
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNaphthalenebisimides as photofunctional surfactants for SWCNTs – towards water-soluble electron donor–acceptor hybrids†
Konstantin
Dirian‡
a,
Susanne
Backes‡
a,
Claudia
Backes
b,
Volker
Strauss
a,
Fabian
Rodler
a,
Frank
Hauke
a,
Andreas
Hirsch
*a and
Dirk M.
Guldi
*a
aDepartment of Chemistry and Pharmacy and Interdisciplinary Center for Molecular Materials, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91058 Erlangen, Germany. E-mail: andreas.hirsch@fau.de; dirk.guldi@fau.de
bSchool of Physics, Trinity College Dublin, Dublin 2, Ireland
First published on 22nd September 2015
Abstract
A water soluble naphthalenebisimide derivative (NBI) was synthesized and probed to individualize, suspend, and stabilize single wall carbon nanotubes (SWCNTs). Besides a comprehensive photophysical and electrochemical characterization of NBI, stable suspensions of SWCNTs were realized in buffered D2O. Overall, the dispersion efficiency of the NBI surfactant was determined by comparison with naphthalene based references. Successful individualization of SWCNTs was corroborated in several microscopic assays. In addition, emission spectroscopy points to the strong quenching of SWCNT centered band gap emission, when NBIs are immobilized onto SWCNTs. The origin of the quenching was found to be strong electronic communication, which leads to charge separation between NBIs and photoexcited SWCNTs, and, which yields reduced NBIs as well oxidized SWCNTs. Notably, electrochemical considerations revealed that the energy content of these charge separated states is one of the highest reported for SWCNT based electron donor–acceptor hybrids so far.
Introduction
Ever since their discovery and large scale production in the 1990's,1 single wall carbon nanotubes (SWCNTs) have emerged as benchmark materials in the fields of materials science,2 molecular electronics,3 catalysis,4 and solar energy conversion.5 High performance SWCNT materials require effort to process, to individualize, and to stabilize them, for example, in the form of suspensions. To date, the aforementioned aspects still remain a challenge, since SWCNTs tend to form bundles/ropes due to strong van der Waals interactions.6 One way to overcome this intrinsic feature requires the covalent attachment of solubilizing and/or individualizing groups by means of chemical functionalization. Covalent functionalization leads, however, to the transformation of sp2 into sp3 hybridized carbons and, in turn, to a notable perturbation of their electronic structure.7 An alternative means to ensure processing, individualization, and stabilization, on the one hand, and preserving the intrinsic properties of SWCNTs, on the other hand, is non-covalent functionalization. In this context, a class of promising materials are amphiphiles such as ionic and non-ionic surfactants.8A careful tuning and/or modification of these amphiphilic building blocks can be achieved through targeting the implementation of additional π-systems, electron donors, electron acceptors, etc. A common denominator of the aforementioned is the gain of control over interactions with SWCNTs. For example, considering the extended π-system of SWCNTs and their p-type semiconducting nature, an amphiphile featuring an extended, electron accepting π-system should enhance/maximize the stability of SWCNT suspensions. Organic rylene derivatives are promising candidates owing to their susceptibility in terms of charge transfer interactions.9 As a matter of fact, perylenebisimides (PBIs) have been successfully utilized towards the realization of SWCNT/PBI based electron donor–acceptor hybrids, for which experimental data corroborates even in the dark, that is, in the ground state, a redistribution of the electron density which evokes the partial oxidation of SWCNTs and reduction of PBIs.10
Contrary to PBIs, naphthalenebisimides (NBIs) as the smallest analogue of the rylene family, have rarely been integrated into organic, functional hybrids based on SWCNTs.11 Despite their smaller size, interesting features of NBIs such as being an n-type semiconductor and exhibiting high π-acidity and chemical versatility renders them promising candidates for novel photoactive SWCNT based hybrids, yielding oxidized SWCNTs as well as reduced NBIs upon illumination or even in the dark.12 Some of these properties have been proven upon implementing NBIs as electron acceptors in photoactive electron donor–acceptor hybrids,13 anion transport14 as well as electron conducting channels15 and layers or even in π-catalysis.16
In this contribution, a water soluble polyanionic NBI (1) was used to prepare photoactive HiPco-SWCNT hybrids in aqueous media. Synergetic effects stemming from hydrophobic/hydrophilic, π–π, and charge transfer interactions, which are operative between 1 and HiPco-SWCNTs, are the basis for individualization of the HiPco-SWCNTs and stabilization of the corresponding HiPco-SWCNT/1 suspensions.17
Results and discussion
The synthesis of 1 and 1a is schematically shown in Scheme 1 and the details are provided in the ESI.†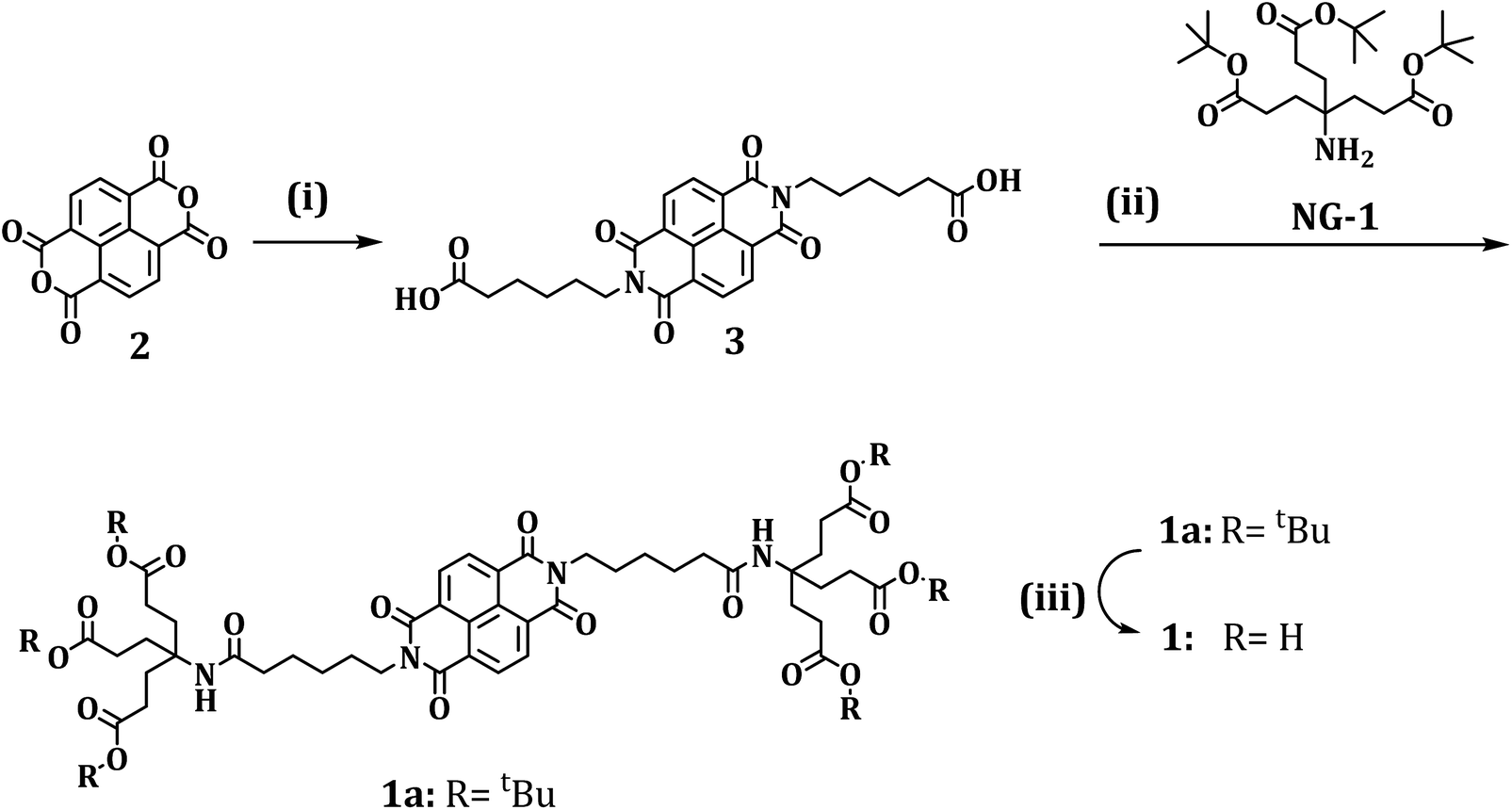 | ||
Scheme 1 Synthesis of NBI 1 (i) 6-aminocapronic acid, toluene/ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 100 °C, o.n.; (ii) 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methoxymorpholinium chloride, THF, NG-1, 5 h; (iii) HCOOH, r.t., 12 h. :1), 100 °C, o.n.; (ii) 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methoxymorpholinium chloride, THF, NG-1, 5 h; (iii) HCOOH, r.t., 12 h. | ||
The first part of our investigation is concerned with the photo- and redoxchemical properties of 1 and its tert-butyl protected analogue 1a as a function of state, that is, monomer versus aggregate, in solution (Fig. 1). Typically, rylenes are prone to aggregation in solution, which has let us to investigate the aggregation of 1 in D2O. In the context of 1, information about its ordering in solution has been gathered in concentration dependent steady state absorption and emission assays.
 | ||
| Fig. 1 Absorption spectrum (black) and fluorescence spectrum (red) of 1 (7.25 × 10−6 M) in D2O/H2PO4−/HPO42− upon 325 nm excitation. | ||
Fig. 1 illustrates that the absorption spectrum of 1 displays – in sound agreement with previous reports – well resolved maxima at 363 and 383 nm and shoulders at 235 and 344 nm with extinction coefficients in the range from 10000 M−1 cm−1 to 20000 M−1 cm−1.12a Interestingly, these features are concentration independent. The latter finding is, however, in stark contrast to investigations with water soluble, H-type aggregate forming perylene bisimides – see Fig. S1.†18 In addition, 325 nm excitation of 1 resulted in two fluorescence maxima at 395 and 413 nm and in low fluorescence quantum yields around 10−4 M.19 Contrary to the ground state absorption, the fluorescence features of 1 give rise to a concentration dependence. At concentrations of less than 10−5 M the fluorescence appears as mirror image of the ground state absorption, which is shown in Fig. 1.
At concentrations of 5 × 10−5 M and higher the long wavelength fluorescence at 413 nm dominates the fluorescence spectrum – Fig. S2.† In the excitation spectra, a good agreement with the absorption features was established, as shown in Fig. S3.† A partial contribution from inner filter effects cannot be ruled out. In additional experiments we probed the influence of ionic strength through the addition of sodium chloride as well as the addition of the non-ionic surfactant Triton X. In line with the aforementioned assays, which focused on concentration dependencies, no appreciable alterations of the spectral shape were noted.18
The energy of the singlet excited state was determined based on the long wavelength absorption and the short wavelength fluorescence maxima as 3.14 eV with a Stokes shift of 0.09 eV. Owing to the well resolved vibronic fine structure in the absorption and fluorescence as well as the small Stokes shift, 1 is unlikely to form H-type aggregates. Instead, J-type aggregates of 1 or even its monomers – both giving rise to an intrinsically low fluorescence quantum yield – have to be taken into consideration. Notably, the weak fluorescence properties of non-core substituted NBIs have recently been reported.20 Unfortunately, fluorescence lifetime measurements by means of single photon counting are rendered impossible due to its low intensity.
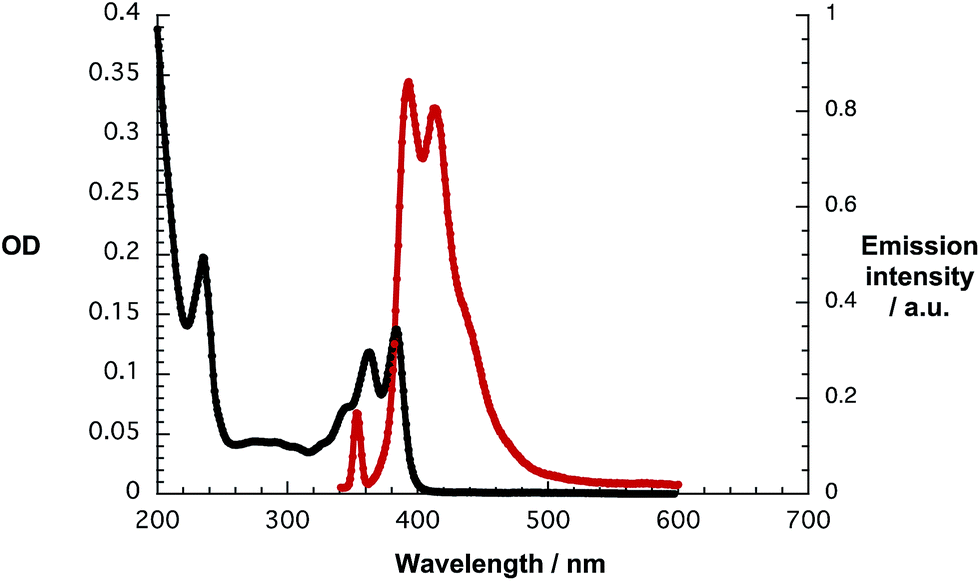
Contrary to the fluorescence, phosphorescence features, which are known to center at 605/665 nm (2.05 eV),21 could not be detected at room temperature during the course of this study. However, upon purging solutions of 1 in D2O/H2PO4−/HPO42− with oxygen the 1O2 fluorescence was discernable at 1270 nm – as shown in Fig. 2. With solutions of C60 in toluene as standard, the triplet quantum yield of 1 was determined to be around 5%.22
 | ||
| Fig. 2 1O2 fluorescence spectra of 1 in D2O/H2PO4−/HPO42− (red) with an OD of 0.1 and D2O/H2PO4−/HPO42− (black) upon 360 nm excitation. | ||
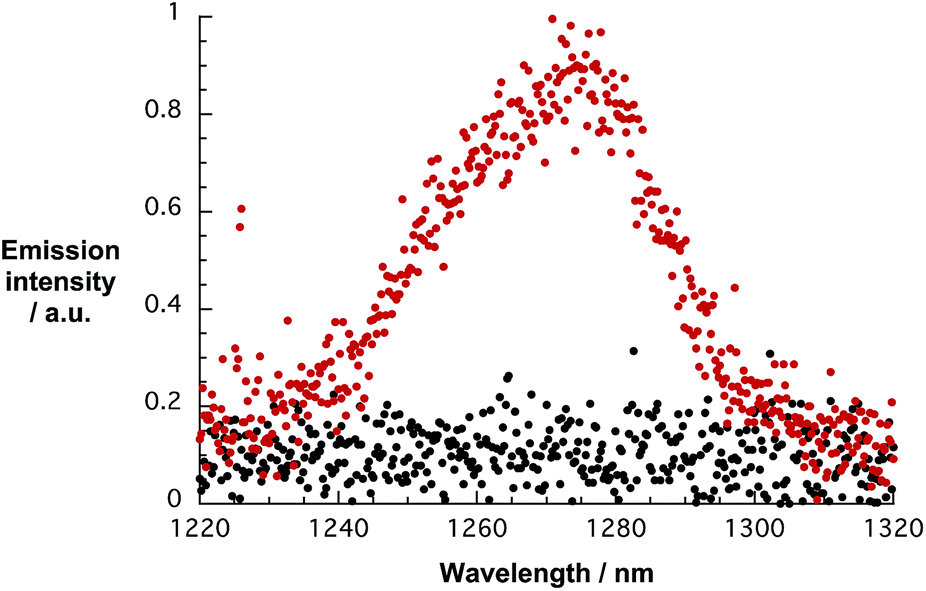
Owing to the electron accepting properties of 1,13a,c we turned to electrochemical and spectroelectrochemical investigations to determine the reduction potentials and the corresponding fingerprints of the one electron reduced form of 1, respectively. Cyclic voltammetric (CV) and square wave voltammetric (SWV) techniques were used to evaluate the reductions of 1 in D2O/H2PO4−/HPO42− and in DMSO. In D2O/H2PO4−/HPO42−, a single, particularly irreversible reduction was noted at around −0.6 V vs. K3[Fe(CN)6]/K4[Fe(CN)6] as shown in Fig. 3. In DMSO, two, reversible reductions were observed at −1.1 and −1.5 V vs. Fc/Fc+ in TBAPF6,15a,23 – Fig. S4.† Importantly, variable scan rates from 25 to 100 mV s−1 lead to identical results – Fig. S4.† In terms of oxidation, no processes were discernible up to 0.8 V in D2O/H2PO4−/HPO42− and up to 1.3 V in DMSO – both vs. Ag-wire.
 | ||
| Fig. 3 Top: CVs of 5 × 10−4 M 1 in D2O/H2PO4−/HPO42− (black) and of D2O/H2PO4−/HPO42− (red) obtained at a 50 mV s−1 scan rate. Bottom: differential absorption spectrum of 1 in D2O/H2PO4−/HPO42− obtained upon electrochemical reduction with an applied potential of −800 mV vs. Ag-wire. | ||
In DMSO, on the one hand, application of −0.3 V vs. Ag-wire results in bleaching of the ground state absorption at around 350 nm and in formation of 476 and 607 nm maxima – Fig. S5.† In D2O/H2PO4−/HPO42−, on the other hand, electrochemical reduction of 1 at −0.8 V vs. Ag-wire is seen to lead to the formation of new maxima in the 400 to 700 nm range, which appear broadened when compared to the DMSO case. An additional feature, which maximizes at 1120 nm, has recently been assigned to arise from π-stacks following reduction – as shown in Fig. 3.24 As Fig. S6† documents, these features are reversible throughout at least 3 electrochemical cycles.
To gather further insight into the excited state dynamics of 1, femtosecond pump probe experiments were performed by exciting 1 in D2O/H2PO4−/HPO42− at 387 nm. The differential absorption spectra of 1 lack any noticeable transient features in the 400 to 1200 nm range, which may be assigned to any singlet excited states, on the femto- and picosecond time scales. When turning, however, to the nano- and microsecond time scales, a transient maximum is noted at 480 nm in stirred and argon purged solutions. Since the corresponding absorption-time profiles reveal decays on the microsecond time range, we tentatively assign the noted transient maximum to the triplet excited state – as shown in Fig. 4. Confirmation for this hypothesis came from probing its oxygen sensitivity. Taking, for example, the 483 nm time absorption profile, a lifetime of 18 μs in argon saturated solution was deduced. Probing oxygen purged solutions resulted in a sufficient quenching of photoexcited 1. Now, the lifetime is 680 ns as determined from the 416 nm kinetics.
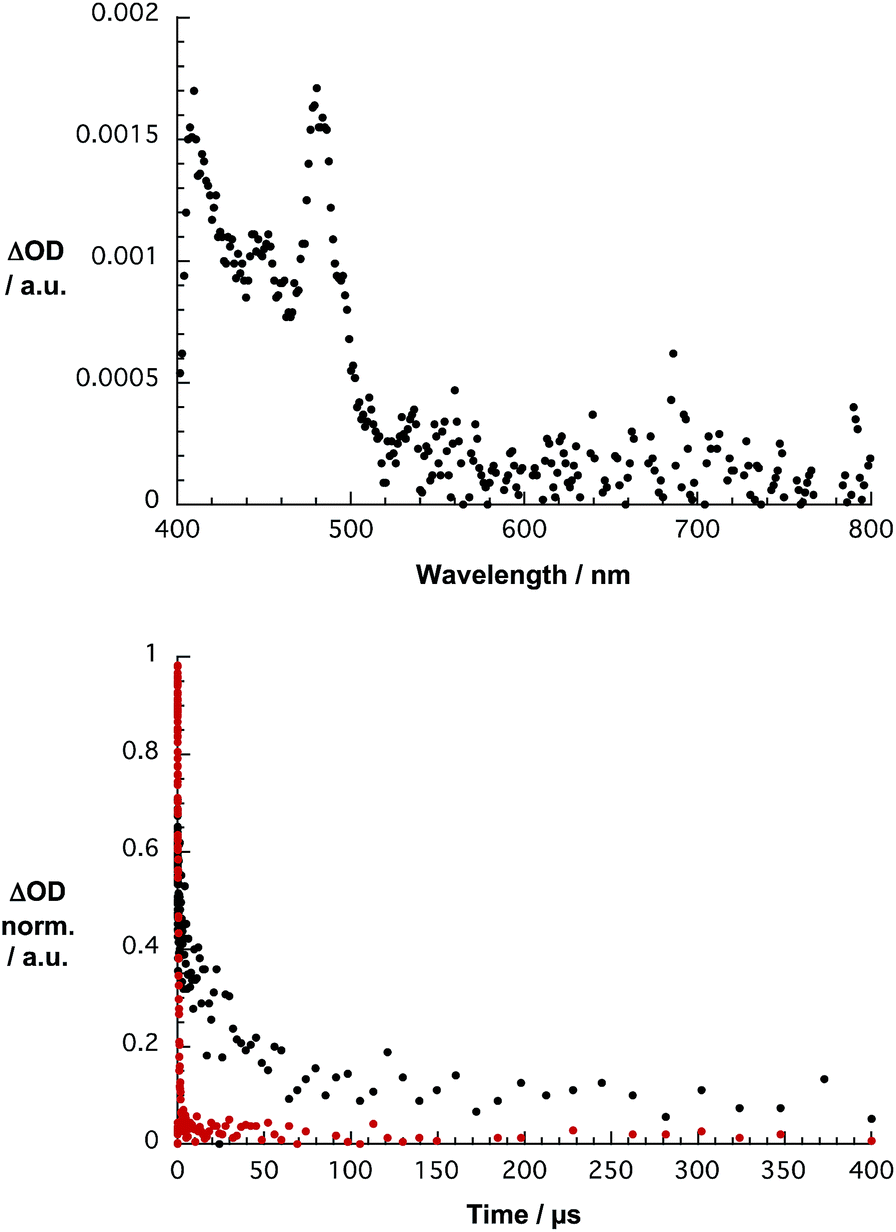 | ||
| Fig. 4 Top: differential absorption spectrum of 1 in stirred D2O/H2PO4−/HPO42− obtained upon femtosecond flash photolysis (387 nm) with a time delay of 250 ns. Bottom: time-absorption profile at 483 nm monitoring in the absence (black) and the presence (red) of molecular oxygen the decay of the triplet excited states. | ||
Reference experiments with 1a in THF produced a rather different picture – Fig. S7.† In particular, a strong transient signature evolves for 1a at 597 nm within the first few picoseconds following 387 nm excitation. These features transform within less than 15 ps into transient signatures at 450 and 480 nm, which are stable on the 7 ns timescale. Similar transient features have been reported in acetonitrile and assigned to the triplet excited states.25 The triplet nature of these excited states was proven by means of quenching experiments with molecular oxygen on the nano- and microsecond time scales. In the absence of molecular oxygen, a monoexponential decay of, for example, the 482 nm kinetics led to a lifetime of 8.4 μs. In the presence of oxygen, the lifetime is only 152 ns. The corresponding spectra are gathered in Fig. S8.† Interestingly, when probing a non-stirred solution of 1 in D2O/H2PO4−/HPO42− the differential absorption spectra with a transient maximum centered at 420 nm and the dynamics with a lifetime of 200 μs – Fig. S9† – differ. A likely rationale is based on a photoreaction with the triplet excited state of 1.
Following the characterization of 1 and 1a, the focus was placed on probing SWCNTs. For the immobilization of 1, HiPco-SWCNTs were utilized. To study the electronic and photophysical properties of SWCNTs with different chiralities and different band gaps, HiPco-SWCNTs were dispersed in D2O via surfactant-stabilization with sodium dodecylbenzenesulfonate (SDBS).
The absorption spectrum is best described as the superimposition of several maxima reaching from the visible to the near-infrared – vide infra (Fig. 8, top). As such, three sets of absorption features are assignable to M11 transitions – at wavelengths shorter than 500 nm – to S22 transitions – dominating the 500 to 700 nm range – and to S11 excitonic transitions – at wavelengths larger than 1000 nm. Assignment of these states to the corresponding SWCNT chiralities is provided in Table S1.† Upon photoexcitation in the visible, distinct fluorescence maxima of the S11 transitions of different nanotube species were detected in the near-infrared – Fig. S10.† These findings are well documented and rationalized on the basis of individualized and stabilized SWCNTs, which are, nevertheless, polydisperse.26
Besides steady-state absorption and fluorescence, Raman spectroscopy is another well established tool to characterize SWCNT dispersions and hybrids.27
For HiPco-SWCNTs solubilized by means of SDBS, Raman analysis upon 1064 nm excitation of the three most important signatures, namely radial breathing modes (RBM), G-, and 2D-modes, was performed and the results were compared to those obtained for solid HiPco-SWCNTs. Importantly, all of the SWCNT modes appear better resolved for HiPco-SWCNT/SDBS. Taking the aforementioned into consideration, we conclude the individualization of HiPco-SWCNTs.28 The absence of notable electronic communication and/or doping is inferred from the absence of notable shifts in the RBM, G-, and 2D-modes – Table S2 and Fig. S11.†
Next, the redox properties of HiPco-SWCNT/SDBS were tested utilizing absorption spectroelectrochemistry. Throughout several electrochemical scans, which were performed in the range from −0.8 to +0.6 V vs. Ag-wire, a quasi-reversible behavior is derived for HiPco-SWCNT/SDBS from the absorption vs. potential dependences – as shown in Fig. 5. Fig. S12† presents the results under oxidative conditions and the resulting potential vs. differential absorption profiles.
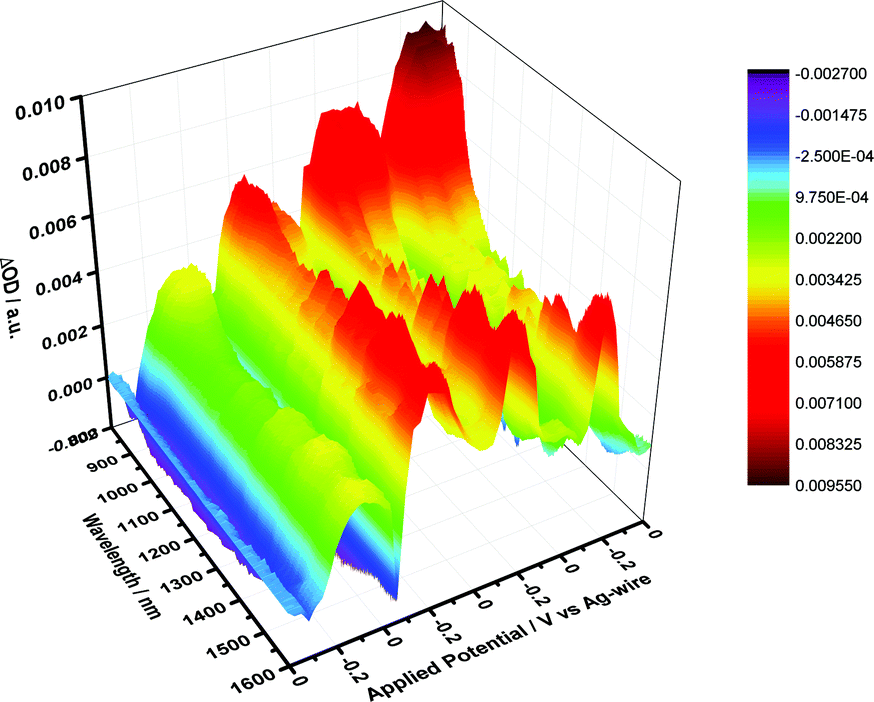 | ||
| Fig. 5 Differential absorption spectra of HiPco-SWCNT/SDBS in D2O/0.05 M NaCl obtained during four electrochemical cycles, that is, from 0 to −800 to +600 mV and back to 0 in 200 mV intervals. | ||
Finally, the excited states of HiPco-SWCNT/SDBS were probed upon 387 nm laser excitation. Immediately after laser excitation, several maxima and minima evolve throughout the visible and near infrared regions. Most prominent is the depopulation of the SWCNT ground state in the form of minima at 1265, 1324, 1378, and 1418 nm as mirror images of the corresponding ground state absorption – S11 – as shown in Fig. 6. From careful kinetic analyses at different wavelengths, a biexponential recovery of the ground state was derived – a short lived, 1 ps component and a longer lived, 50 ps component. Since excitation at 387 nm leads to the population of S22 excitonic states, we assign the short lived component to interband relaxation, whereas the long lived component relates to an excitonic recombination in individualized SWCNTs.29
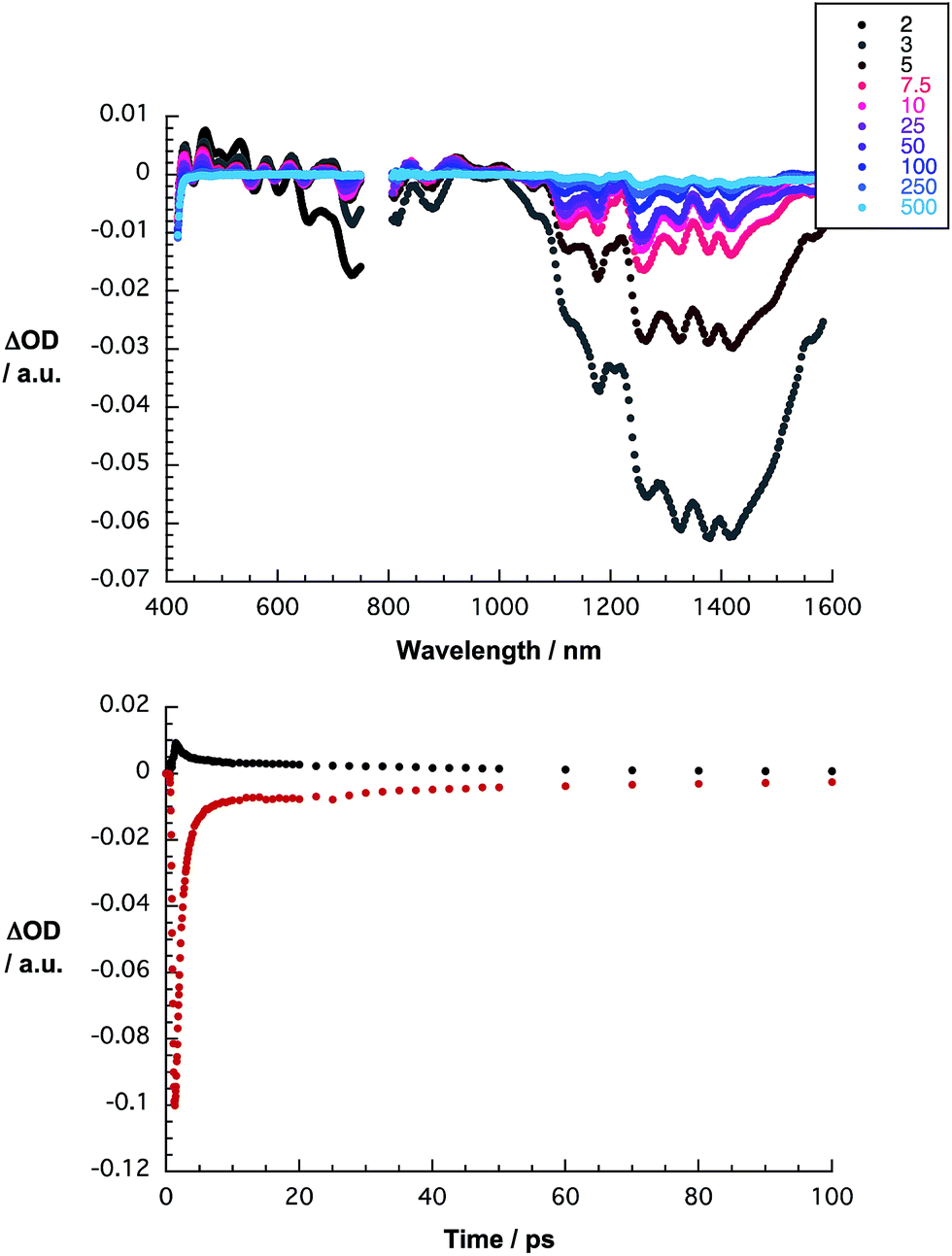 | ||
| Fig. 6 Top: differential absorption spectra of HiPco-SWCNT/SDBS in D2O obtained upon femtosecond flash photolysis (387 nm) with time delays between 2 (black) and 500 ps (blue). Bottom: time absorption profiles at 470 nm (black), and 1323 nm (red), monitoring the excited state decay. | ||
Following the characterization of 1, 1a, and well debundled HiPco-SWCNT, we started optimizing the conditions to individualize and to disperse HiPco-SWCNT in the form of HiPco-SWCNT/1 rather than SWCNT/SDBS. To shed light on the dispersion efficiency of 1, amphiphilic NBI 4 as well as the corresponding naphthalenemonoimide (NMI, 5) were probed for comparison – as shown in Fig. 7. Synthesis details are given in the ESI.†
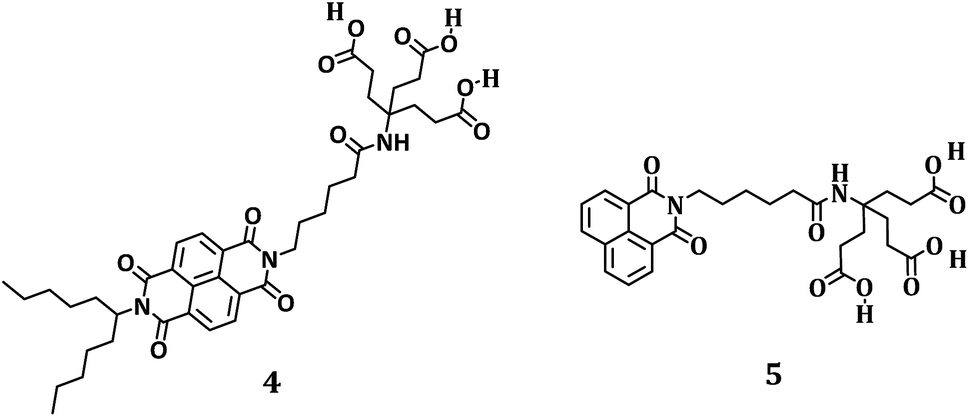 | ||
| Fig. 7 Structures of 4 and 5. | ||
In perfect consistency with our previous studies on PBI, we find the dispersion efficiency to be significantly higher in buffered systems.30 This is attributed to a higher packing density of the dyes on the nanotube scaffolds due to charge screening by the counterions.30
In the following, we have therefore focused on the investigation of 1, 4 and 5 in buffered media, which are either neutral (pH 7, D2O/H2PO4−/HPO42−) or alkaline (pH 10, H2O). Notably, decomposition of 1 caused by sonication is ruled out based on NMR data – Fig. S13.† Stable hybrid materials were, however, obtained only with HiPco-SWCNTs, since attempts to create stable hybrids with CoMoCAT-SWCNTs failed.
A likely rationale is the imbalance between the bulky dendron and the small π skeleton of 1, reducing π–π interactions between 1 and smaller diameter SWCNTs. Upon comparing the absorption spectra of 1 and HiPco-SWCNT/1 no appreciable shifts in, for example, the visible range, where NBI centered features dominate, are noted – as shown in Fig. 8. In the near infrared, a region where SWCNT centered transitions are expected, those for HiPco-SWCNT/1 are strongly broadened and red shifted relative to HiPco-SWCNT/SDBS. The resulting maxima at 1171, 1317, and 1425 nm are assigned to the S11 transitions (8,6), (9,4), (8,7), (7,6), and (11,1) of HiPco-SWCNT, which are broadened and superimposed as a consequence of the interactions with 1.
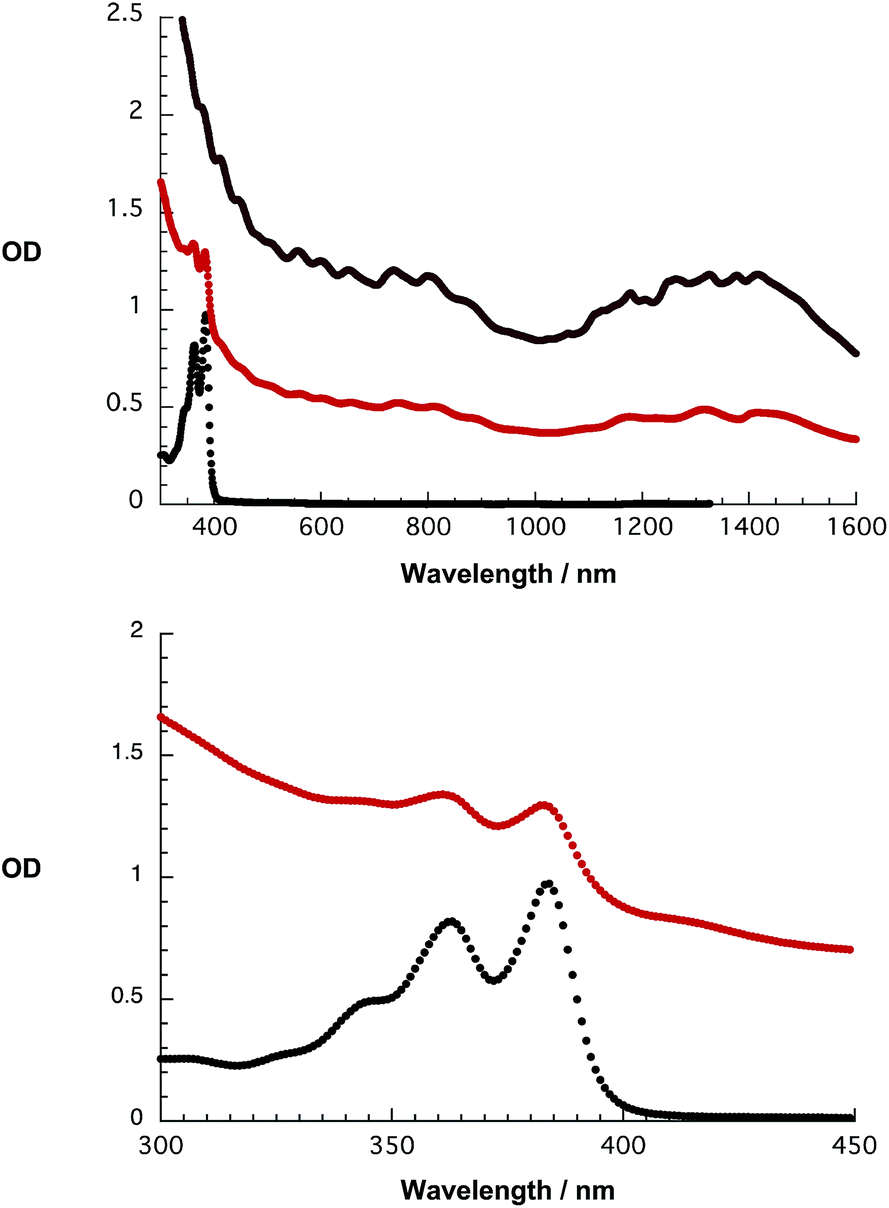 | ||
| Fig. 8 Top: absorption spectra of 1 (black), HiPco-SWCNT/1 (red) and HiPco-SWCNT/SDBS (grey) recorded in D2O/H2PO4−/HPO42−. Bottom: a zoom of the 300–450 nm range with focus on the absorption features of 1 and HiPco-SWCNT/1. | ||
In a complementary approach, the dispersion efficiency of 1 was correlated with that of 4 and 5, respectively. At pH 10, dispersion efficiencies are derived from the SWCNT concentration in the stable supernatant dispersion – based on the extinction coefficient previously determined31 – and the initial SWCNT concentrations. Both 1 and 4 are potent surfactants yielding dispersions, in which typically 60% of all SWCNTs are effectively dispersed. The related naphthalene monoimide 5 exhibits a significantly lower dispersion efficiency of only 5% as shown in Fig. S14.† As a complement to the near infrared absorptions, the near infrared fluorescence of HiPco-SWCNT/1 was investigated. All throughout the excitation range, that is, from 550 to 775 nm, no appreciable SWCNT related band gap emission evolves. This is consistent with our previous data on perylene bisimide surfactants and documents that the SWCNT fluorescence is quenched and red-shifted when dispersed with 1 and 4.17,30,32 This is, nevertheless, in stark contrast to perylene imidodiesters, for which the SWCNT fluorescence is not subject to any appreciable quenching.33 An illustration for HiPco-SWCNT/SDBS and HiPco-SWCNT/1 without and with additional SDBS is shown in Fig. 9, which documents the overall quenching.
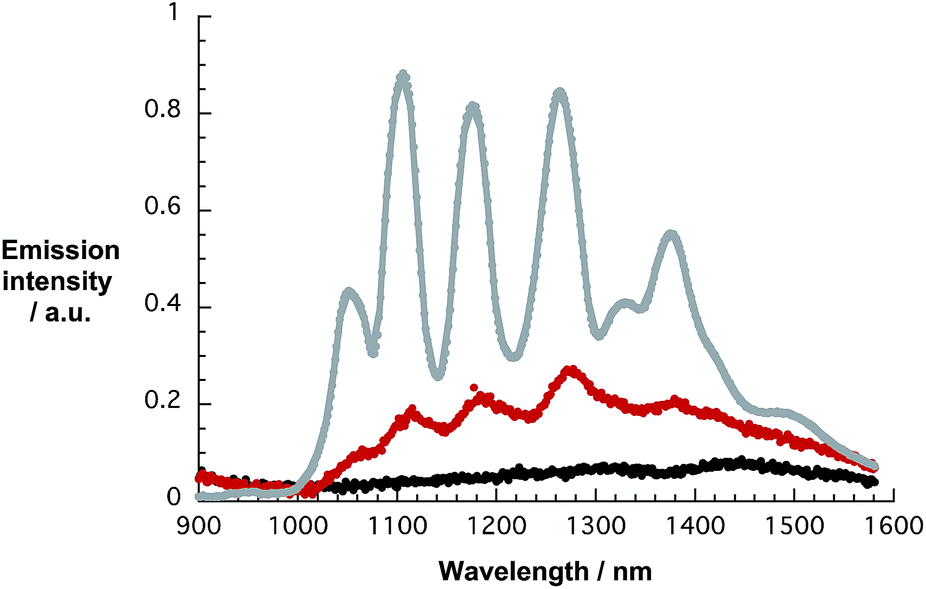 | ||
| Fig. 9 Fluorescence spectra, with an equal optical density of about 0.4 at the excitation wavelength (724 nm), of HiPco-SWCNT/SDBS (grey), HiPco-SWCNT/1 (black), and HiPco-SWCNT/1 after addition of SDBS (red) recorded in D2O/H2PO4−/HPO42−. The black and red spectra have been amplified by a factor of 5. | ||
Considering that both effects, namely broadened/red shifted absorption features, on the one hand, and strongly quenched fluorescence, on the other hand, may be caused by insufficient debundling34 and/or strong electronic communication10a between HiPco-SWCNT and 1, transmission electron microscopy (TEM) was used to probe the degree of stable individualization of HiPco-SWCNT/1. To this end, HiPco-SWCNT/1 was applied on an ultrathin carbon film covered TEM grid and scanned for individual and bundled objects. The majority of the SWCNTs were found to be individualized throughout the entire specimen – as shown in Fig. 10. To probe the morphology of HiPco-SWCNT/1, a highly concentrated dispersion of HiPco-SWCNT/1 was applied on a lacey carbon film and scanned for freestanding SWCNT networks. As shown in Fig. 10 and S15,† the freestanding individual SWCNTs feature an amorphous coating that is typical for functionalized or wrapped SWCNTs.
 | ||
| Fig. 10 Top: TEM images (80 kV) of freestanding HiPco-SWCNT/1 (left) and on ultrathin carbon film (right) processed from D2O/H2PO4−/HPO42−. Bottom: AFM image of HiPco-SWCNT/1 processed from H2O/pH 10. | ||
Simultaneously, we performed statistical atomic force microscopy (AFM) analysis of HiPco-SWCNT/1 alkaline dispersions to corroborate our TEM assays. As shown in Fig. 10, the SWCNTs are predominantly individualized. Considering that all of the one-dimensional objects with AFM heights <1.5 nm relate to individualized SWCNTs, the degree of individualization is >65%. Notably, this is significantly higher than what is typically found when employing SDBS.17b,31 From the latter we conclude that the reduced fluorescence intensities in the HiPco-SWCNT/1 dispersions are due to intermolecular interactions rather than a lack of efficient individualization. The aforementioned assays corroborate the HiPco-SWCNT exfoliation and, in turn, aggregation induced quenching of the fluorescence is ruled out. Consequently, strong electronic communication between HiPco-SWCNT and 1 in the form of, for example, charge transfer, is likely to be in operation.
Further support for this notion came from Raman spectroscopy. Table S2,† which lists Raman data for pristine HiPco-SWCNT, HiPco-SWCNT/SDBS, and HiPco-SWCNT/1 taken on alumina substrates, reveal notable differences. Firstly, two Lorentzian functions are required to appropriately fit the G-mode in HiPco-SWCNT/1. Secondly, the 2D mode – Fig. 11 – of HiPco-SWCNT/1 is shifted to 2553 ± 2 cm−1 compared to 2550 cm−1 for HiPco-SWCNT/SDBS, a finding that infers, for example, a shift of electron density from the electron donating HiPco-SWCNT to the electron accepting 1, which is in support of charge transfer interactions. For a more comprehensive understanding, Raman spectra, including RBM analysis, of pristine HiPco-SWCNT, HiPco-SWCNT/SDBS and HiPco-SWCNT/1 are shown in Fig. S16.†35
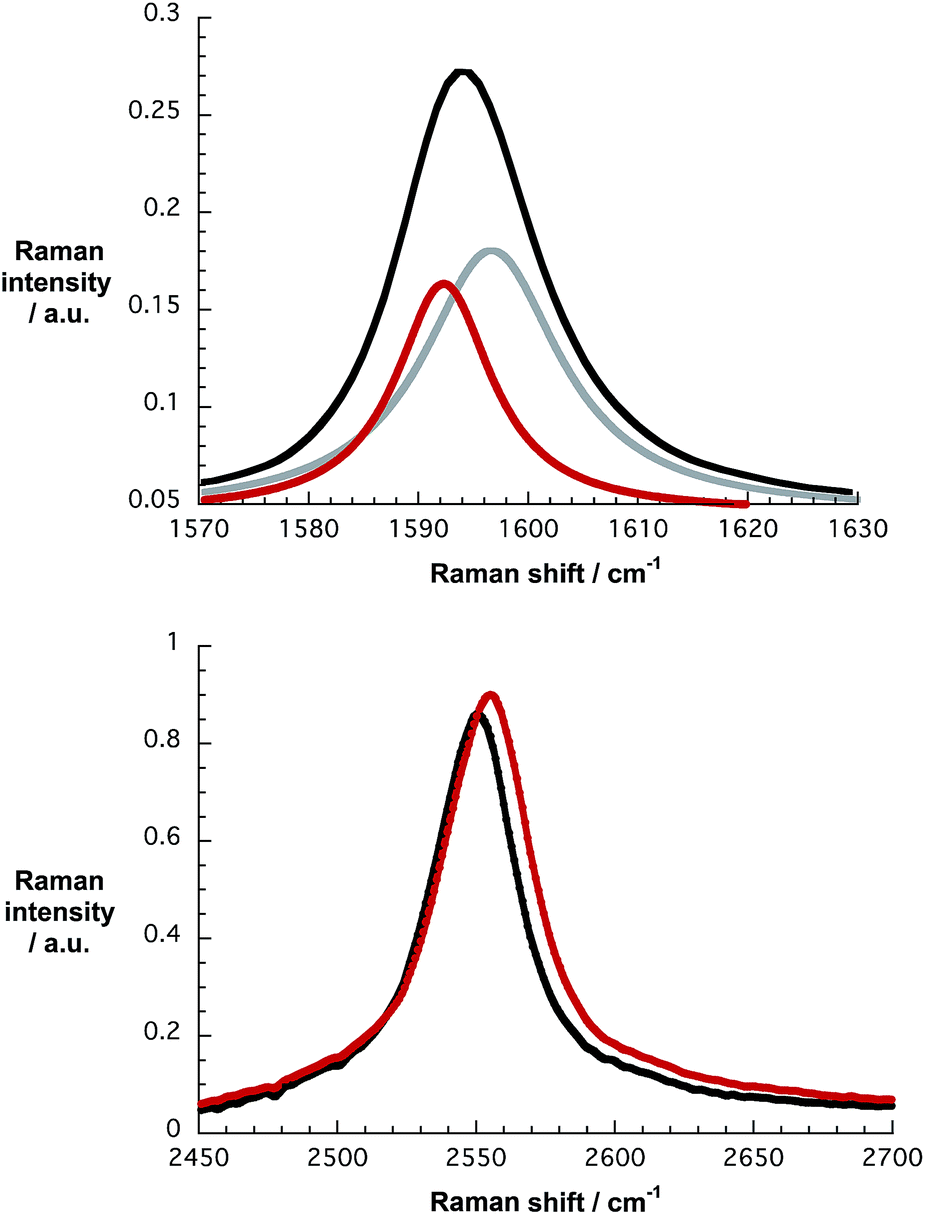 | ||
| Fig. 11 Top: G-band of HiPco-SWCNT/1 upon 1064 nm excitation on alumina substrates and the corresponding fit by using two Lorentzian functions. Bottom: comparison of the 2D-band of HiPco-SWCNT/SDBS (black) and HiPco-SWCNT/1 (red) upon 1064 nm excitation on alumina substrates. | ||
To shed light on the nature of the interactions, which are operative between HiPco-SWCNT and 1, transient absorption spectroscopic measurements were performed. Immediately following 387 nm pulses, differential absorptions evolve, which differ significantly from those seen for HiPco-SWCNT/SDBS – as shown in Fig. 12. For HiPco-SWCNT/1, maxima were registered in the visible range at 431, 468, 500, 530, 580, 626, and 680 nm. In stark contrast, for HiPco-SWCNT/SDBS, no minima are, however, discernible in this spectral region. In this context it is important that our spectroelectrochemical investigations – vide supra – help to document the spectroscopic fingerprints of the one electron reduced form of 1 in this range of the spectrum. The superimposition of these features with the characteristics of HiPco-SWCNT is the origin of the overall spectral shape. Additional evidence for an ultrafast charge transfer was derived from the transient features in the near infrared. In particular, minima, which are noted at 890, 1327, and 1451 nm, are a reflection of the ground state absorption. As time progresses, the aforementioned minima blue shift, that is, from 1327 to 1311 nm and from 1451 to 1442 nm and the underlying transformation is likely to be due to a charge separation.10,35 Thus, we postulate that photoexcitation of HiPco-SWCNT/1 is followed by the formation of a charge separated state comprising one electron reduced 1 and oxidized HiPco-SWCNTs. Biexponential kinetic analyses at different wavelengths yields lifetimes of 1 and 41 ps, which are assigned to charge separation and charge recombination, respectively – as shown in Fig. 12.
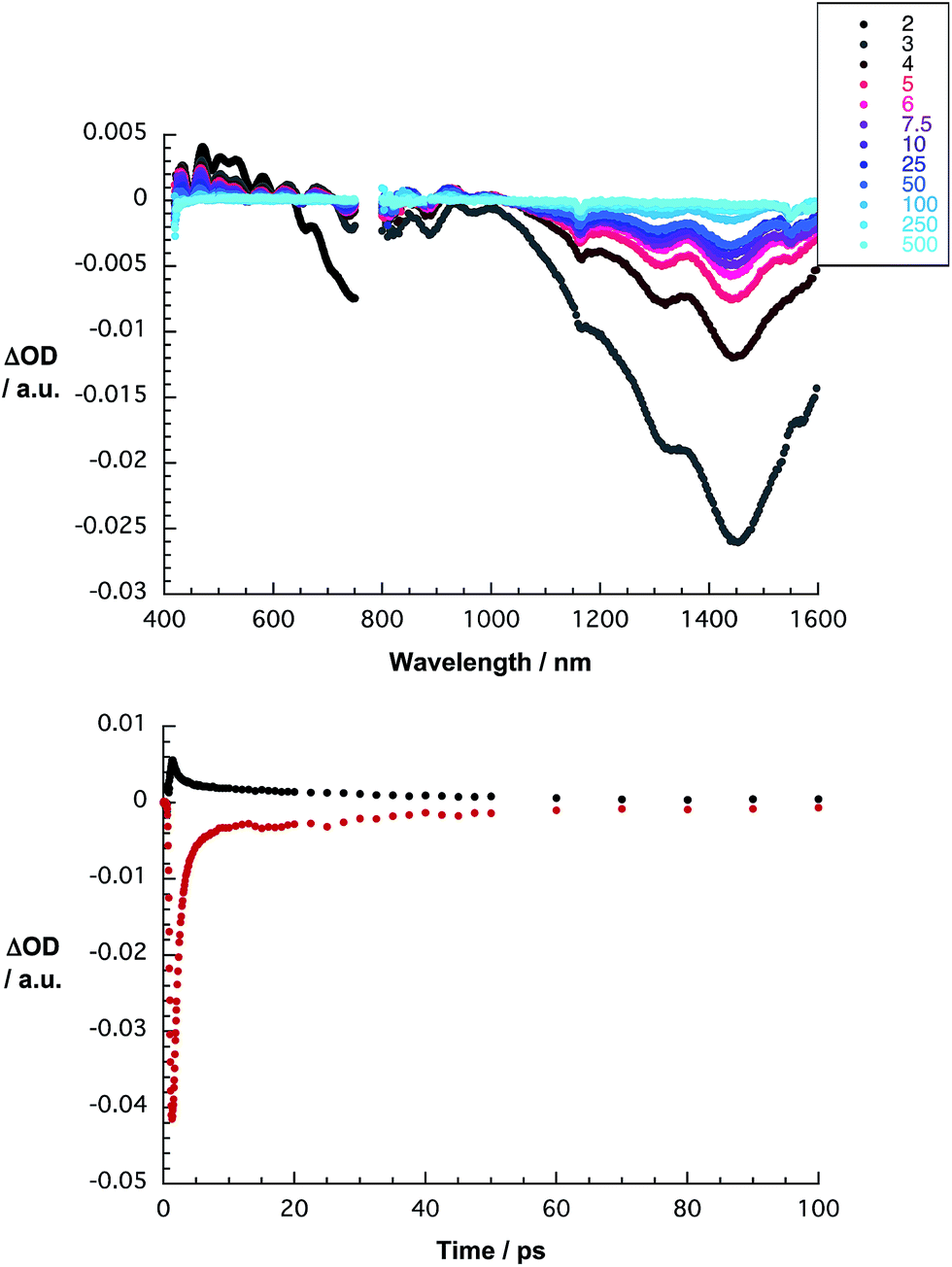 | ||
| Fig. 12 Top: differential absorption spectra of HiPco-SWCNT/1 in D2O/H2PO4−/HPO42− obtained upon femtosecond flash photolysis (387 nm) with time delays between 2 (black) and 500 ps (blue). Bottom: time absorption profiles at 470 nm (black) and 1435 nm (red) monitoring the charge separation and charge recombination. | ||
The electrochemical/spectroelectrochemical experiments assisted in determining the energy of the charge-separated state relative to the ground state. For example, the characteristic features of the one electron reduced form of 1 appear at a potential of −800 mV, whereas the onset of the oxidatively induced changes of the E11 and E22 transitions appears in a potential range of +200 and +600 mV, in line with published reports.10a,36 Consequently, the energy of the charge separated state ranges from 1.0 to 1.4 eV. Besides localized singlet excited states, which are either HiPco-SWCNT or 1 centered, a charge transfer state is likely to be the starting point for the charge separation. The corresponding energy values are 3.1 eV for 1 and 1.8–2.4 eV for HiPco-SWCNT. We conclude from these considerations that both the reduction of 1 and the oxidation of SWCNTs are feasible with driving forces of 1.7–2.1 and 0.4–1.4 eV, respectively – as shown in Fig. 13. In line with these thermodynamic arguments is the fact that no evidence is found for any triplet excited state.37
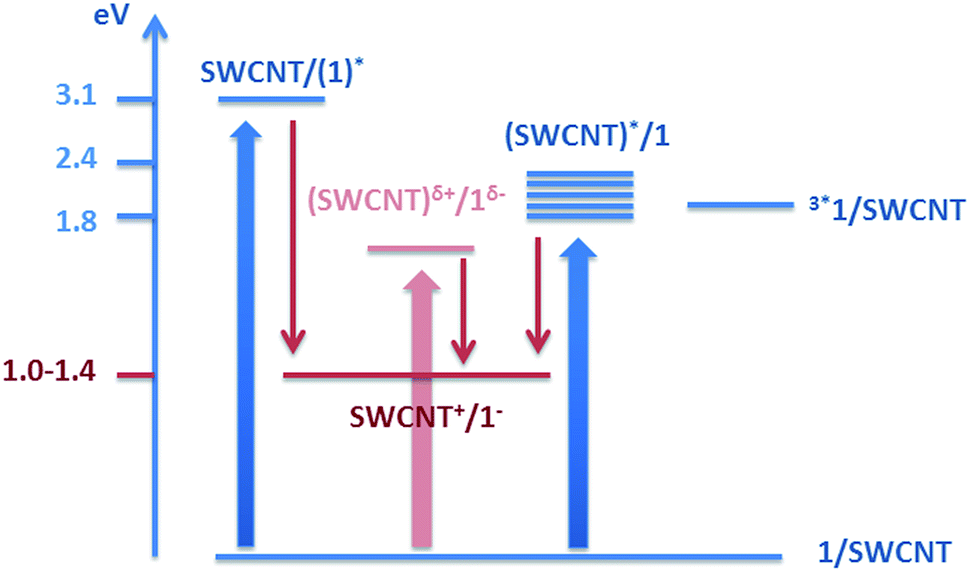 | ||
| Fig. 13 Energy diagram of HiPco-SWCNT/1. | ||
We rounded off our investigation with the selective deimmobilization/removal of either 1 or 4 from SWCNTs. To this end, solid SDBS was added to stable dispersions of HiPco-SWCNT/1 or HiPco-SWCNT/4 and the corresponding spectral changes were monitored by means of steady state absorption, fluorescence, and transient absorption measurements. For a detailed description and analysis see the ESI,† where data from the steady state absorption/emission measurements as well as transient absorption assays are gathered – Fig. S17–21.†
Materials and methods
Chemicals
TBAPF6, SDBS and THF were purchased from Sigma Aldrich and used without further purification. DMSO was purchased from Roth chemicals and used without further purification. D2O was provided by Deutero. To adjust the pH 7 of the solutions 726 mg of Na2HPO4 and 352 mg of KH2PO4 were added to 100 mL of D2O.Spectroscopy
1H and 13C NMR spectra were recorded with Jeol JNM EX 400, Jeol JNM GX (400 MHz for 1H and 100 MHz for 13C) and Jeol Bruker Avance 300 (300 MHz for 1H and 75 MHz for 13C) spectrometers. Chemical shifts (δ) are reported in ppm at room temperature (r.t.) using CDCl3 as solvent and internal standard unless otherwise indicated. IR spectra were recorded with an ASI React IR 1000 spectrometer. Mass spectrometry was carried out on a Micromass Zabspec (Cs+) spectrometer with 3-nitrobenzylalcohol (NBA) as the matrix in fast atom bombardement (FAB) or on an AXIMA Confidences (Shimadzu) matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) apparatus. The measurements were carried out in reflectron mode with sin (sinapinic acid) or dhb (2,5-dihydroxybenzoic acid) as matrices. For elemental analyses (EA) a CE instrument EA 1110 CHNS was used. UV/Vis/nIR measurements were performed using either a Perkin Elmer Lambda 2 spectrometer or a Cary 5000 spectrometer (Varian). Emission was detected in the visible range using a Flouromax3 spectrometer (Horiba Scientific) and in the near infrared using a Floruolog 3 (Horiba Scientific) equipped with an InGaAs symphony as the detection unit.Transient absorption spectra on the pico- and nanosecond timescales were performed with output from CPA2110 or CPA 2101 Ti/sapphire laser systems from Clark-MXR Inc. (775 nm, 1 kHz, 150 fs FWHM pulses). 387 nm excitation pulses were generated by second harmonic generation. For measurements with pico- and nanosecond time resolutions a Helios transient absorption spectrometer was used. For the corresponding measurements on the microsecond time scale, an EOS transient absorption spectrometer was used. Both systems were provided by Ultrafast Systems. All measurements were carried out in 2 mm OS quartz cuvettes. Raman spectra were recorded from solid/solid films deposited on alumina pellets. A Bruker Raman spectrometer RFS 100 (nitrogen cooled Ge-detector, λex = 1064 nm) was used for all of the experiments.
Microscopy
Transmission electron microscopy was conducted on a Zeiss Leo EM912 Omega with an acceleration voltage of 80 kV. Specimens were prepared through dip casting of either lacey carbon/copper or lacey carbon supported ultrathin carbon/copper grids into a suspension of SWCNT/1 or SWCNT/SDBS. The sample grids were dried at 80 °C for 5 h prior to microscopy.Atomic force microscopy tapping mode images were recorded on a Solver Pro scanning probe microscope (NT-MDT) equipped with a Sony Exwave HAD camera optical zoom (6.5). The carbon nanotube material was spread on oxidized silicon wafers with a 200 nm thermally grown oxide layer through spin coating (100 rps).
Electrochemistry
Cyclic voltammetry and square wave voltammetry were performed using a Methrom μAutlobabFraIII with an incorporated impedance unit. Measurements were performed in a homemade cell (V = 5 mL) with a glassy carbon working electrode (3 mm diameter), silver wire acting as the pseudo reference electrode and Pt wire serving as the counter electrode. NOVA 1.10© from Methrom served as the control software.Spectroelectrochemical measurements were performed in a homemade three neck cell equipped with Pt-mesh as the transparent working electrode, Ag-wire as the pseudo-reference electrode and a Pt plate as the counter electrode. A PGStat 101 from Methrom was used as the potentiostat. The latter was controlled using the software NOVA 1.10© provided by Methrom.
Conclusions
In summary, we have demonstrated that water soluble NBI 1 is a powerful surfactant for the individualization, stabilization, and photo-/redoxchemical modulation of HiPco-SWCNTs in aqueous media. Our concept to combine π–π, hydrophobic/hydrophilic, and charge transfer interactions, which was earlier established for both perylenebismide/CoMoCAT-SWCNT and perylenebismide/HiPco-SWCNT hybrids,10,17 was successfully transferred for the first time to naphthalenebisimides yielding stable HiPco-SWCNT/1 hybrids in 0.1 M H2PO4−/HPO42−/D2O. In stark contrast, we failed to generate stable hybrids based on CoMoCAT-SWCNTs due to an unsuitable balance between the size of the Newkome dendron and that of the naphthalene core. Sequentially, detailed replacement studies with HiPco-SWCNT/1 as well as reference experiments with HiPco-SWCNT/4 and SDBS point to a preferential stabilization of larger diameter SWCNTs with 1 as a result of the packing densities – vide supra – as shown in Fig. 14 and S18.†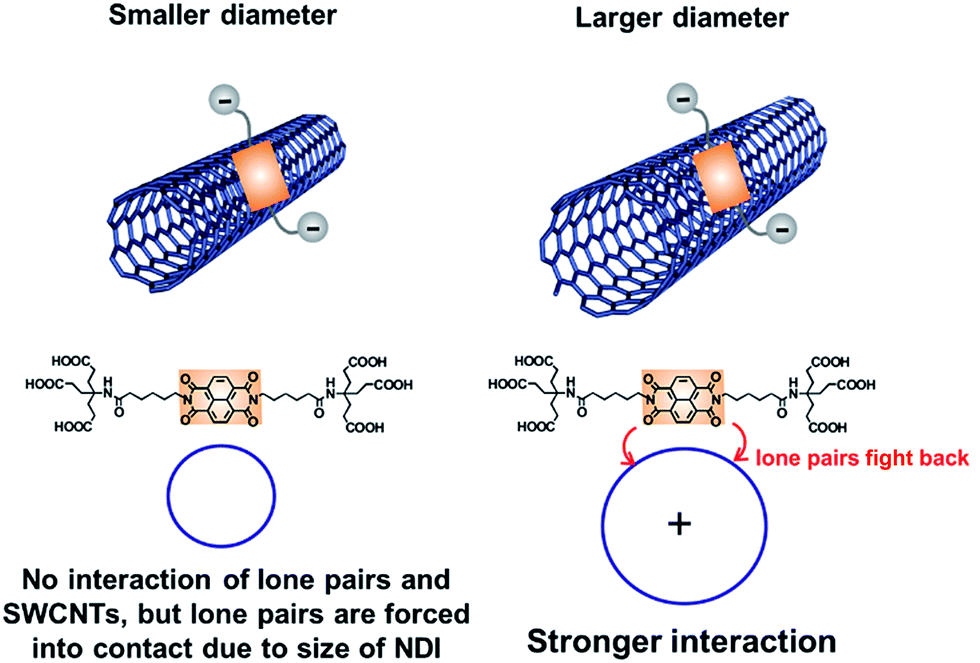 | ||
| Fig. 14 Schematic representation of the mutual orientation of 1 onto HiPco-SWCNT and the resulting interactions. | ||
Besides the comprehensive characterization of 1, in which we corroborated the high energetics, but found only moderately emitting excited states together with a strong electron accepting character, the ground and excited state interactions between HiPco-SWCNTs and 1 were at the focal point of our investigations. With the help of an arsenal of spectroscopic and microscopic techniques, the formation of a charge separated state – comprising oxidized HiPco-SWCNTs and the one electron reduced form of 1 – was corroborated. The latter decays on the picosecond time scale by reinstating the singlet ground state. Notable is the fact that NBIs are among the smallest molecular building blocks which have ever been immobilized onto SWCNTs, and, which afford stable SWCNT suspensions. In light of their high lying and accessible excited states, 1 forms with HiPco-SWCNTs highly energetic charge separated states (1.2–1.4 eV).
Acknowledgements
This work was carried out with support from the DFG through SFB 953 “Synthetic Carbon Allotropes”, the Bavarian state through the “Solar Technologies Go Hybrid” network and the Graduate School of Molecular Science (GSMS) of the University of Erlangen-Nürnberg. Volker Strauss was supported by the “Universität Bayern e.V”.Notes and references
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS PubMed.
- (a) Y. Li-Li, I. A. Kinloch and A. H. Windle, Science, 2004, 304, 276 CrossRef PubMed; (b) S. Nardecchia, D. Carriazo, M. L. Ferrer, M. C. Gutierrez and F. del Monte, Chem. Soc. Rev., 2013, 42, 794 RSC.
- (a) Z. Wang, S. Mohammadzadeh, T. Schmaltz, J. Kirschner, A. Khassanov, S. Eigler, U. Mundloch, C. Backes, H.-G. Steinrück, A. Magerl, F. Hauke, A. Hirsch and M. Halik, ACS Nano, 2013, 7, 11427 CrossRef CAS PubMed; (b) P. Avouris, Z. Chen and V. Perebeinos, Nat. Nanotechnol., 2007, 2, 605 CrossRef CAS PubMed; (c) H. Park, A. Afzali, S.-J. Han, G. S. Tulevski, A. D. Franklin, J. Tersoff, J. B. Hannon and W. Haensch, Nat. Nanotechnol., 2012, 7, 787 CrossRef CAS PubMed.
- F. M. Toma, A. Sartorel, M. Iurlo, M. Carraro, P. Parisse, C. Maccato, S. Rapino, B. Rodriguez Gonzalez, H. Amenitsch, T. Da Ros, L. Casalis, A. Goldoni, M. Marcaccio, G. Scorrano, G. Scoles, F. Paolucci, M. Prato and M. Bonchio, Nat. Chem., 2010, 2, 826 CrossRef CAS PubMed.
- (a) J. Bartelmess, B. Ballesteros, G. de la Torre, D. Kiessling, S. Campidelli, M. Prato, T. Torres and D. M. Guldi, J. Am. Chem. Soc., 2010, 132, 16202 CrossRef CAS PubMed; (b) S. Campidelli, B. Ballesteros, A. Filoramo, D. D. Diaz, G. de la Torre, T. Torres, G. M. A. Rahman, C. Ehli, D. Kiessling, F. Werner, V. Sgobba, D. M. Guldi, C. Cioffi, M. Prato and J.-P. Bourgoin, J. Am. Chem. Soc., 2008, 130, 11503 CrossRef CAS PubMed; (c) M. Ince, J. Bartelmess, D. Kiessling, K. Dirian, M. V. Martınez-Diaz, T. Torres and D. M. Guldi, Chem. Sci., 2012, 3, 1472 RSC; (d) D. M. Guldi and V. Sgobba, Chem. Commun., 2011, 47, 606 RSC; (e) A. S. D. Sandanayaka, E. Maligaspe, T. Hasobe, O. Ito and F. D'Souza, Chem. Commun., 2010, 46, 8749 RSC; (f) S. K. Das, N. K. Subbaiyan, F. D'Souza, A. S. D. Sandanayaka, T. Hasobe and O. Ito, Energy Environ. Sci., 2011, 4, 707 RSC; (g) K. Dirian, A. M. Herranz, G. Katsukis, J. Malig, L. Rodriguez-Perez, C. Romero-Nieto, V. Strauss, N. Martin and D. M. Guldi, Chem. Sci., 2013, 4, 4335 RSC.
- A. Thess, R. Lee, P. Nikolaev, H. Dai, P. Petit, J. Robert, C. Xu, Y. Hee Lee, S. G. Kim, A. G. Rinzler, D. T. Colbert, G. E. Scuseria, D. Tomanek, J. E. Fischer and R. E. Smalley, Science, 1996, 273, 483 CAS.
- (a) A. Hirsch, Angew. Chem., Int. Ed., 2002, 41, 1853 CrossRef CAS; (b) C. Jiang, A. Saha, C. Xiang, C. C. Young, J. M. Tour, M. Pasquali and A. A. Marti, ACS Nano, 2013, 7, 4503 CrossRef CAS PubMed; (c) P. Singh, S. Campidelli, S. Giordani, D. Bonifazi, A. Bianco and M. Prato, Chem. Soc. Rev., 2009, 38, 2214 RSC; (d) F. Hof, S. Bosch, S. Eigler, F. Hauke and A. Hirsch, J. Am. Chem. Soc., 2013, 135, 18385 CrossRef CAS PubMed.
- (a) V. C. Moore, M. S. Strano, E. H. Haroz, R. H. Hauge and R. E. Smalley, Nano Lett., 2003, 3, 1379 CrossRef CAS; (b) M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Karoz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kitrell, J. Ma, R. H. Hauge, R. B. Weizman and R. E. Smalley, Science, 2002, 297, 593 CrossRef PubMed; (c) O. Matarredona, H. Rhoads, Z. Li, J. H. Harwell, L. Balzano and D. E. Resasco, J. Phys. Chem. B, 2003, 107, 13357 CrossRef CAS.
- (a) T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Muellen, Angew. Chem., Int. Ed., 2010, 49, 9068 CrossRef CAS PubMed; (b) F. N. Castellano, Dalton Trans., 2012, 41, 8493 RSC; (c) X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268 CrossRef CAS PubMed.
- (a) C. Ehli, C. Oelsner, D. M. Guldi, A. Mateo-Alonso, M. Prato, C. Schmidt, C. Backes, F. Hauke and A. Hirsch, Nat. Chem., 2009, 1, 243 CrossRef CAS PubMed; (b) C. Oelsner, C. Schmidt, F. Hauke, M. Prato, A. Hirsch and D. M. Guldi, J. Am. Chem. Soc., 2011, 133, 4580 CrossRef CAS PubMed.
- Z. Hu, G. D. Pantoş, N. Kuganathan, R. L. Arrowsmith, R. M. J. Jacobs, G. Kociok-Köhn, J. O'Byrne, K. Jurkschat, P. Burgos, R. M. Tyrrell, S. M. Botchway, J. K. M. Sanders and S. I. Pascu, Adv. Funct. Mater., 2012, 22, 503 CrossRef CAS PubMed.
- (a) N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225 RSC; (b) S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331 RSC; (c) S. Guha, F. S. Goodson, S. Roy, L. J. Corson, C. A. Gravenmier and S. Saha, J. Am. Chem. Soc., 2011, 133, 15256 CrossRef CAS PubMed; (d) S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679 CrossRef CAS PubMed.
- (a) H. Imahori, H. Yamada, D. M. Guldi, Y. Endo, A. Shimomura, S. Kundu, K. Yamada, T. Okada, Y. Sakata and S. Fukuzumi, Angew. Chem., Int. Ed., 2002, 41, 2344 CrossRef CAS; (b) M. E. El-Khouly, C. A. Wijesinghe, V. N. Nesterov, M. E. Zandler, S. Fukuzumi and F. D'Souza, Chem.–Eur. J., 2012, 18, 13844 CrossRef CAS PubMed; (c) N. Banerji, S. V. Bhosale, I. Petkova, S. J. Langford and E. Vauthey, Phys. Chem. Chem. Phys., 2011, 13, 1019 RSC; (d) F. Chaignon, M. Falkenström, S. Karlsson, E. Blart, F. Odobel and L. Hammarström, Chem. Commun., 2007, 64 RSC; (e) R. T. Hayes, M. R. Wasieleweski and D. Gosztola, J. Am. Chem. Soc., 2000, 122, 5563 CrossRef CAS; (f) C. Liao, J. E. Yarnell, K. D. Glusac and K. S. Schanze, J. Phys. Chem. B, 2010, 114, 14763 CrossRef CAS PubMed.
- (a) A. V. Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791 CrossRef PubMed; (b) P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2005, 127, 6528 CrossRef CAS PubMed; (c) S. Bhosale, A.-L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84 CrossRef CAS PubMed.
- (a) R. S. K. Kishore, O. Kel, N. Banerji, D. Emery, G. Bollot, J. Mareda, A. Gomez-Casado, P. Jonkheijm, J. Huskens, P. Maroni, M. Borkovec, E. Vauthey, N. Sakai and S. Matile, J. Am. Chem. Soc., 2009, 131, 11106 CrossRef CAS PubMed; (b) R. Bhosale, J. Mısek, N. Sakai and S. Matile, Chem. Soc. Rev., 2010, 39, 138 RSC; (c) M. Lista, J. Areephong, N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 15228 CrossRef CAS PubMed.
- Y. Zhao, C. Beuchat, Y. Domoto, J. Gajewy, A. Wilson, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 2101 CrossRef CAS PubMed.
- (a) C. Backes, F. Hauke and A. Hirsch, Adv. Mater., 2011, 23, 2588 CrossRef CAS PubMed; (b) C. Backes, C. D. Schmidt, F. Hauke, C. Böttcher and A. Hirsch, J. Am. Chem. Soc., 2009, 131, 2172 CrossRef CAS PubMed.
- C. D. Schmidt, C. Böttcher and A. Hirsch, Eur. J. Org. Chem., 2007, 5497 CrossRef CAS PubMed.
- Pyrene in THF was utilized as standard.
- (a) T. C. Barros, S. Brochsztain, V. G. Toscano, P. B. Filho and M. J. Politi, J. Photochem. Photobiol., A, 1997, 111, 97 CrossRef CAS; (b) G. Andric, J. F. Boas, A. M. Bond, G. D. Fallon, K. P. Ghiggino, C. F. Hogan, J. A. Hutchinson, M. A.-P. Lee, S. J. Langford, J. R. Pilbrow, G. J. Troup and C. P. Woodward, Aust. J. Chem., 2004, 57, 1011 CrossRef CAS.
- S. Green and M. A. Fox, J. Chem. Phys., 1995, 99, 14752 CrossRef CAS.
- J. R. Hurst, J. D. McDonald and G. B. Schuster, J. Am. Chem. Soc., 1982, 104, 2065 CrossRef CAS.
- The redox potential of Fc/Fc+ was determined in a separate measurement and used as calibration for the silver wire acting as reference electrode.
- J. F. Penneau, B. J. Stallman, P. H. Kasai and L. L. Miller, Chem. Mater., 1991, 3, 791 CrossRef CAS.
- B. M. Aveline, S. Matsugo and R. W. Redmond, J. Am. Chem. Soc., 1997, 119, 11785 CrossRef CAS.
- (a) S. M. Bachilo, M. S. Strano, C. Kitrell, R. H. Hauge, R. E. Smalley and R. B. Weiszman, Science, 2002, 298, 2361 CrossRef CAS PubMed; (b) J. G. Duque, C. G. Densmore and S. K. Doorn, J. Am. Chem. Soc., 2010, 132, 16165 CrossRef CAS PubMed; (c) J. G. Duque, A. N. G. Parra-Vasquez, N. Behabtu, M. J. Green, A. L. Higginbotham, B. K. Price, A. D. Leonard, H. K. Schmidt, B. Lounis, J. M. Tour, S. K. Doorn, L. Cognet and M. Pasquali, ACS Nano, 2010, 4, 3063 CrossRef CAS PubMed.
- (a) M. S. Dresselhaus, G. Dresselhaus, R. Saito and A. Jorio, Phys. Rep., 2005, 409, 47 CrossRef PubMed; (b) M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751 CrossRef CAS PubMed.
- F. Schoeppler, N. Ruehl and T. Hertel, Chem. Phys., 2013, 413, 112 CrossRef CAS PubMed.
- (a) L. J. Carlson and T. D. Krauss, Acc. Chem. Res., 2008, 41, 235 CrossRef CAS PubMed; (b) G. N. Ostojic, S. Zaric, J. Kono, M. S. Strano, V. C. Moore, R. H. Hauge and R. E. Smalley, Phys. Rev. Lett., 2004, 92, 117402 CrossRef CAS.
- C. Backes, F. Hauke and A. Hirsch, Phys. Status Solidi B, 2013, 250, 2592 CrossRef CAS PubMed.
- C. Backes, C. D. Schmidt, K. Rosenlehner, F. Hauke, J. N. Coleman and A. Hirsch, Adv. Mater., 2010, 22, 788 CrossRef CAS PubMed.
- (a) C. Backes, U. Mundloch, C. D. Schmidt, J. N. Coleman, W. Wohlleben, F. Hauke and A. Hirsch, Chem.–Eur. J., 2010, 16, 13185 CrossRef CAS PubMed; (b) C. Backes, C. D. Schmidt, F. Hauke and A. Hirsch, Chem.–Asian. J., 2011, 6, 438 CrossRef CAS PubMed.
- (a) F. Ernst, T. Heek, A. Setaro, R. Haag and S. Reich, Adv. Funct. Mater., 2012, 22, 3921 CrossRef CAS PubMed; (b) F. Ernst, T. Heek, R. Haag, S. Reich and A. Setaro, Phys. Status Solidi B, 2012, 249, 2465 CrossRef CAS PubMed.
- J. Crochet, M. Clemens and T. Hertel, J. Am. Chem. Soc., 2007, 129, 8058 CrossRef CAS PubMed.
- C. Romero-Nieto, R. Garcia, M. A. Herranz, L. Rodriguez-Perez, M. Sanchez-Navarro, J. Rojo, N. Martin and D. M. Guldi, Angew. Chem., Int. Ed., 2013, 52, 10216 CrossRef CAS PubMed.
- D. Paolucci, M. M. Franco, M. M. Iurlo, M. Marcaccio, M. Prato, F. Zerbetto, A. Penicaud and F. Paolucci, J. Am. Chem. Soc., 2008, 130, 7393 CrossRef CAS PubMed.
- (a) J. Park, P. Deria and M. Therien, J. Am. Chem. Soc., 2011, 133, 17156 CrossRef CAS PubMed; (b) D. Stich, F. Spaeth, H. Kraus, A. Sperlich, V. Dyakonov and T. Hertel, Nat. Photonics, 2014, 8, 139 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc02944a |
| ‡ K. Dirian and S. Backes contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |