechanisms might exist whereby the substrate undergoes a C–C bond cleavage without its direct coordination to vanadium. Two outer sphere mechanisms in the T1 state were examined (Fig. S16†); however, these reactions were not as favourable as the reaction presented in Fig. 8. Hence, the reaction of the vanadium complex considered here seems to be distinct from the cases of other photosensitizers that rely on an outer sphere electron transfer mechanism.19,34,35 We also explored if an equatorial oxo arrangement is possible for the catalyst 2 or RC in Fig. 8a. However, in the former case, the axial isomer was more stable, and the equatorial isomer is possible only when vanadium has a methanol ligand. The equatorial isomer could not be located on the potential energy surface for RC without a methanol ligand. These additional calculations are presented in Fig. S17.† In the presence of substrates 13 and 15, the yields of products are diminished over the course of 24 h irradiation, but the absence of the primary alcohol in both substrates does not completely shut down the activity. As shown in Fig. S20,† the highest energy transition state over the two steps in Fig. 8a and b is lowest for 11 (6.1 kcal mol−1) compared to 13 (7.3 kcal mol−1) and 15 (8.6 kcal mol−1), lending support to a kinetic origin for the lower product yields from 13 and 15. For phenolic substrates like 31, the photocatalytic reactivity is completely absent. The greater acidity of a phenolic group allows the phenoxide to bind strongly to VV oxo. However, efficient LMCT-driven C–C cleavage, as depicted in Fig. 9, is not possible in this case. Indeed, the aliphatic C–C bond cleavage along the T1 energy surface has a high energy barrier (Fig. S18†), suggesting that for such substrates, even photoexcitation will not provide a viable route for aliphatic C–C cleavage. This result highlights the need to pre-process natural lignin19,23,25,34 to passivate all phenolic groups, before a photocatalytic approach using 2 can be applied for the depolymerization reaction. By leveraging on the lower pKa of phenols compared to aliphatic alcohols, soluble forms of lignin can be selectively esterified or alkylated by using weaker bases such as amines and carbonates. Overall, the theoretical calculations provide satisfying explanations for the selective photocatalytic reactions, which also allude to a general strategy to effect aliphatic C–C activation reactions in a broader range of organic substrates, by using dark excited states generated after photoirradiation.
Conclusions
We have prepared a photocatalyst consisting of an earth-abundant VV oxo complex supported by a redox non-innocent salicylaldimine ligand 1. A catalytic amount of 2 can effect the chemoselective, aliphatic C–C bond cleavage of representative lignin model compounds under ambient conditions with visible light (>420 nm) irradiation at moderate to high yields. The product analysis is supported by isotope labeling studies, which confirm that the lignin model compounds cleave regioselectively into two primary degradation products. The resulting aryl aldehyde and aryl formates are important building blocks in organic synthesis, since they possess highly reactive and versatile aldehyde groups. Theoretical calculations at the DFT level confirm that 2 has unusual, facile mechanistic pathways on the T1 energy surface generated only via photoexcitation, and exhibits greater selectivity for C–C bond cleavage over HAT by the VV oxo. Our fundamental study demonstrates a unique and eco-friendly approach to harvest solar energy to perform C–C activation reactions, which can potentially be employed in late-stage transformations of complex organic molecules or disassembly and valorization of (bio)macromolecules in the future.Acknowledgements
This work was supported by a NTU start-up grant (M4081012, M4080551), the Nanyang Assistant Professorship (M4081154, M4080755), an MOE Tier 1 grant (M4081144), and the Singapore-Berkeley Research Initiative for Sustainable Energy (SinBeRISE) CREATE Programme. This research programme is funded by the National Research Foundation (NRF), Prime Minister's Office, Singapore under its Campus for Research Excellence and Technological Enterprise (CREATE). W. K. H. N. and H. H. thank the High Performance Computing Centre of Nanyang Technological University for computer resources. The authors thank Dr. Subas K. Muduli for assistance with TEM measurements. We are also grateful to Dr Xiangyang Wu and Prof. Edwin Yeow for help with TCSPC experiments.Notes and references
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS PubMed.
- T. P. Yoon, M. A. Ischay and J. N. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- Z. W. Zuo, D. T. Ahneman, L. L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- H. S. Soo, A. C. Komor, A. T. Iavarone and C. J. Chang, Inorg. Chem., 2009, 48, 10024–10035 CrossRef CAS PubMed.
- H. S. Soo, M. T. Sougrati, F. Grandjean, G. J. Long and C. J. Chang, Inorg. Chim. Acta, 2011, 369, 82–91 CrossRef CAS PubMed.
- S. K. Muduli, S. L. Wang, S. Chen, C. F. Ng, C. H. A. Huan, T. C. Sum and H. S. Soo, Beilstein J. Nanotechnol., 2014, 5, 517–523 CrossRef CAS PubMed.
- H. S. Soo, A. Agiral, A. Bachmeier and H. Frei, J. Am. Chem. Soc., 2012, 134, 17104–17116 CrossRef CAS PubMed.
- A. Agiral, H. S. Soo and H. Frei, Chem. Mater., 2013, 25, 2264–2273 CrossRef CAS.
- M. L. Macnaughtan, H. S. Soo and H. Frei, J. Phys. Chem. C, 2014, 118, 7874–7885 CAS.
- H. S. Soo, M. L. Macnaughtan, W. W. Weare, J. Yano and H. Frei, J. Phys. Chem. C, 2011, 115, 24893–24905 CAS.
- C. O. Tuck, E. Perez, I. T. Horvath, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. Jongerius and B. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 709 CrossRef CAS PubMed.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- P. C. Bruijnincx and B. M. Weckhuysen, Nat. Chem., 2014, 6, 1035–1036 CrossRef CAS PubMed.
- J. D. Nguyen, B. S. Matsuura and C. R. Stephenson, J. Am. Chem. Soc., 2014, 136, 1218–1221 CrossRef CAS PubMed.
- H. Michel, Angew. Chem., Int. Ed., 2012, 51, 2516–2518 CrossRef CAS PubMed.
- K. Barta and P. C. Ford, Acc. Chem. Res., 2014, 47, 1503–1512 CrossRef CAS PubMed.
- D. Mohan, C. U. Pittman and P. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef CAS PubMed.
- J. Nichols, L. Bishop, R. Bergman and J. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS PubMed.
- T. H. Vom Stein, T. Den Hartog, J. Buendia, S. Stoychev, J. Mottweiler, C. Bolm, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2015, 54, 5859–5863 CrossRef CAS PubMed.
- S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott and D. L. Thorn, Inorg. Chem., 2010, 49, 5611–5618 CrossRef CAS PubMed.
- S. K. Hanson, R. L. Wu and L. A. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410–3413 CrossRef CAS PubMed.
- S. Son and F. D. Toste, Angew. Chem., Int. Ed., 2010, 49, 3791–3794 CrossRef CAS PubMed.
- G. Zhang, B. L. Scott, R. Wu, L. A. Silks and S. K. Hanson, Inorg. Chem., 2012, 51, 7354–7361 CrossRef CAS PubMed.
- B. Sedai, C. Diaz-Urrutia, R. T. Baker, R. L. Wu, L. A. Silks and S. K. Hanson, ACS Catal., 2013, 3, 3111–3122 CrossRef CAS.
- B. Sedai, C. Diaz-Urrutia, R. T. Baker, R. L. Wu, L. A. Silks and S. K. Hanson, ACS Catal., 2011, 1, 794–804 CrossRef CAS.
- D. W. Cho, R. Parthasarathi, A. S. Pimentel, G. D. Maestas, H. J. Park, U. C. Yoon, D. Dunaway-Mariano, S. Gnanakaran, P. Langan and P. S. Mariano, J. Org. Chem., 2010, 75, 6549–6562 CrossRef CAS PubMed.
- S. Lim, K. Nahm, C. Ra, D. Cho, U. Yoon, J. Latham, D. Dunaway Mariano and P. Mariano, J. Org. Chem., 2013, 78, 9431–9443 CrossRef CAS PubMed.
- J. M. W. Chan, S. Bauer, H. Sorek, S. Sreekumar, K. Wang and F. D. Toste, ACS Catal., 2013, 3, 1369–1377 CrossRef CAS.
- M. E. Brown and M. C. Y. Chang, Curr. Opin. Chem. Biol., 2014, 19, 1–7 CrossRef CAS PubMed.
- L. Wang, W. Qin, X. Tang, W. Dou and W. Liu, J. Phys. Chem. A, 2011, 115, 1609–1616 CrossRef CAS PubMed.
- T. K. Kirk and R. L. Farrell, Annu. Rev. Microbiol., 1987, 41, 465–505 CrossRef CAS PubMed.
- P. Pullanikat, J. H. Lee, K. S. Yoo and K. W. Jung, Tetrahedron Lett., 2013, 54, 4463–4466 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2009.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- D. Jacquemin, E. A. Perpete, G. E. Scuseria, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2008, 4, 123–135 CrossRef CAS.
- E. A. B. Kantchev, T. B. Norsten and M. B. Sullivan, Org. Biomol. Chem., 2012, 10, 6682–6692 CAS.
- R. F. Li, J. J. Zheng and D. G. Truhlar, Phys. Chem. Chem. Phys., 2010, 12, 12697–12701 RSC.
- J. J. De Voss and M. J. Cryle, in The Ubiquitous Roles of Cytochrome P450 Proteins, John Wiley & Sons, Ltd., 2007, pp. 397–435, DOI:10.1002/9780470028155.ch13.
- J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterization, photoluminescence and TCSPC, TEM, as well as DFT calculations. CCDC 1058817–1058819. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02923f |
| This journal is © The Royal Society of Chemistry 2015 |