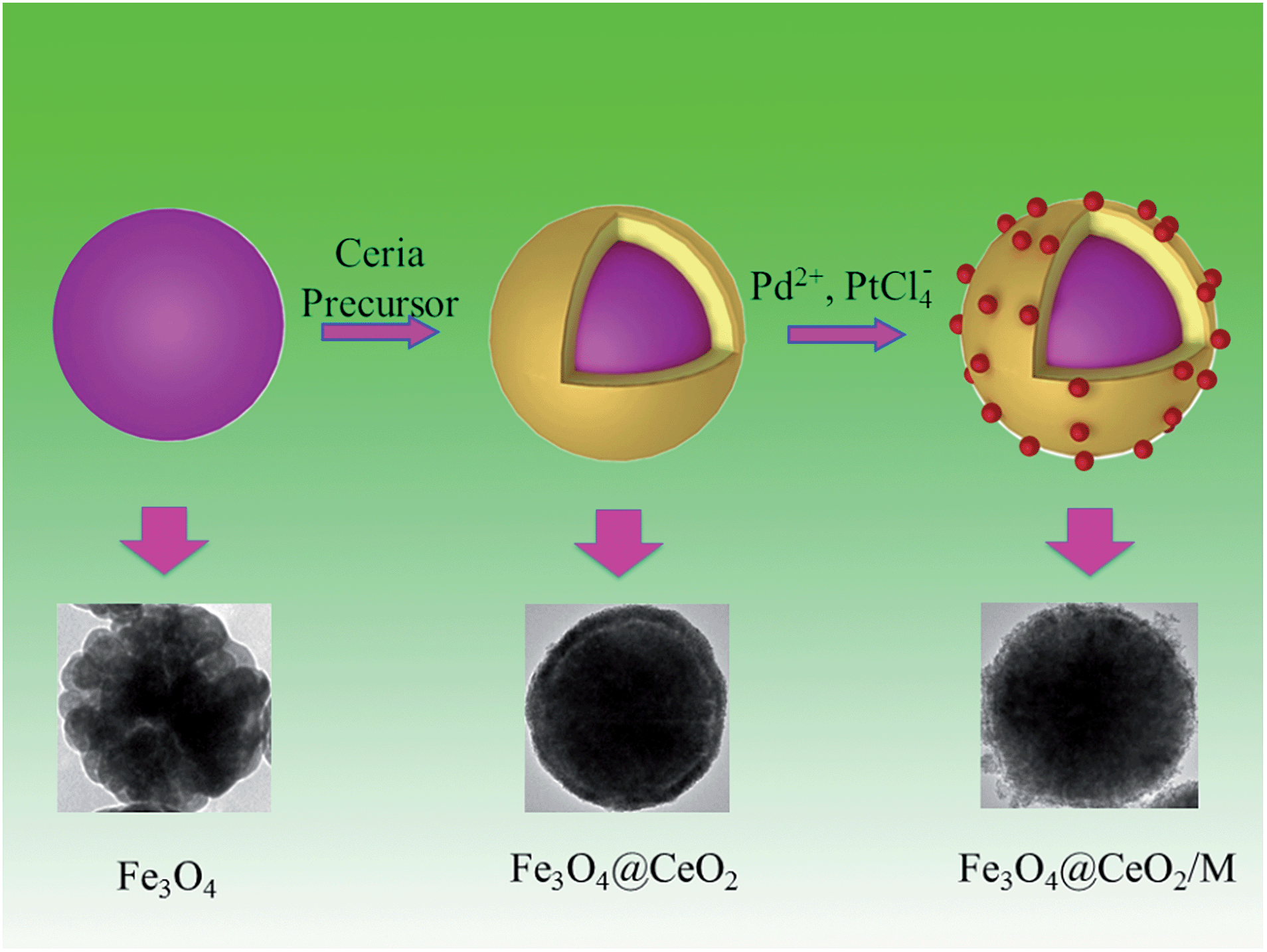
Novel recyclable dual-heterostructured Fe3O4@CeO2/M (M = Pt, Pd and Pt–Pd) catalysts: synergetic and redox effects for superior catalytic performance†
Received
23rd October 2014
, Accepted 4th November 2014
First published on 4th November 2014
Abstract
We report the creation of highly efficient, dual-heterostructured catalysts consisting of magnetic Fe3O4 cores and CeO2 shells coated with noble metal nanoparticles (NMNPs) (Pt, Pd and Pt–Pd nanoparticles). These intriguing dual-heterostructural features that favor reactant diffusion and exposure of active sites can enhance synergistic catalytic effects between the NMNPs and CeO2 nanoparticles. The synergistic catalytic effect and redox effect features between Fe3O4 and CeO2 are beneficial for superior catalytic and electrochemical performance. Impressively, the resultant Fe3O4@CeO2/Pd (3 wt%) and Fe3O4@CeO2/Pt (20 wt%) catalysts manifest superior catalytic efficiency and electrocatalytic reactivity toward the reduction of 4-nitrophenol and oxidation of methanol in alkaline solution, demonstrating the significance of the dual heterostructure of these recyclable catalysts. This synthetic strategy provides a new methodology for the fabrication of other high-performance and multifunctional catalysts, which would be very useful in various catalytic reactions.
Introduction
With economic globalization, phenolic wastewater pollution has become one of many strategic issues.1–3 Catalytic hydrogenation that can be conducted under relatively mild conditions without contaminant by-products is a promising method for phenolic wastewater treatment. Heterostructural catalysts composed of diverse metals and metal oxides into an integrated structure are a fascinating class of materials that may maintain the properties of each component, as well as show higher catalytic performance due to their structural features.4–8 However, heterostructural catalysts exhibit relatively high reaction rates in catalytic water treatment; difficulties in recycling them from treated water may cause redispersion into the water environment. Therefore, these nanocatalysts are sometimes used via complex methods, such as tedious centrifugation and filtration. In this increasingly environmentally conscious age, the search for an efficient recoverable catalyst has become inexorable. Magnetic separation might be a more efficient technique to remove these nanocatalysts from the water environment. In this regard, integrating magnetic Fe3O4 NPs into heterostructural materials as a magnetically recoverable catalyst could be a promising method. Fe3O4 NPs have gained much attention due to their rapid response to an applied magnetic field, which are often hybridized with the catalysts to form magnetic nanocatalysts that make it possible to realize convenient catalyst recycling.9–15
Noble metal nanoparticles (NMNPs), especially Pt and Pd, have a wide range of applications such as catalysis, electrochemical, energy processing, and pollution control.16,17 To enhance their catalytic activity and stability, lots of oxides have been used as catalyst supports for the deposition of these NMNPs. CeO2, which has a Ce4+/Ce3+ redox cycle, is an ideal candidate due to its low cost, high oxygen storage capability and high oxygen mobility.18,19 However, due to its poor conductivity, it is necessary to modify the CeO2 nanoparticles to increase electrochemical conductivity and subsequently catalytic performance. Loading NMNPs onto cerium oxide can dramatically improve catalytic and electrochemical activities owing to synergetic catalytic effects between CeO2 nanoparticles and NMNPs.20
Herein, we report a novel dual-heterostructured Fe3O4@CeO2/M (M = Pt, Pd and Pt–Pd) catalysts, which are composed of magnetic Fe3O4 cores and porous CeO2 shells and coated with NMNPs. These dual-heterostructural features that favor reactant diffusion and exposure of active sites can dramatically enhance synergistic catalytic effects between NMNPs and CeO2 shells. The synergistic catalytic and redox effect features between Fe3O4 and CeO2 are beneficial for superior catalytic and electrochemical activities. The catalytic activity of the dual-heterostructural catalysts was systematically tested using the 4-NP reduction and oxidation of methanol in alkaline solution as model reactions. Results indicated that the catalytic efficiency varies with the dual heterostructure and the kinds of NMNPs. Impressively, the resultant Fe3O4@CeO2/Pd (3%) catalyst exhibits superior catalytic performance toward catalytic reduction of 4-NP to 4-AP. The Fe3O4@CeO2/Pt (20%) catalyst manifests superior electrochemical activities toward methanol oxidation in alkaline solution. Our results therefore provide a general approach based on a dual heterostructure for preparation and discovery of highly efficient and magnetic nanocatalysts, which would be very useful in various catalytic reactions.
Experimental
Synthesis of magnetite microspheres
The magnetite microspheres were prepared according to the following method. Typically, FeCl3·6H2O (1.35 g, 5 mmol), polyethylene glycol (1.0 g) and NaAc (3.6 g) were dissolved in 40 mL of ethylene glycol with stirring for 30 min to form a clear solution. Then the obtained solution was transferred to a 50 mL Teflon-lined stainless steel autoclave. The autoclave was heated at 200 °C for 8 h. The precipitate was collected by centrifuge and then washed with ethanol and deionized water several times.
Synthesis of ceria precursor
The ceria precursor was prepared by the solvothermal method. 1 g Ce (NO3)3·6H2O and 1 g NaOH were separately dissolved in 20 mL of ethanol. The mixture was stirred vigorously for 24 h at 50 °C. Hydrogen peroxide (30% H2O2, 0.05 mL) was taken into the mixture solution, stirred for 2 h. The precipitate was washed and collected by centrifugation before drying at 60 °C for 4 h. 1 g of precipitate was then dispersed in 20 mL distilled water under continuous stirring. Then, the pH of the solution was adjusted to 0.1 by adding concentrated nitric acid. The reaction was allowed to proceed at 40 °C for 2 h under continuous stirring. After the solution was cooled to room temperature naturally, the transparent light-yellow solution was obtained.
Synthesis of Fe3O4@CeO2/Pd spheres
The Fe3O4@CeO2 microspheres were prepared in accordance with the following procedure: 0.1 g of Fe3O4 nanoparticles was dispersed in a mixture containing 30 mL distilled water and 3 mL of ceria precursor through an ultrasonic treatment process for 15 min. Subsequently, 0.5 mol L−1 ammonium hydroxide solution was added to adjust the pH to 6.8. The final mixed solution was agitated vigorously by stirring for 4 h at a constant temperature of 60 °C. After the reaction was completed, the particles were collected through an external magnet and washed thoroughly with ethanol before drying at 60 °C overnight. An aqueous solution of PdCl2 (10 mL) and polyvinyl alcohol (PVA, 1 wt%, 0.72 mL) were diluted by water (120 mL). After stirring for 30 min, 0.1 M NaBH4 (1.98 mL) was added to form a dark brown sol. Subsequently, sulfuric acid solution was added to adjust the pH to 1. Then, 0.2 g of Fe3O4@CeO2 was ultrasonically dispersed in the mixture under vigorous stirring. After 2 h, the resulting slurry was filtered, washed three times with distilled water and absolute alcohol to remove the surface-adsorbed PVA on the surface of the samples, and finally dried in a vacuum oven at 60 °C overnight. Finally, the as-prepared nanoparticles were calcined in N2 at 300 °C for 2 h.
Synthesis of Fe3O4@CeO2/Pt spheres
Pt nanoparticles were supported on Fe3O4@CeO2 nanocrystals by using the sol-immobilization method. An aqueous solution of H2PtCl6·6H2O (16.0 mL) and polyvinyl alcohol (PVA, 1 wt%, 0.72 mL) was diluted by water (120 mL). After stirring for 30 min, 0.1 M NaBH4 (1.98 mL, NaBH4/Pt = 5) was added to form a dark brown sol. Subsequently, sulfuric acid solution was added to adjust the pH to 1. Then, 0.2 g of Fe3O4@CeO2 was ultrasonically dispersed in the mixture under vigorous stirring. After 2 h, the resulting slurry was filtered, washed several times with distilled water and absolute alcohol to remove the surface-adsorbed PVA from the sample, and finally dried in a vacuum oven at 60 °C overnight. Finally, the resultant samples were calcined in N2 at 300 °C for 2 h.
Synthesis of Fe3O4@CeO2/Pt–Pd spheres
An aqueous solution of H2PtCl6·6H2O (8.0 mL) and PdCl2 (5.0 mL) and polyvinyl alcohol (PVA, 1 wt%, 0.72 mL) were diluted by water (120 mL). After stirring for 30 min, 0.1 M NaBH4 (1.98 mL, NaBH4/Pt = 5) was added to form a dark brown sol. Subsequently, sulfuric acid solution was added to adjust the pH to 1. Then, 0.2 g of Fe3O4@CeO2 was ultrasonically dispersed in the mixture under vigorous stirring. After 2 h, the resulting slurry was filtered, washed several times with distilled water and absolute alcohol to remove the surface-adsorbed PVA on the sample surface, and finally dried in a vacuum oven at 60 °C overnight. Finally, the resultant samples were calcined in N2 at 300 °C for 2 h.
Characterization
Transmission electron microscopy (TEM) characterization was performed on a JEM-2010 system and a HITACHI 800 system operated at acceleration voltages of 120 kV and 200 kV, respectively. Powder X-ray diffraction (XRD) measurements were performed using a PuXi XD3 diffractometer (Cu Kα radiation, λ = 0.15406 nm). Magnetic measurements were carried out using a vibrating sample magnetometer (VSM; Digital Measurement System JDM-13). X-ray photoelectron spectroscopy (XPS) measurements were performed in a VG Scientific ESCALAB Mark II spectrometer equipped with two ultrahigh-vacuum (UHV) chambers. Surface area measurements were performed on an ASAP 2010 Brunauer–Emmett–Teller (BET) analyzer. The absorption spectra were recorded using a SPECORD.50 UV-vis spectrophotometer (Analytikjena). Actual metal content in the catalysts was estimated with ICP-mass spectroscopy (Varian Vista-MPX).
Catalytic tests
The catalytic performance of the catalysts was tested by employing the reduction of 4-nitrophenol to 4-aminophenol as a model reaction. Typically, the catalyst (2 mg) was added to deionized water (40 mL) to form a homogeneous suspension by ultrasonication. Then NaBH4 aqueous solution (0.5 M, 0.5 mL) was added to the above suspension, and the suspension was stirred at room temperature for 10 min. Next, 4-nitrophenol (0.012 M, 0.25 mL) was infused into the above suspension. After stirring for several seconds, the mixture was rapidly transferred to the quartz cell to monitor the reaction progress by measuring the UV-vis absorption spectra of the mixture to evaluate the catalytic activity and stability of the catalysts.
Preparation of catalyst-modified GCE
Before modification, a glassy carbon electrode (GCE) (3.0 mm in diameter) was polished with 0.05 μm α-Al2O3 powder, and then ultrasonically rinsed with ethanol and ultrapure water, and dried in N2 at room temperature. To prepare a catalyst-coated working electrode, the catalyst was dispersed in a mixture of solvents containing water, ethanol and Nafion® (5%) (v/v/v = 15/5/1), and formed a 10 mg mL−1 suspension. Next, 0.01 mL catalyst ink was dropped onto the GCE surface and dried at room temperature to finish the modification. Commercial Pt/C (10% Pt) catalyst was prepared the same way and used for the contrast experiment.
Electrochemical measurements
Voltammetric measurements were carried out with a CHI 750D electrochemical workstation. Ag/AgCl and Pt wire were used as the reference electrode and counterelectrode, respectively. The electrodes were immersed in the nitrogen-saturated 0.5 M H2SO4 solution (25 °C), and the potential was scanned from −0.2 to 0.3 V at a scan rate of 50 mV s−1. The scan was repeated several times to ensure that a stable cyclic voltammogram (CV) was obtained. The CVs were used to estimate the electrochemically active surface area (EASA) of the catalyst by calculating the hydrogen under potential desorption (Hupd) area of the catalyst. CVs for MOR were conducted in 0.5 M KOH and 1.0 M methanol from −0.9 V to 0.5 V at a scan rate of 50 mV s−1. The amperometric current density–time (i–t) curves were measured at a fixed potential for 3600 s in 0.5 M KOH and 1.0 M CH3OH. For each catalyst, the current was normalized to the loading of noble metals (Pt) to obtain mass activity. All experiments were conducted at room temperature.
Results and discussion
The synthesis procedures of Fe3O4@CeO2/M heterostructural nanocatalysts involved several steps (Scheme 1). As illustrated in Scheme 1, the synthesis of these microspheres involves three steps, and each step is well controlled. After coating, the NMNPs were uniformly distributed on the surface of the oxide microspheres.
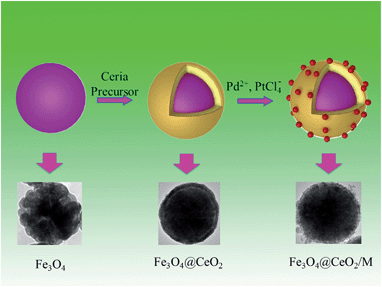 |
| | Scheme 1 Schematic illustration of preparation procedures for heterostructural microspheres and corresponding TEM images. | |
The crystal structures of the as-synthesized nanocatalysts were examined using X-ray diffraction (XRD). Fig. S1† shows the XRD patterns of typical nanocatalysts. The diffraction peaks corresponding to the lattice planes of (111), (220) (2θ = 47.5°) and (311) (2θ = 57°) are clearly discerned and can be readily indexed to the pure cubic fluorite structure of CeO2 with a lattice constant of a = 5.410 Å, a value very close to the reported data (JCPDS no. 34-0394, a = 5.411 Å).21 The (220) (2θ = 30°), (311) (2θ = 35.4°), (400), (511), and (440) planes can be indexed to the face-centered cubic phase of Fe3O4 with a lattice constant of a = 8.392 Å, which is very close to the reported data (JCPDS no. 85-1436, a = 8.393 Å).22 The positions and their relative intensities of reflection peaks agree well with those of CeO2 and Fe3O4 nanocrystals reported in the literature. No noble metal signals were detected due to low actual content loading on the surface of the Fe3O4@CeO2 supports (actual content of Pt and Pd are 1.36 wt% and 1.62 wt%, respectively).
The morphologies and sizes of Fe3O4@CeO2/M (M = Pt, Pd and Pt–Pd) catalysts are shown in Fig. 1. It reveals that the obtained Fe3O4@CeO2/M catalysts with sizes in the range of 300–350 nm, and CeO2 nanospheres were composed of a large number of tiny nanoclusters assembled in situ to form the nanocrystals. The NMNPs, with average diameter of about 3 nm, were well-dispersed on the surface of the CeO2 nanocrystals, and no obvious aggregation was observed. The HAADF-STEM and elemental mapping were also performed to reveal the element distribution in the heterostructure microspheres. It reveals that the NMNPs observed as white dots were remarkably well-dispersed on the surface of Fe3O4@CeO2 microspheres (Fig. 2). Mapping results (Fig. 2e–l) indicate that the elements Fe, Ce, O, Pt and Pd were spread evenly in the whole spheres and the Ce and NMNPs were almost located on the outermost surface of the microspheres, which confirms the dual heterostructure of Fe3O4@CeO2/M as obtained by the step-by-step assembly procedures illustrated in Scheme 1.
 |
| | Fig. 1 TEM and HR-TEM images: Fe3O4@CeO2, (a) and (e); Fe3O4@CeO2/Pt (3 wt%) catalyst, (b) and (f); Fe3O4@CeO2/Pt–Pd (3 wt%) catalyst, (c) and (g); and Fe3O4@CeO2/Pd (3 wt%) catalyst, (d)–(h). | |
 |
| | Fig. 2 HAADF-STEM images of (a) Fe3O4@CeO2, (b) Fe3O4@CeO2/Pt (3 wt%), (c) Fe3O4@CeO2/Pd–Pt (3 wt%), and (d) Fe3O4@CeO2/Pd (3 wt%); and mapping results of elements (e) Fe, (f) O, (g) Ce, (h) Cu of Fe3O4@CeO2, and (i) Pt, (j) Pd, (k) Pt, and (l) Pd corresponding to Fe3O4@CeO2/Pt (3 wt%) (i), Fe3O4@CeO2/Pd–Pt (3 wt%) (j and k), and Fe3O4@CeO2/Pd (3 wt%) (l), respectively. | |
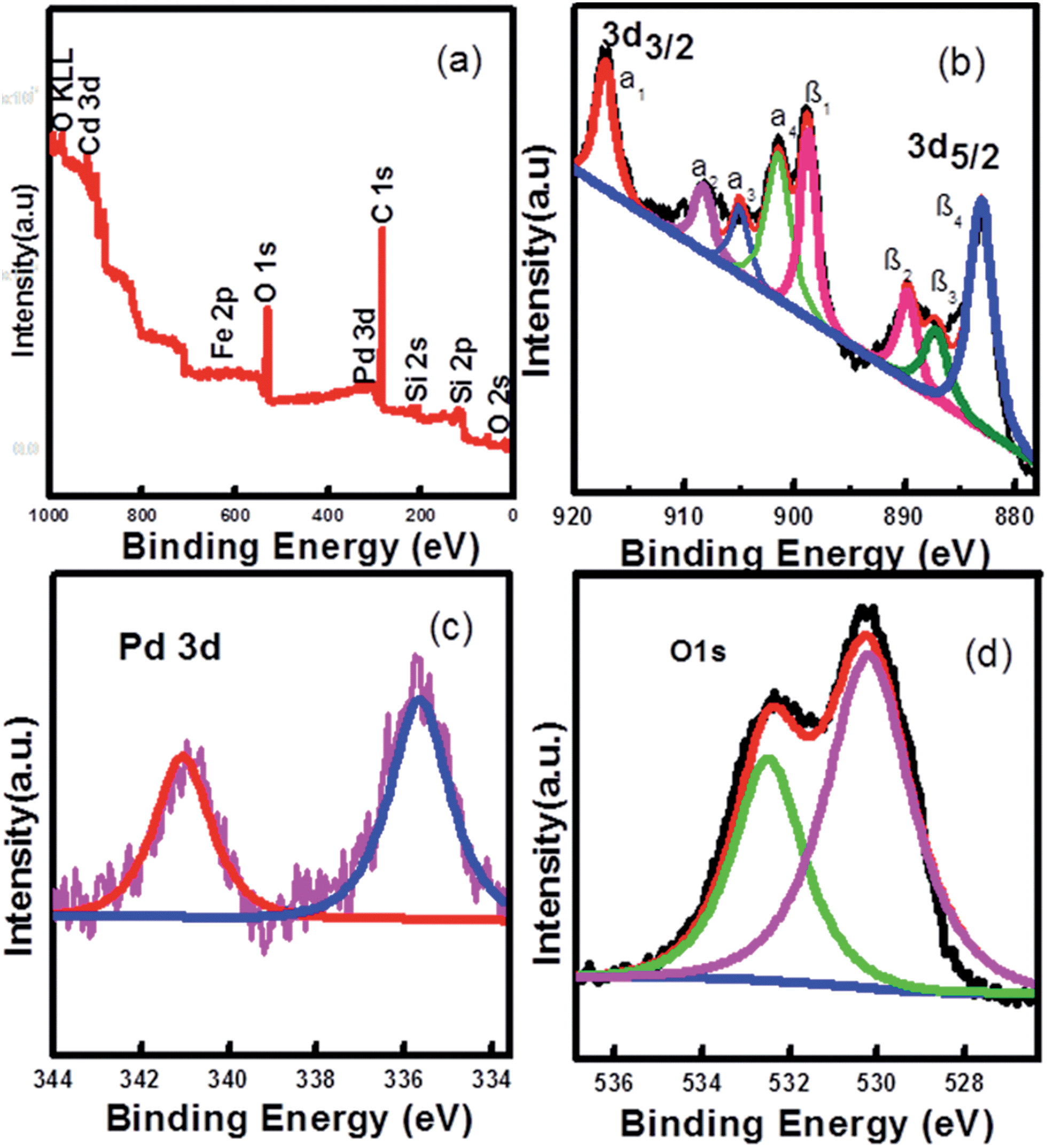
X-ray photoelectron spectra (XPS) were also employed to examine surface elements and their valence states (Fig. 3). The XPS elemental survey scan of the surface of the Pd-loaded Fe3O4@CeO2 microspheres reveals that oxygen, cerium, carbon and palladium are present in the samples, as show in Fig. 3a. Regarding the typical signals of Ce3d spectra, the four main 3d3/2 peaks featured at around 917.4 eV, 908.5 eV, 905.2 eV and 901.7 eV corresponded to the α1, α2, α3 and α4 components, while the Ce3d5/2 located at 898.8 eV, 889.9 eV, 887.3 eV, and 883.1 eV corresponded to β1, β2, β3, and β4, respectively (Fig. 3b). The signals α3 and β3, characteristics of Ce3+, were found in the Fe3O4@CeO2/Pd catalyst, while other peaks corresponded to the Ce4+. The presence of Ce3+ contributed to the interaction between ceria and the surrounding palladium and oxygen atoms that may have led to a transfer of lattice oxygen in the CeO2 lattice. In Fig. 3c, the regional XPS spectrum of Fe3O4@CeO2/Pd exhibited two peaks centered at 341.1 and 335.7 eV, which were assigned to Pd(0) of 3d3/2, and 3d5/2, respectively. The regional XPS spectrum of Pd indicated that the Pd(0) species were formed on the surface of Fe3O4@CeO2 microspheres. The spectra for the O1s ionization feature were fitted with Gaussian–Lorenz features as shown in Fig. 3d. The primary peak located at around 532.3 eV denoted as adsorption oxygen (Oads) was ascribed to the chemisorbed oxygen caused by the adsorption of H2O and CO2 molecules to the surface. The additional peak features at 530.3 eV denoted as lattice oxygen (Olat) was mainly ascribed to Fe3O4 and ceria support.
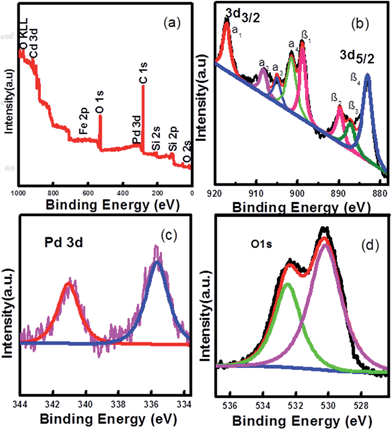 |
| | Fig. 3 Experimental and fixed XPS spectra of Fe3O4@CeO2/Pd catalyst: (a) full spectrum, (b) Ce3d, (c) Pd3d, and (d) O1s. | |
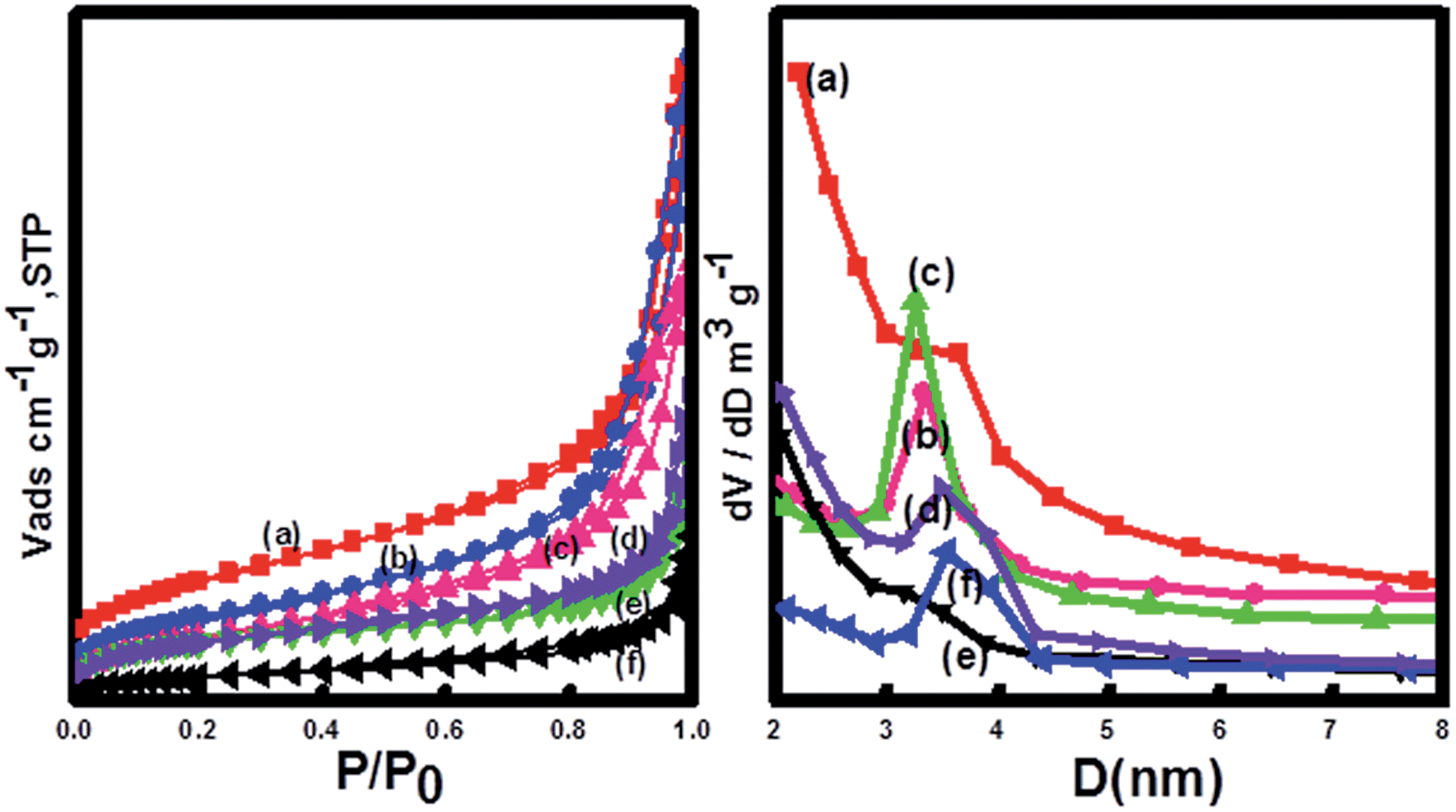
The mesoporous structure was evaluated by measurement of N2 adsorption–desorption isotherms and Brunauer–Emmett–Teller (BET) surface areas (Fig. 4). The results indicated that the adsorption–desorption isotherm of the Fe3O4@CeO2/M catalysts was type IV-like with an apparent H3 hysteresis loop at relatively higher P/P0, indicating the presence of mesoporous structure. The BET surface areas of the Fe3O4@CeO2/Pd, Fe3O4@CeO2/Pt and Fe3O4@CeO2/Pd–Pt catalysts are 96.2 m2 g−1, 81.6 m2 g−1, and 84.7 m2 g−1, respectively.
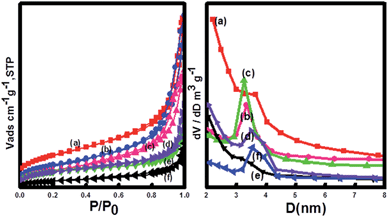 |
| | Fig. 4 N2 adsorption–desorption isotherms and BJH measurements of (a) CeO2, (b) Fe3O4@CeO2, (c) Fe3O4@CeO2/Pd, (d) Fe3O4@CeO2/Pt–Pd, (e) Fe3O4@CeO2/Pt, and (f) Fe3O4. | |
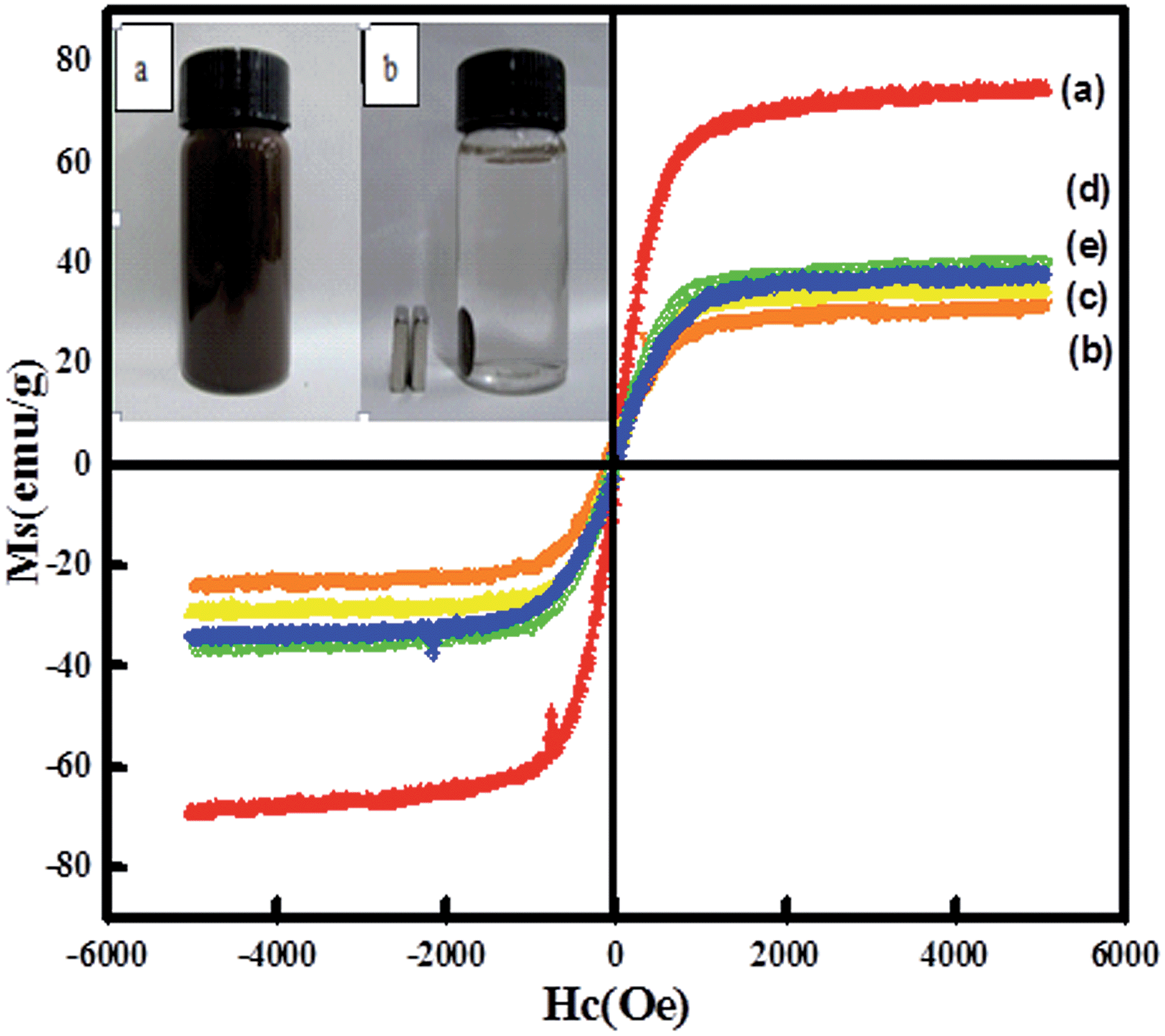
Magnetic properties of the nanocatalysts were investigated via a vibrating sample magnetometer (VSM) at room temperature. Fig. 5 shows the presence of hysteresis loops, revealing a strong magnetism with negligible coercivity and remanence magnetization at room temperature. For Fe3O4, Fe3O4@CeO2, and Fe3O4@CeO2/Pd microspheres, the saturation magnetization (Ms) values were 74.7, 31.3, and 34.2 emu g−1, respectively. The differences in Ms were attributed to the existence of CeO2 and Pd nanoparticles, dual heterostructure and magnetocrystalline anisotropy. Thus, the Fe3O4@CeO2/M can easily disperse in the absence of a magnetic field, and can be rapidly separated from the mixture within 10 s with the help of the magnet (inset in Fig. 5). This excellent magnetic response behavior of Fe3O4@CeO2/M microspheres contributed to rapid and high throughput enrichment and separation.
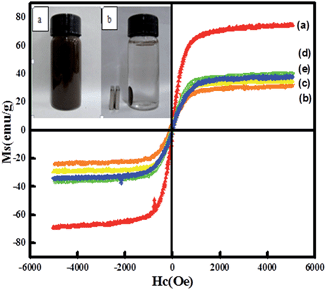 |
| | Fig. 5 Magnetization curves of samples measured at 298 K: (a) Fe3O4, (b) Fe3O4@CeO2, (c) Fe3O4@CeO2/Pd, (d) Fe3O4@CeO2/Pt, (e) Fe3O4@CeO2/Pd–Pt. Photographs (inset) of samples dispersed in aqueous solution with and without an external magnetic field. | |
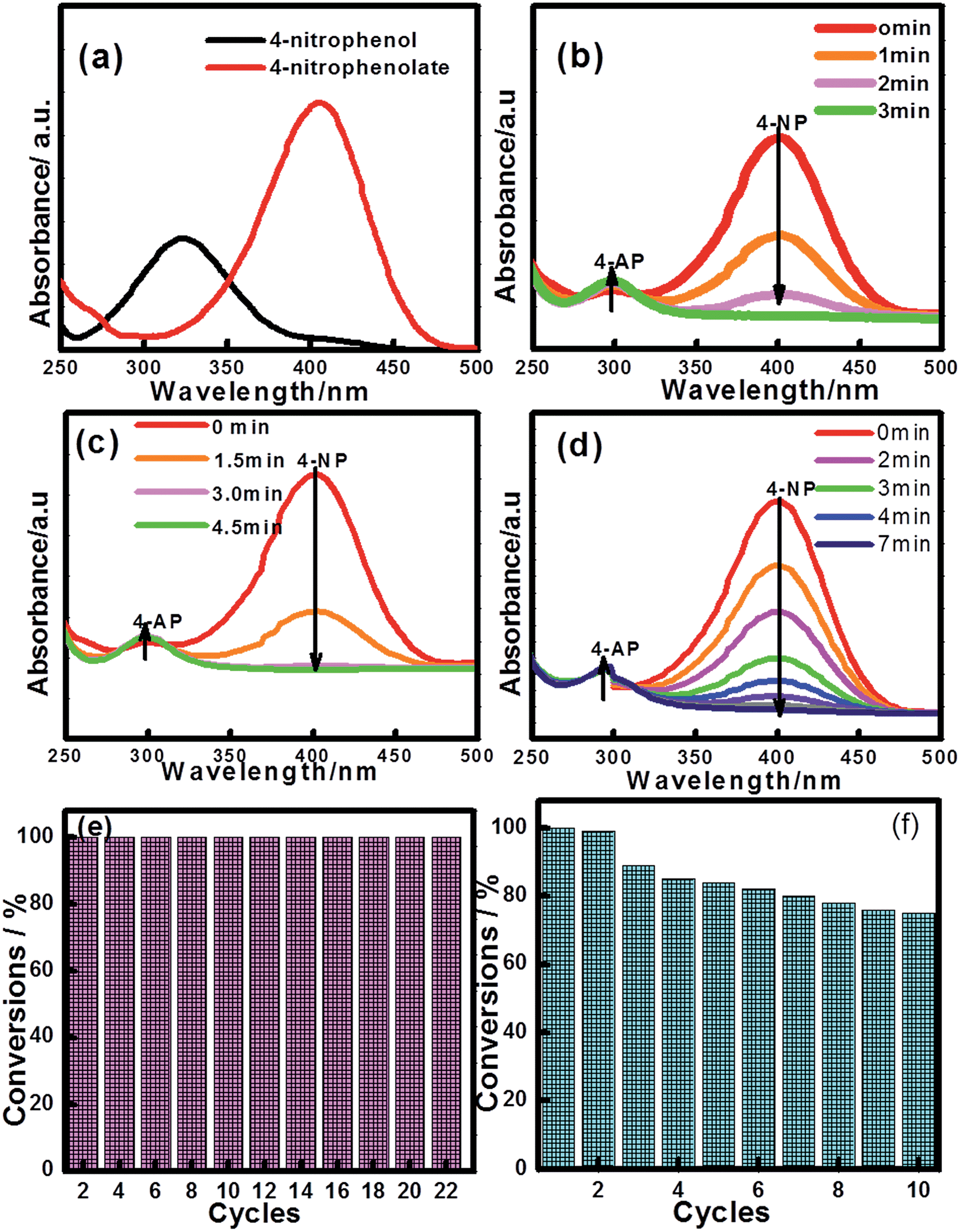
The reduction of 4-NP to 4-AP by NaBH4 over various nanocatalysts was used as a model reaction to evaluate the catalytic activity, and further to study the relationship between the various hybrid structures and their catalytic hydrogenation performance. The evolution process with reaction time for reduction of 4-NP to 4-AP over different catalysts was monitored by UV-vis spectra (Fig. 6). In a typical catalytic reaction, upon the addition the NaBH4, the absorption peak of 4-NP underwent a red shift from 317 nm to 400 nm due to the formation of 4-nitrophenolate (Fig. 6a). After adding 1 mg of catalyst to the system, the reduction of 4-NP was started and the absorption peak at 400 nm gradually decreased in intensity along with the increase of a new absorption peak at about 317 nm, indicating the formation of 4-AP. The results of reduction reaction over the Fe3O4@CeO2/Pd (3 wt%) catalyst indicated that the CeO2–Pd catalytic system exhibited superior catalytic activity and that the 4-NP can be completely reduced into 4-AP within 3 min. The catalytic performance of the Fe3O4@CeO2/Pd catalyst was superior to that of Fe3O4@CeO2/Pt and Fe3O4@CeO2/Pt–Pd catalysts. This result indicated that loading Pd nanoparticles onto Fe3O4@CeO2 nanoparticles can dramatically improve catalytic activity. The superior catalytic activity of Fe3O4@CeO2/Pd catalyst was mainly due to their unique dual heterostructures. First, the mesoporous CeO2 shells coated onto the surface of the magnetite favor the fast diffusion of reactants and the more active sites exposure, which are beneficial for heterogeneous catalysis. Second, the hierarchical structure and synergistic effect between the NMNPs and the CeO2 shells will speed up the rate of charge transfer and effectively accelerate the reduction of 4-NP to 4-AP. Third, a competitive redox reaction between CeO2 and Fe3O4 nanoparticles in BH4− systems, which is thermodynamically favored, can also speed up the rate of charge transfer. And finally, the CeO2 shells can restrict the NMNPs from growing larger during the synthesis and expose more active sites, thus improving the catalytic efficiency.23,24 The possible schematic diagram of the reaction mechanism is shown in Scheme 2. The catalytic performance towards the reduction of 4-nitrophenol over Fe3O4@CeO2/Pd and Fe3O4@CeO2/Pt catalysts are both higher than others samples, suggesting the existence of synergistic catalytic effect between the Pd or Pt NPs and the CeO2 shells. The catalytic stability tests of Fe3O4@CeO2/Pd, Fe3O4@CeO2/Pt microspheres are shown in Fig. 6e and f. The Fe3O4@CeO2/Pd catalyst exhibits relatively stable catalytic performance even after reaction running for more than 20 cycles. And the catalytic activity of Fe3O4@CeO2/Pt decreased by 20% after reaction running for more than 10 cycles. Because the structural features of CeO2 shell encapsulation, thin mesoporous shells on Fe3O4@CeO2/M microspheres favored reactant diffusion and exposure of active sites, leading to improvement of catalytic activity and stability. Notably, the Fe3O4@CeO2/Pd catalyst exhibited stable catalytic performance even after running for more than 20 cycles. This may be due to competitive redox reactions between CeO2 and Fe3O4 nanoparticles in BH4− systems. The standard redox potential of Ce4+/Ce3+ is 1.44 V, while that of Fe3+/Fe2+ is 0.77 V; hence, the transfer of electrons from Fe2+ to Ce4+ is thermodynamically favored. Cerium (4+) is capable of redox cycling in the presence of BH4− and produces Fe2+, behaving similarly to iron in a Fenton-like reaction. The redox reactions follow:
| | | Fe2+ + Ce4+ → Fe3+ + Ce3+ | (2) |
| | | Fe2+ + Ce4+ → Fe3+ + Ce3+ | (4) |
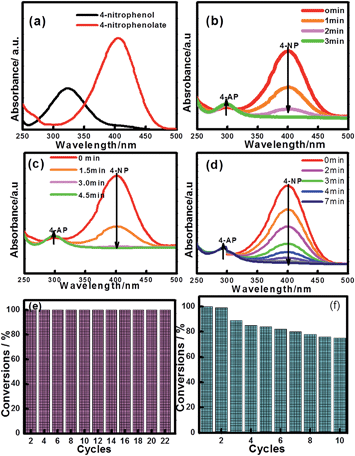 |
| | Fig. 6 UV-vis spectra of 4-nitrophenol before and after adding NaBH4 solution (a), and the successive reduction of 4-NP to 4-AP over various catalysts: (b) Fe3O4@CeO2/Pd (3 wt%), (c) Fe3O4@CeO2/Pt (3 wt%), (d) Fe3O4@CeO2/Pd–Pt (3 wt%); and catalytic stability tests over (e) Fe3O4@CeO2/Pd and (f) Fe3O4@CeO2/Pt catalysts. | |
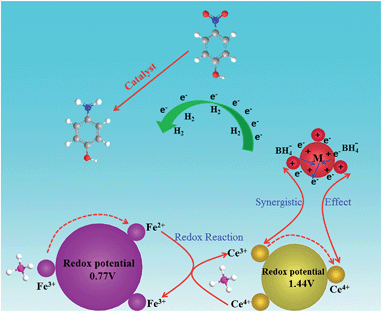 |
| | Scheme 2 Schematic diagram of the reaction mechanism of the Fe3O4@CeO2/M catalysts. | |
Since the amount of NaBH4 was greatly exceeded, the reductions are assumed to be a pseudo-first-order to 4-NP concentration. Therefore, the reaction kinetics can be described as follows:
where
k is the apparent first-order rate constant (min
−1), and
t is the reaction time.
Ct and
C0 are concentrations of the 4-NP at time
t and 0, respectively. As seen from Fig. S2,
† all plots show a straight line, indicating good coincidence with the pseudo-first-order equation. Based on the plots, the reaction rate constant per unit mass
k for Fe
3O
4@CeO
2/Pd (3 wt%) was calculated to be 1.29 min
−1, suggesting that the Fe
3O
4@CeO
2/Pd catalyst possessed greater catalytic activity and efficiency. The actual Pd and Pt contents loaded in various catalysts and corresponding values of specific surface area (
SBET), saturation magnetization (
Ms), and the reaction-rate-constant catalyzed reduction of 4-nitrophenol are shown in Table S1.
†
CeO2 is a wide-band gap insulator that adopts a cubic fluorite structure with lattice parameter, a, of 5.41 Å (Fig. 7a and c). The introduction of Pd2+ ions onto CeO2 surface required the formation of an equivalent number of O vacancies. The Pd atom substituted for a Ce atom in a C2v symmetric surface site, with subsequent charge-compensating vacancy formation, similar to the calculations of Hermansson and co-workers.25,26Fig. 7b and d clearly show that the lattice distortion force existing in CeO2 crystal lattices may have placed the Pd ions in a square planar coordination environment between four lattice oxygen. The Pd–O–Ce superbonds were formed due to chemical adsorption interaction between the adsorption oxygen and Pd and Ce ions.27 The surface side views (Fig. 7e and f) clearly show that the tendency of Pd2+ to form square-planar complexes underpinned the stability of the active Pd–O–Ce surface superstructure. Despite reported improvements in CeO2–Pd catalytic activity, the underlying reasons for superior catalytic performance of the Fe3O4@CeO2/Pd catalyst continue to be examined.
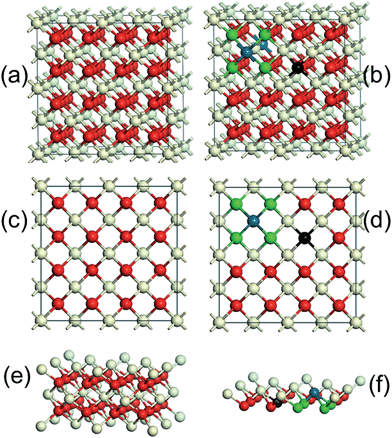 |
| | Fig. 7 Schematic diagram of the CeO2 cubic crystal lattice, (a) and (c). Pd dopant onto CeO2 surface sitting in a square planar coordination between four lattice oxygen, (b) and (d). Side view of the equilibrium surface Ce–O structure, (e). Side view of the equilibrium surface Pd–O–Ce structure (f). Blue spheres denote Pd ions, and grey and red spheres denote Ce and O ions, respectively. Black spheres represent the position of the charge-compensating vacancy, and green spheres represent lattice oxygen. | |
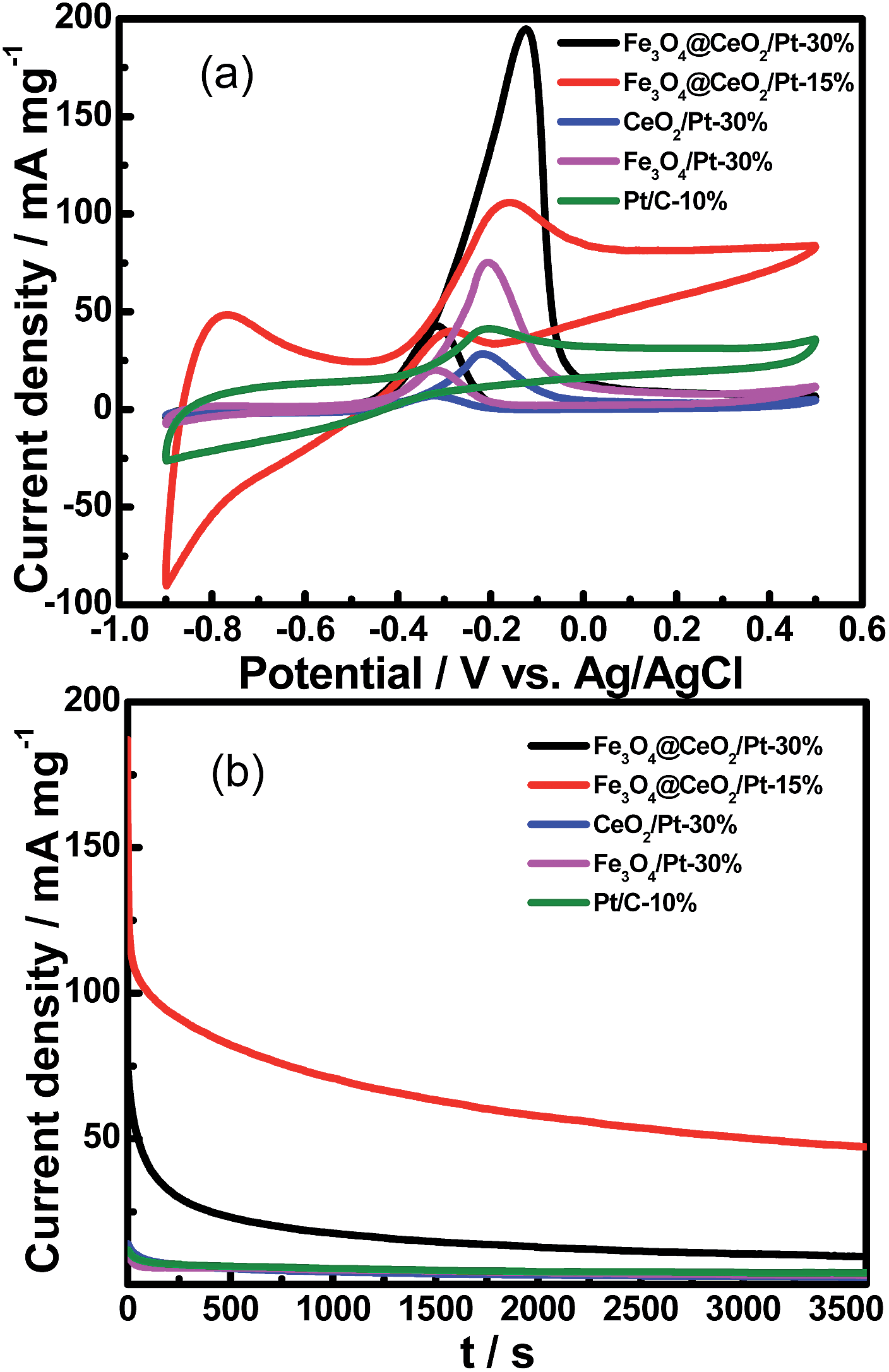
Cyclic voltammetry (CV) and chronoamperometric (CA) were used to evaluate the comparative electrochemical properties of these heterostructure nanocatalysts in alkaline solution. The electrochemical activity was evaluated and normalized to the real surface area per mass Pt. Fig. 8 shows the CVs and CA results of the Fe3O4@CeO2/Pt (10–30 wt%) catalysts. It reveals that the methanol current density (jmassp) of Fe3O4@CeO2/Pt (20 wt%) catalyst (273.0 mA mg−1) is higher than that of Fe3O4@CeO2/Pt (10 wt%) (26.3 mA mg−1) and Fe3O4@CeO2/Pt (30 wt%) (196.4 mA mg−1). This indicates that the electrocatalytic activity of Fe3O4@CeO2/Pt (20 wt%) is obviously higher than that of other catalysts. The higher methanol oxidation current density over Fe3O4@CeO2/Pt nanocatalysts were further confirmed by the CA measurements performed in 0.5 M KOH and 1.0 M methanol for 1 h (Fig. 8B). We can observe that the current intensity decay of the Fe3O4@CeO2/Pt (20 wt%) is much lower than those of the Fe3O4@CeO2/Pt (10 wt%) and Fe3O4@CeO2/Pt (30 wt%) during the entire time range. It reveals that after loading Pt nanoparticles onto the Fe3O4@CeO2 surface, the electrical conductivity of catalysts can be significantly improved due to the good electrical conductivity of Pt and strong synergism interactions between CeO2 and Pt nanoparticles.28–30 And this can be proved by the HR-TEM and XPS results of these catalysts.
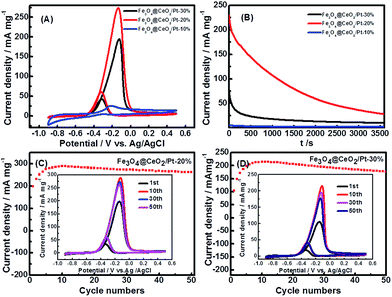 |
| | Fig. 8 (A) CVs of Fe3O4@CeO2/Pt (10 wt%), Fe3O4@CeO2/Pt (20 wt%), Fe3O4@CeO2/Pt (30 wt%) in N2-saturated 0.5 M KOH and 1.0 M methanol (scan rate: 50 mV s−1). (B) Chronoamperometric (CA) results of methanol oxidation for Fe3O4@CeO2/Pt (10 wt%) catalysts in 0.5 M KOH and 1.0 M methanol at constant voltage of −0.20 V for 1 h. CA curves of Fe3O4@CeO2/Pt (20 wt%) and Fe3O4@CeO2/Pt (30 wt%) were recorded at −0.12 V. Plots of the forward peak current densities with the number cycles in 0.5 M KOH and 1.0 M methanol over the (C) Fe3O4@CeO2/Pt (20 wt%) and (D) Fe3O4@CeO2/Pt (30 wt%) catalyst-modified electrodes, respectively. Insets (C and D) are corresponding 1st, 10th, 30th and 50th cyclic voltammograms at a scan rate of 50 mV s−1. | |
Fig. S5† shows the HAADF-STEM images of the Fe3O4@CeO2/Pt (10–30 wt%) catalysts and their corresponding mapping results of Pt nanoparticles. Notably, it can be seen in Fig. S5(b) and (e)† that the Pt NMNPs observed as bright dots are remarkably well-dispersed on the surface of Fe3O4@CeO2 microspheres over the Fe3O4@CeO2/Pt (20 wt%) catalysts. The synergism interactions between CeO2 and Pt nanoparticles became weaker because of the obvious aggregation on the surface of Fe3O4@CeO2 microspheres over the Fe3O4@CeO2/Pt (30 wt%) catalyst. Therefore, the electrocatalytic performance of Fe3O4@CeO2/Pt (20 wt%) is better than the Fe3O4@CeO2/Pt (30 wt%) catalyst. Fig. S6† is the experimental and fixed XPS spectra of the Fe3O4@CeO2/Pt (10–30 wt%) catalysts (Pt4f7/2). The XPS spectra show that the Pt4f7/2 peaks of Fe3O4@CeO2/Pt (30 wt%) are located at 72.1 eV, which are higher compared with 71.9 eV for Fe3O4@CeO2/Pt (20 wt%). A different degree of upshift for the peak positions of Pt is observed, which can be attributed to the change of the electronic structure of Pt nanoparticles (Fig. S6†).31 Furthermore, the ratio of the forward oxidation current density to the backward current density (If/Ib) are 5.40, 4.50, and 4.60 for the Fe3O4@CeO2/Pt (10 wt%), Fe3O4@CeO2/Pt (20 wt%) and Fe3O4@CeO2/Pt (30 wt%) catalyst, respectively, which are higher than the Pt–Pd nanodendrites (3.5), Pt/C (4.60) and Pt black (4.50) catalysts.32
These results indicate that the Fe3O4@CeO2/Pt catalyst system manifests superior anti-poisoning abilities. Actual Pt contents loaded in various catalysts for methanol electro-oxidation and their corresponding CV parameters (peak potential (Ep), mass-normalized current densities (jgeomp and jmassp)), electrochemically active surface area (EASA), and the ratio of the forward oxidation current density to the backward current density (If/Ib) are shown in Table 1.
Table 1 Actual Pd and Pt contents loaded in various catalysts for methanol electro-oxidation and corresponding CV parametersa
| Samples |
Actual Pt content (wt%) |
E
p (V) |
EASA (m2 g−1) |
j
geomp (mA cm−2) |
j
massp (mA mg−1) |
I
f/Ib |
|
E
p, peak potential; jgeomp and jmassp, mass-normalized current densities; EASA, electrochemically active surface area; If/Ib, ratio of forward oxidation current density to backward current density.
|
| Fe3O4@CeO2/Pt-10% |
5.0 |
−0.2 |
— |
0.61 |
26.3 |
5.4 |
| Fe3O4@CeO2/Pt-20% |
16.6 |
−0.12 |
61.6 |
21.6 |
273.0 |
4.5 |
| Fe3O4@CeO2/Pt-30% |
18.3 |
−0.12 |
11.4 |
17.4 |
196.4 |
4.6 |
| Fe3O4@CeO2/Pt-15% |
6.7 |
−0.15 |
— |
3.5 |
105.5 |
2.7 |
| Fe3O4@CeO2/Pt–Pd-30% |
12.4 |
−0.2 |
5.1 |
4.2 |
— |
4.3 |
| Fe3O4/Pt-30% |
12.1 |
−0.2 |
4.9 |
4.4 |
75.3 |
3.7 |
| CeO2/Pt-30% |
9.0 |
−0.2 |
— |
1.2 |
29.1 |
3.8 |
| Pt/C |
10.0 |
−0.2 |
— |
2.0 |
42.4 |
4.6 |
Fig. 9 shows the CV curves of various catalysts for methanol electro-oxidation in 0.5 M KOH and 1.0 M methanol at the scan rate of 50 mV s−1. The electrochemical activity was evaluated and normalized to the real surface area per mass Pt. It is clear that the methanol current density of Fe3O4@CeO2/Pt (30 wt%) (196.4 mA mg−1) and Fe3O4@CeO2/Pt (15 wt%) (105.5 mA mg−1) catalysts are all higher than that of Fe3O4/Pt (30 wt%) (75.3 mA mg−1), CeO2/Pt (30 wt%) (29.1 mA mg−1) and commercial Pt/C (10 wt%) catalysts (42.4 mA mg−1). This indicates that the electrocatalytic activities of Fe3O4@CeO2/Pt (15–30 wt%) are obviously higher than that of other catalysts. In addition, the current intensity decay of the Fe3O4@CeO2/Pt (15 wt%) is much lower than those of the Fe3O4/Pt (30 wt%), CeO2/Pt (30 wt%) and commercial Pt/C (30 wt%) during the entire time range.
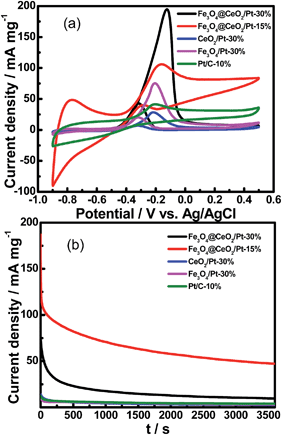 |
| | Fig. 9 (a) CVs of Fe3O4@CeO2/Pt (30 wt%), Fe3O4@CeO2/Pt (15 wt%), Fe3O4/Pt (30 wt%), CeO2/Pt (30 wt%) and commercial Pt/C (10 wt%) in N2-saturated 0.5 M KOH and 1.0 M methanol (scan rate: 50 mV s−1); (b) CA results of methanol oxidation of catalysts in 0.5 M KOH and 1.0 M methanol at constant voltage of −0.20 V for 1 h. | |
Conclusions
In summary, a novel dual-heterostructural nanocatalyst with high catalytic efficiency was fabricated by a self-assembly strategy, which is composed of magnetic Fe3O4 cores and mesoporous CeO2 shells and coated with NMNPs. By rational design of the proper assembled structures, it shows an excellent catalytic performance. These dual-heterostructural features, which favor reactant diffusion and active sites exposure, can dramatically enhance synergistic catalytic effects between the NMNPs and CeO2 nanoparticles. The Fe3O4@CeO2/Pd (3 wt%) nanocatalysts manifest superior catalytic performance compared to other catalysts towards the catalytic reduction of 4-NP to 4-AP. And the Fe3O4@CeO2/Pt (20 wt%) catalyst exhibits superior electrochemical activity towards the oxidation of methanol in alkaline solution. The synthetic strategy may be an effective method for preparation of highly efficient dual-heterostructure mesoporous catalysts. These dual heterostructures will have great applications not only as catalysts but also in environmental and biological chemistry.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21201097, 21467019, 21261011), Key Project of Inner Mongolia National Natural Science Foundation (2010Zd03), Application Program from Inner Mongolia Science and Technology Department (113307), Program for New Century Excellent Talents in University (NCET-10-0907), and Natural Science Foundation of Inner Mongolia Autonomous Region of China (2014MS0506).
Notes and references
- L. Q. Jing, W. Zhou, G. H. Tian and H. G. Fu, Chem. Soc. Rev., 2013, 42, 9509 RSC.
- C. D. Wu, X. H. Liu, D. B. Wei, J. C. Fan and L. S. Wang, Water Res., 2001, 35, 3927 CrossRef CAS.
- B. Iurascu, I. Siminiceanu, D. Vione, M. A. Vicente and A. Gil, Water Res., 2009, 43, 1313 CrossRef CAS.
- T. Zeng, X. L. Zhang, Y. R. Ma, H. Y. Niu and Y. Q. Cai, J. Mater. Chem., 2012, 22, 18658 RSC.
- K. C. Leung, S. H. Xuan, X. M. Zhu, D. W. Wang, C. Chak, S. Lee, W. K.-W. Ho and B. C.-T. Chung, Chem. Soc. Rev., 2012, 41, 1911 RSC.
- L. J. Xu and J. L. Wang, Environ. Sci. Technol., 2012, 46, 10145 CAS.
- X. Wang, D. P. Liu, S. Y. Song and H. J. Zhang, Chem.–Eur. J., 2013, 19, 5169 CrossRef CAS.
- Q. Wang, W. J. Jia, B. C. Liu, A. Dong, X. Gong, C. Y. Li, P. Jing, Y. J. Li, G. R. Xu and J. Zhang, J. Mater. Chem. A, 2013, 1, 12732 CAS.
- R. B. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752 RSC.
- K. Jiang, H. X. Zhang, Y. Y. Yang, R. Mothes, H. Lang and W. B. Cai, Chem. Commun., 2011, 47, 11924 RSC.
- S. K. Li, F. Z. Huang, Y. Wang, Y. H. Shen, L. G. Qiu, A. J. Xie and S. J. Xu, J. Mater. Chem., 2011, 21, 7459 RSC.
- H. F. Li, S. Y. Gao, Z. L. Zheng and R. Cao, Catal. Sci. Technol., 2011, 1, 1194 CAS.
- Z. J. Wu, C. X. Sun, Y. Chai and M. H. Zhang, RSC Adv., 2011, 1, 1179 RSC.
- W. Z. Sun, Q. Li, S. A. Gao and J. K. Shang, J. Mater. Chem. A, 2013, 1, 9215 CAS.
- K. S. Lee, R. M. Anisur, K. W. Kim, W. S. Kim, T. Park, E. J. Kang and I. S. Lee, Chem. Mater., 2012, 24, 682 CrossRef CAS.
- A. J. Amali and R. K. Rana, Green Chem., 2009, 11, 1781 RSC.
- C. Lian, H. Q. Liu, C. Xiao, W. Yang, K. Zhang, Y. Liu and Y. Wang, Chem. Commun., 2012, 48, 3124 RSC.
- S. Tsunekawa, K. Ishikawa, Z.-Q. Li, Y. Kawazoe and A. Kasuya, Phys. Rev. Lett., 2000, 85, 3440 CrossRef CAS.
- Y. Y. Chu, Z. B. Wang, Z. Z. Jiang, D. M. Gu and G. P. Yin, Adv. Mater., 2011, 23, 3100 CrossRef CAS PubMed.
- X. Wang, D. P. Liu, S. Y. Song and H. J. Zhang, J. Am. Chem. Soc., 2013, 135, 15864 CrossRef CAS.
- Q. Wang, W. J. Jia, B. C. Liu, W. Z. Zhao, C. Y. Li, J. Zhang and G. Xu, Chem.–Asian J., 2012, 7, 2258 CrossRef CAS.
- Q. Wang, A. B. Wu, L. X. Yu, Z. L. Liu, W. Xu and H. Yang, J. Phys. Chem. C, 2009, 113, 19875 CAS.
- B. C. Liu, S. L. Yu, Q. Wang, W. T. Hu, P. Jing, Y. Liu, W. J. Jia, Y. X. Liu, L. X. Liu and J. Zhang, Chem. Commun., 2013, 49, 3757 RSC.
- B. C. Liu, Q. Wang, S. L. Yu, T. Zhao, J. X. Han, P. Jing, W. T. Hu, L. X. Liu, J. Zhang, L. D. Sun and C. H. Yan, Nanoscale, 2013, 5, 9747 RSC.
- Z. X. Yang, G. X. Luo, Z. S. Lu and K. Hermansson, J. Chem. Phys., 2007, 127, 074704 CrossRef.
- Z. X. Yang, G. X. Luo, Z. S. Lu, T. K. Woo and K. Hermansson, J. Phys.: Condens. Matter, 2008, 20, 035210 CrossRef.
- S. Colussi, A. Gayen, M. F. Camellone, M. Boaro, J. Llorca, S. Fabris and A. Trovarelli, Angew. Chem., 2009, 121, 8633 CrossRef.
- Z. R. Shen, J. Liu, F. Y. Hu, S. Liu, N. Cao, Y. Sui, Q. D. Zeng and Y. T. Shen, CrystEngComm, 2014, 16, 3387 RSC.
- S. K. Meher and G. R. Rao, ACS Catal., 2012, 2, 2795 CrossRef CAS.
- R. F. B. De Souza, A. E. A. Flausino, D. C. Rascio, R. T. S. Oliveira, E. Teixeira Neto, M. L. Calegaro and M. C. Santos, Appl. Catal., B, 2009, 91, 516 CrossRef CAS PubMed.
- X. J. Liu, C. H. Cui, M. Gong, H. H. Li, Y. Xue, F. J. Fan and S. H. Yu, Chem. Commun., 2013, 49, 8704 RSC.
- J. J. Lv, J. N. Zheng, S. S. Li, L. L. Chen, A. J. Wang and J. J. Feng, J. Mater. Chem. A, 2014, 2, 4384 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta05691d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.